Introduction
Pheochromocytoma (PCC) is regarded as a form of
neuroendocrine tumor (NET) (1,2). PCC and
NETs are extremely similar in terms of tumor cell origin, clinical
and pathological features, and malignant potential (1,2). PCC
arises from chromaffin cells in the adrenal medulla and typically
produces catecholamines, resulting in characteristic symptoms,
including hypertension, hyperglycemia and headaches (1). PCC without symptoms or secretion of
catecholamines, which has been generally termed nonfunctional PCC,
is extremely rare (3).
PCC has malignant potential similar to other NETs
(4). Generally, ~10% of all PCC cases
develop metastases in other areas of the body such as lymph node,
bone, liver and lung, and are commonly diagnosed as malignant at
the time that metastases appear (1,4). It is
difficult to diagnose malignant PCC prior to the appearance of
metastases due to the similarity of pathological features between
benign and malignant PCC (5,6). Thus, the diagnosis of malignancy and
classification of PCC have remained under discussion.
The present case described a 72-year-old man with
locally advanced, carcinoma-like, nonfunctional PCC of the adrenal
gland, which appeared to be more similar to a neuroendocrine
carcinoma (NEC) than a malignant PCC. The current study also
discussed the malignant potential of PCC and NETs, and examined the
potential application of the gastroenteropancreatic NET (GEP-NET)
classification to the adrenal gland in evaluation of
malignancy.
Case report
A 72-year-old man, who was followed up for
non-muscle-invasive bladder cancer [urothelial carcinoma, grade
3>2, pT1, according to World Health Organization (WHO) 1973
classification system (7)] with no
recurrence or metastasis for 2 years, was referred to Saitama Red
Cross Hospital (Saitama, Japan) in August 2012, presenting with a
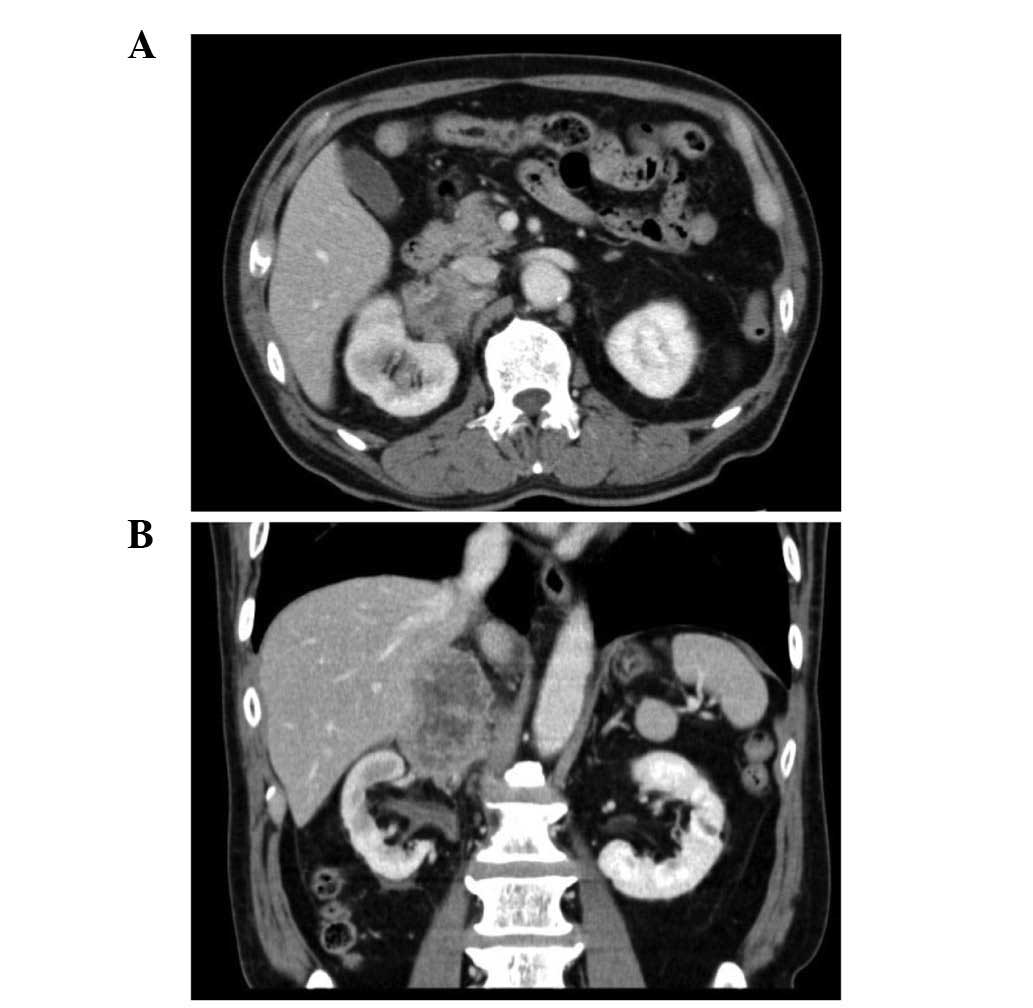
2-month history of right flank pain. A computed tomography scan
(Aquilion; Toshiba Medical Systems Co., Ltd., Otawara, Japan)
revealed a right adrenal tumor, measuring 6.0 cm in diameter, with
an irregular and partially nodular outline. The tumor extended to
the inferior vena cava (IVC), hilum of the right kidney and liver
(Fig. 1). Iodine-123
metaiodobenzylguanidine (MIBG) scintigraphy results were negative,
and all serum adrenal hormone levels were within normal limits;
however, a slightly elevated level of the tumor marker
neuron-specific γ-enolase (25.7 ng/ml; normal range, <10 ng/ml)
was observed. These imaging and laboratory findings suggested the
presence of adrenal gland cancer without metastasis. Radical
surgery was performed with en bloc resection of the tumor by
removing the right kidney, and adjacent areas of the liver and IVC.
The specimen was fixed with 20% formalin and embedded in paraffin.
Sections (3-µm thick) were cut and stained with hematoxylin and
eosin. Immunohistochemistry was performed using the
avidin-biotin-peroxidase-complex method with antibodies against
cytokeratin (CK) AE1/AE3 (mouse monoclonal antibody, diluted 1:1;
Nichirei Biosciences Inc., Tokyo, Japan), chromogranin A (rabbit
polyclonal antibody, diluted 1:1; Nichirei Biosciences Inc.),
synaptophysin (mouse monoclonal antibody, diluted 1:1; catalogue
number 27G12; Nichirei Biosciences Inc.), cluster of
differentiation (CD) 56 (mouse monoclonal antibody, diluted 1:1;
catalogue number 1B6; Nichirei Biosciences Inc.) and Ki-67 (rabbit
polyclonal antibody, diluted 1:1; catalogue number SP6; Nichirei
Biosciences Inc.). Pathological examination revealed that the tumor
cells were arranged in cord-like and alveolar patterns, and
exhibited a high nucleus-to-cytoplasm ratio with abundant nuclear
chromatin. Immunohistochemical staining was positive for CKAE1/AE3,
chromogranin A, synaptophysin, CD56 and Ki-67. The mitotic count
was 27/10 high-power fields (HPFs), and the Ki-67 index was 80%
(Fig. 2). The pathological diagnosis
was confirmed as carcinoma-like PCC or NEC of the right adrenal
gland. The tumor also invaded the IVC and metastasized to the lymph
nodes surrounding the IVC. Adjuvant chemotherapy for this type of
tumor is not known to be effective (8); therefore, the patient was observed
postoperatively and experienced no recurrence after 9 months with
follow-ups every 3 months. Informed consent was obtained from the
patient for publication of the present case report.
Discussion
NETs may appear in a number of locations in the
body, and are commonly observed in the lungs, pancreas and
gastrointestinal tract (9). NETs
originate from neuroendocrine cells, and the overproduction of
hormones within the tumor results in characteristic symptoms, which
are dependent on the unique effect of each hormone (2). NETs have been considered as malignant
tumors as they are able to metastasize long after treatment
(10). However, a number of different
organ-specific diagnostic systems have been developed for each NET
origin; therefore, the staging and grading of NETs remains unclear
(11).
PCC also has specific grading systems for
malignancy. The diagnosis of malignant PCC prior to the appearance
of metastases is considered difficult due to the histological
similarities with benign PCC (5,6). There are
certain grading systems, including Pheochromocytoma of the Adrenal
Scaled Score (5) and Grading system
for Adrenal Pheochromocytoma and Paraganglioma (6), used to characterize the malignancy of
PCC (4,5). Generally, scoring systems are very
useful for risk assessment, particularly for predicting the
probability of recurrence or metastasis. However, they are a little
cumbersome as diagnostic tools to discriminate malignancy from
benign tumor in a clinical setting, since they require 12 or 6
pathological factors for their individual scoring systems.
Furthermore, these scoring systems fully depend on the pathological
findings, and this is the cause of variance in judging each feature
among pathologists (12). Therefore,
they have not yet had a significant impact on changes in
pathological diagnosis and follow-up management. Previous studies
have suggested that the Ki-67 index may be valuable in the
diagnosis of malignant PCC (13–15). The
classification using the Ki-67 index alone may be considered as
simple and easy to apply in the clinical setting. However, the
majority of malignant PCCs exhibit lower Ki-67 index than general
malignant tumors, and setting a clear cut-off value within a narrow
range (<5%) appears to be difficult (16). The cut-off value of Ki-67 index
remains controversial. Therefore, a Ki-67 index-based
classification of PCC would require careful consideration prior to
application.
NEC is a rare form of NET with a high grade of
malignancy (17). However, the
definition of NEC remains unclear due to its rarity and diversity
(17). NECs include a number of
different tumors that originate in various organs and occasionally
co-exist with another malignancy such as adenocarcinoma or
urothelial carcinoma in the same specimen, which makes it difficult
to clearly categorize (18). However,
to the best of our knowledge, the majority of NEC cases are small
cell carcinoma (SCC), and ~95% of all SCCs originate from the lungs
(19). Therefore, extrapulmonary NECs
are extremely rare.
In 2010, the WHO advocated a simple and high-impact
classification of GEP-NETs (17). The
classification system categorized GEP-NET into three groups
according to mitotic count and Ki-67 index as follows: ii) NET
grade 1 (low-grade malignancy); ii) NET grade 2 (intermediate-grade
malignancy); and iii) NEC (high-grade malignancy, described as
carcinoma) (17). NEC is defined by a
mitotic count of >20/10 HPFs or a Ki-67 index of >20%
(17). This classification system is
simple and has been proven useful for predicting survival;
therefore, it has been gradually accepted in pathology, digestive
internal medicine and surgery (20).
NETs in various organs share a number of common pathological
features; therefore, whether the GEP-NET classification can be
applied to other organs is now under careful consideration
(12).
The present case was considered to be more similar
to NEC of the adrenal gland than malignant PCC. Although the tumor
localization and the immunohistochemical staining indicated that
this tumor had arisen from the adrenal medulla, the tumor lacked
several clinical features of PCC, such as an accumulation on
iodine-123 MIBG scintigraphy and overproduction of catecholamines.
Scintigraphy using iodine-123 MIBG, which mimics noradrenaline, is
generally able to detect PCC (1), but
the current case demonstrated negative results. Nonfunctional PCC,
which refers to non-secreting PCC in the present study, is known to
exist, but is extremely rare (3).
Nonfunctional PCC, which may be associated with succinate
dehydrogenase subunit B mutations (3), has not yet been clearly defined. Certain
authors understand that nonfunctional PCC means “PCC without any
symptoms”, regardless of the overexpression of catecholamine, and
it may be occasionally described as asymptomatic PCC or unsuspected
PCC. Furthermore, a number of oncocytic PCCs may be regarded as
nonfunctional PCC, and nonfunctional adenomas that are not resected
or biopsied may contain nonfunctional PCCs. Therefore, it is not
possible to clearly state the definite number of cases of
“nonfunctional (non-secreting) PCC”, and only a few studies have
reported cases of nonfunctional or non-secreting PCC (21–23).
Altogether, nonfunctional PCC itself is extremely rare, and
malignant change or anomaly of nonfunctional PCC is no longer
inconceivable; therefore, the present case may not be classified as
malignant PCC.
NEC may also be diagnosed following identification
of distinctive immunohistochemical staining, including chromogranin
A, synaptophysin and CD56. NEC is reportedly strongly positive for
CKs such as CK18, AE1/AE3 and CAM 5.2, while PCC is typically
negative (24,25). In the present case, all CK and
neuroendocrine markers were positive. This pathological finding
provided support for the diagnosis of NEC, and the tumor in the
present case appears to be different from the malignant PCC
generally observed among 10% of all PCCs. Furthermore, the present
case of carcinoma-like PCC may be regarded as a type of SCC of the
adrenal gland. However, SCC may account for a large number of NEC
cases. Therefore, the tumor in the present case was preferably
described as NEC of the adrenal gland, since PCC is a form of
NET.
To the best of our knowledge, only 2 cases of NEC of
the adrenal gland have been reported (26,27). If
the GEP-NET classification system is applied to adrenal medulla
tumors, then PCC would be equivalent to NET grade 1, malignant PCC
would be equivalent to NET grade 2 and carcinoma-like tumors (as in
the current case) would be equivalent to NEC. The versatility of
this NET classification requires further examination and
clarification in future investigations.
In conclusion, the current study presented an
extremely rare case of carcinoma-like, nonfunctional PCC, which may
have been considered as more similar to NEC of the adrenal gland.
To the best of our knowledge, there are currently no reports
clearly indicating that NECs are the same as carcinoma-like PCC.
However, small cell carcinoma may account for a large number of NEC
cases, and carcinoma-like PCC may be a form of small cell carcinoma
of the adrenal gland; this appears different from the malignant PCC
generally observed among 10% of all PCCs. Therefore, the tumor in
the present case was diagnosed as a carcinoma-like PCC, but was
preferably described as NEC of the adrenal gland. Such cases are
extremely rare, and conclusive evidence was not available in the
current case; however, it is considered that these findings will
contribute to further investigation of malignancies in the adrenal
gland.
References
|
1
|
Strosberg JR: Update on the management of
unusual neuroendocrine tumors: Pheochromocytoma and paraganglioma,
medullary thyroid cancer and adrenocortical carcinoma. Semin Oncol.
40:120–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kulke MH, Shah MH, Benson AB III,
Bergsland E, Berlin JD, Blaszkowsky LS, Emerson L, Engstrom PF,
Engstrom PF, Fanta P, et al: National comprehensive cancer network:
Neuroendocrine tumors, version 1.2015. J Natl Compr Canc Netw.
13:78–108. 2015.PubMed/NCBI
|
|
3
|
Mannelli M, Lenders JW, Pacak K, Parenti G
and Eisenhofer G: Subclinical phaeochromocytoma. Best Pract Res
Clin Endocrinol Metab. 26:507–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Korevaar TI and Grossman AB:
Pheochromocytomas and paragangliomas: Assessment of malignant
potential. Endocrine. 40:354–365. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Modlin IM, Oberg K, Chung DC, Jensen RT,
de Herder WW, Thakker RV, Caplin M, Delle Fave G, Kaltsas GA,
Krenning EP, et al: Gastroenteropancreatic neuroendocrine tumours.
Lancet Oncol. 9:61–72. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thompson LD: Pheochromocytoma of the
Adrenal gland Scaled Score (PASS) to separate benign from malignant
neoplasms: A clinicopathologic and immunophenotypic study of 100
cases. Am J Surg Pathol. 26:551–566. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mostofi FK, Sobin LH and Torloni H:
Histological typing of urinary bladder tumours. International
Histological Classification of Tumours. 10:World Health
Organization. (Geneva). 1973.
|
|
8
|
Strosberg JR, Coppola D, Klimstra DS, Phan
AT, Kulke MH, Wiseman GA and Kvols LK: North American
Neuroendocrine Tumor Society (NANETS): The NANETS consensus
guidelines for the diagnosis and management of poorly
differentiated (high-grade) extrapulmonary neuroendocrine
carcinomas. Pancreas. 39:799–800. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kimura N, Watanabe T, Noshiro T, Shizawa S
and Miura Y: Histological grading of adrenal and extra-adrenal
pheochromocytomas and relationship to prognosis: A
clinicopathological analysis of 116 adrenal pheochromocytomas and
30 extra-adrenal sympathetic paragangliomas including 38 malignant
tumors. Endocr Pathol. 16:23–32. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Massironi S, Sciola V, Peracchi M,
Ciafardini C, Spampatti MP and Conte D: Neuroendocrine tumors of
the gastro-entero-pancreatic system. World J Gastroenterol.
14:5377–5384. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klimstra DS, Modlin IR, Coppola D, Lloyd
RV and Suster S: The pathologic classification of neuroendocrine
tumors: A review of nomenclature, grading, and staging systems.
Pancreas. 39:707–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu D, Tischler AS, Lloyd RV, DeLellis RA,
de Krijger R, van Nederveen F and Nosé V: Observer variation in the
application of the Pheochromocytoma of the Adrenal Gland Scaled
Score. Am J Surg Pathol. 33:599–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clarke MR, Weyant RJ, Watson CG and Carty
SE: Prognostic markers in pheochromocytoma. Hum Pathol. 29:522–526.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van der Harst E, Bruining HA, Jaap Bonjer
H, van der Ham F, Dinjens WN, Lamberts SW, de Herder WW, Koper JW,
Stijnen T, Proye C, et al: Proliferative index in
phaeochromocytomas: Does it predict the occurrence of metastases? J
Pathol. 191:175–180. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boltze C, Mundschenk J, Unger N,
Schneider-Stock R, Peters B, Mawrin C, Hoang-Vu C, Roessner A and
Lehnert H: Expression profile of the telomeric complex
discriminates between benign and malignant pheochromocytoma. J Clin
Endocrinol Metab. 88:4280–4286. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Watanabe M: Malignant pheochromocytoma -
Difficulties for diagnosis of malignancy. Official Journal of the
Japan Association of Endocrine Surgeons and the Japanese Society of
Thyroid Surgery. 30:41–44. 2013.(In Japanese).
|
|
17
|
Chang K, Dai B, Kong YY, Qu YY, Gan HL, Gu
WJ, Ye DW, Zhang HL, Zhu Y and Shi GH: Genitourinary small-cell
carcinoma: 11-year treatment experience. Asian J Androl.
16:705–709. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Richardson RL and Weiland LH:
Undifferentiated small cell carcinomas in extrapulmonary sites.
Semin Oncol. 9:484–496. 1982.PubMed/NCBI
|
|
19
|
Bosman FT, Carneiro F, Hruban RH and
Theise ND: WHO Classification of Tumours of the Digestive System.
IARC Press. Lyon: 2010.
|
|
20
|
Jann H, Roll S, Couvelard A, Hentic O,
Pavel M, Müller-Nordhorn J, Koch M, Röcken C, Rindi G, Ruszniewski
P, et al: Neuroendocrine tumors of midgut and hindgut origin:
Tumor-node-metastasis classification determines clinical outcome.
Cancer. 117:3332–3341. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gong J, Wang X, Chen X, Chen N, Huang R,
Lu C, Chen D, Zeng H and Zhou Q: Adrenal and extra-adrenal
nonfunctioning composite pheochromocytoma/paraganglioma with
immunohistochemical ectopic hormone expression: Comparison of two
cases. Urol Int. 85:368–372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maurea S, Lastoria S, Cuocolo A, Celentano
L and Salvatore M: The diagnosis of nonfunctioning
pheochromocytoma. The role of I-123 MIBG imaging. Clin Nucl Med.
20:22–24. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mannelli M, Pupilli C, Lanzillotti R,
Ianni L, Amorosi A, Credi G and Pratesi C: A nonsecreting
pheochromocytoma presenting as an incidental adrenal mass. Report
on a case. J Endocrinol Invest. 16:817–822. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gould VE, Wiedenmann B, Lee I,
Schwechheimer K, Dockhorn-Dworniczak B, Radosevich JA, Moll R and
Franke WW: Synaptophysin expression in neuroendocrine neoplasms as
determined by immunocytochemistry. Am J Pathol. 126:243–257.
1987.PubMed/NCBI
|
|
25
|
Chetty R, Pillay P and Jaichand V:
Cytokeratin expression in adrenal phaeochromocytomas and
extra-adrenal paragangliomas. J Clin Pathol. 51:477–478. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ochiai T, Komiyama S, Ikoma H, Kubota T,
Nakanishi M, Ichikawa D, Kikuchi S, Fujiwara H, Sakakura C, Kokuba
Y, et al: A case report of metastatic neuroendocrine carcinoma of
the right adrenal gland successfully treated with chemotherapy and
surgery. Int J Clin Oncol. 15:423–427. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Juarez D, Brown RW, Ostrowski M, Reardon
MJ, Lechago J and Truong LD: Pheochromocytoma associated with
neuroendocrine carcinoma. A new type of composite pheochromocytoma.
Arch Pathol Lab Med. 123:1274–1279. 1999.PubMed/NCBI
|