Introduction
Primary intracranial chondrosarcoma is an extremely
rare malignant tumor of the central nervous system, which accounts
for <0.16% of all primary intracranial tumors (1). The tumor most commonly arises from the
skull base, however, cases originating from the choroid plexus,
dura matter and brain parenchyma have also been reported (2). As a result of their slow growth rate,
intracranial chondrosarcomas do not usually metastasize until the
very late stages of the disease. The symptoms vary among patients,
although a long-standing history of headaches and signs associated
with intracranial pressure are the main symptoms. Furthermore,
dizziness, tinnitus, sensory disturbances of the face, and
decreased visual acuity have been reported in some cases (3).
Cranial computed tomography (CT) and magnetic
resonance imaging (MRI) can aid the diagnosis of these tumors,
although a clinical pathological diagnosis is the gold-standard.
Radical excision is the standard treatment for intracranial
chondrosarcoma, and postoperative adjuvant radiotherapy is the
preferred treatment for remnants of the lesion (4). The prognosis of patients with
intracranial chondrosarcoma is strongly influenced by a number of
factors, including the use of postoperative adjuvant radiotherapy,
pathological patterns, previous treatment (surgery or radiotherapy)
and the extent of tumor removal. The overall 5-year mortality rate
among patients in a previous study was 11%, with an average
survival time of 53.7 months (5).
Histologically, three variants of chondrosarcoma
have been defined: Myxoid, mesenchymal and classic chondrosarcoma.
In this study, the case of a patient with low-grade, classic
intracranial chondrosarcoma in the left frontoparietal region,
which was misdiagnosed as meningioma preoperatively, is
presented.
Case report
A 40-year-old male patient was admitted to The
Department of Neurology, Tianjin Huanhu Hospital (Tianjin, China)
on February 24, 2014 with a progressive headache and dizziness that
had lasted for 2 years. Neurological examinations (including for
hemiparesis and sensory disturbance) were normal and pathological
signs (such as the Babinski sign) were negative. All laboratory
tests (including routine blood count tests and assessments of liver
and kidney, immune and blood coagulation functions) were within the
normal ranges. The patient had no history of trauma and no family
history of any hereditary illness. Cranial CT and an MRI scan
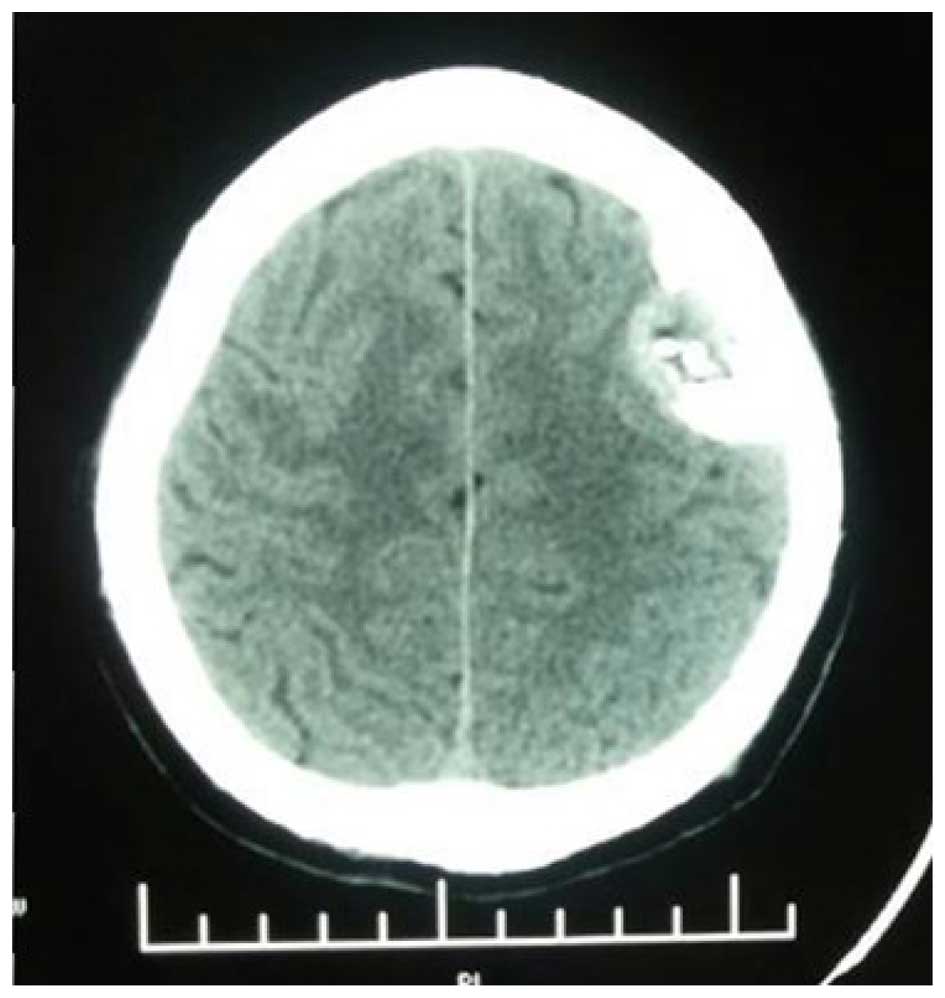
revealed a mass in the left frontal region. CT scan revealed an
iso-/hyperdense mass with unclear boundaries located in the left
frontoparietal region (Fig. 1).
Multiple patchy calcification shadows were present at the margins
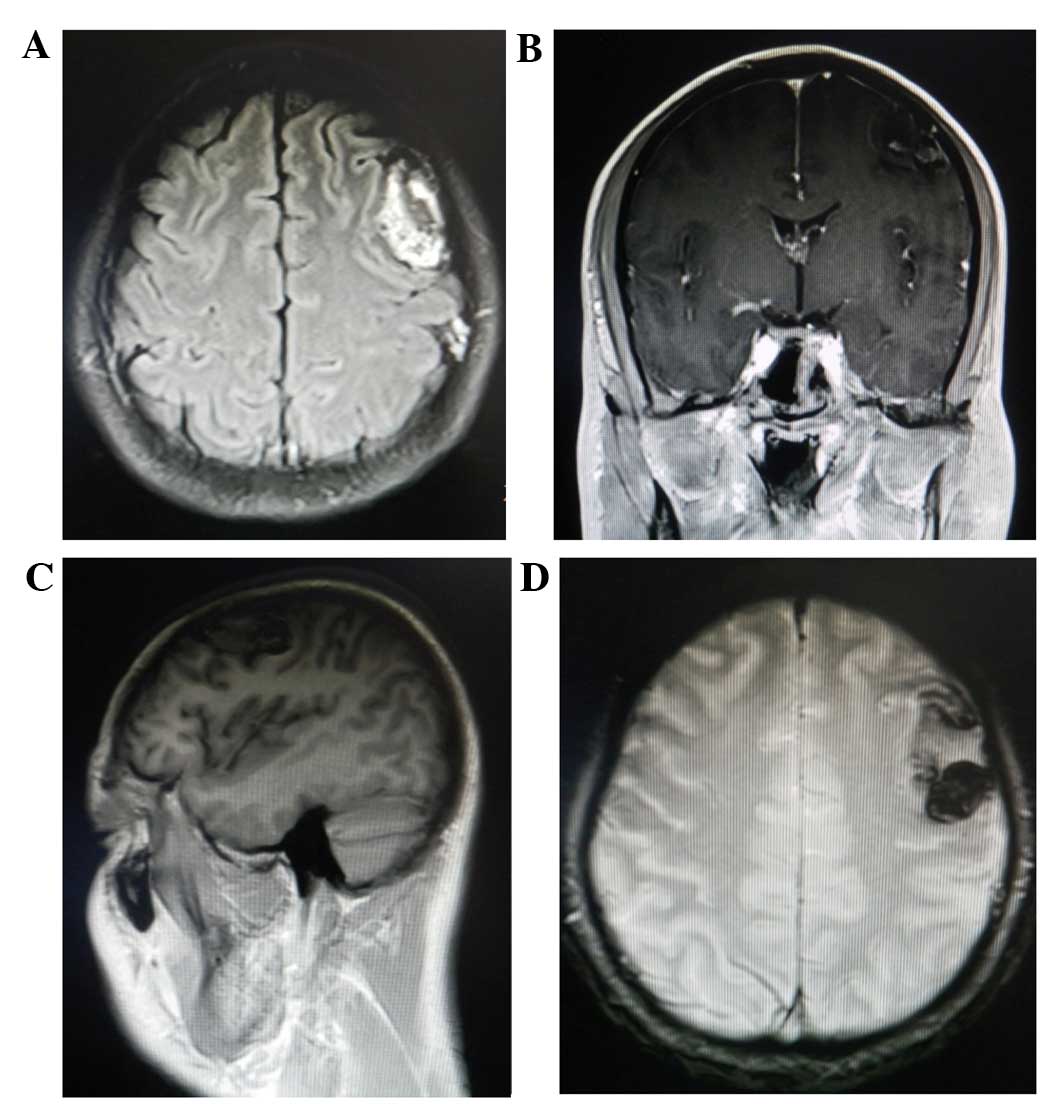
of the mass. Volumetric contrast-enhanced MRI revealed a
heterogeneous extra-axial mass with clear margins (Fig. 2). The superficial margin of the mass
was broad and based along the inner table of the skull; the center
of the mass exhibited hyperostosis. The lesion exhibited a signal
intensity that was higher than the grey matter on T1-weighted
images and T2-weighted images. Based on the results of preoperative
imaging, left frontal meningioma was suspected. The patient
underwent tumor resection using the left frontal approach on
February 28, 2014. Grossly, the tumor was attached to the tentorium
and adjacent dura and was brown in color, hypervascularized and
hard with clear boundaries. A dural incision was made along the
tumor-brain interface and the involved dura and tumor were
completely removed. Analysis of a frozen tumor biopsy showed that
the tumor tissues were lobulated, contained blood vessels and
exhibited nuclear enlargement, which indicated meningioma.
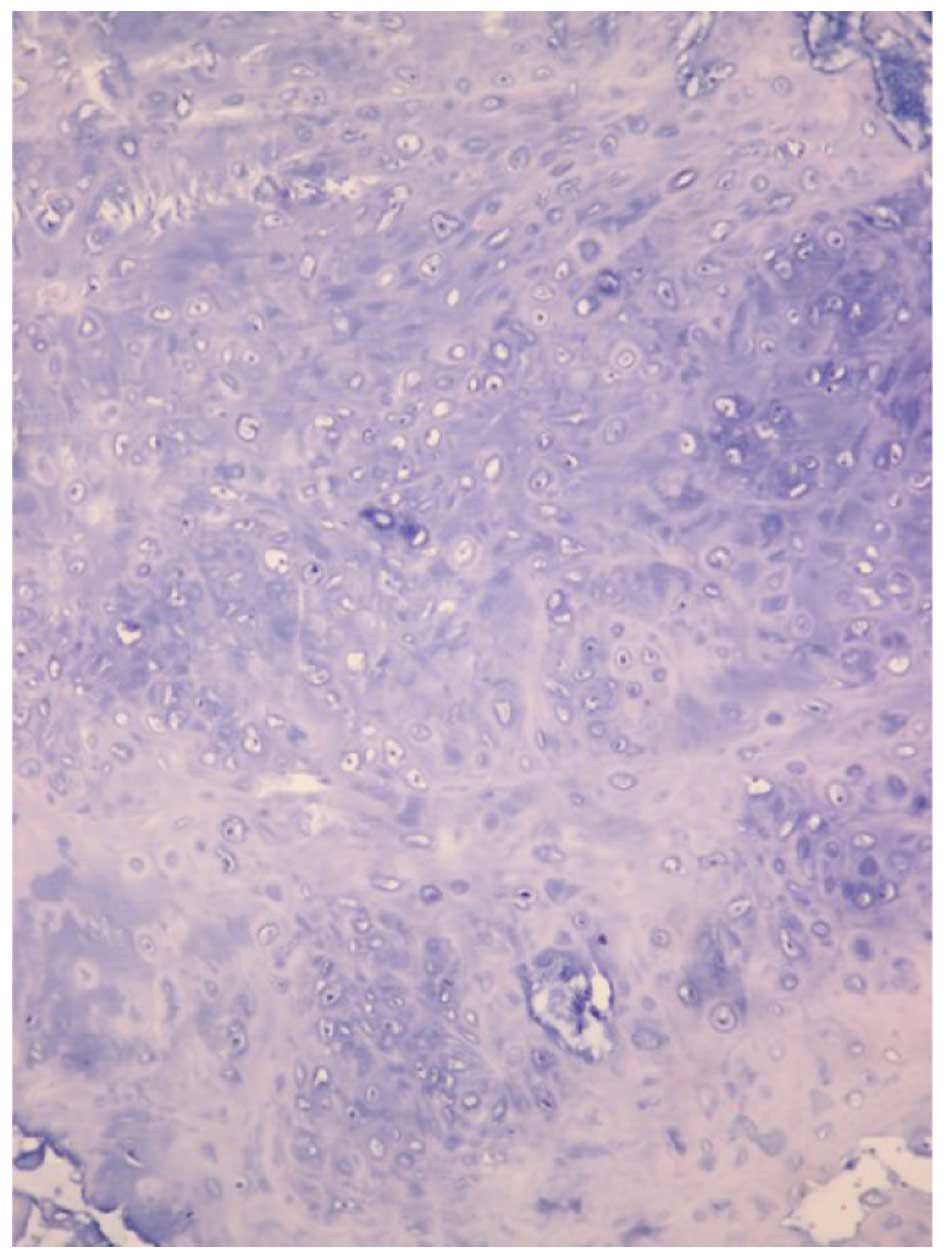
On histopathological evaluation, the mass was
composed of undifferentiated round or spindle-shaped cells and
mature cartilaginous tissue (Fig. 3).
Immunohistochemical examination revealed that the
well-differentiated round cells were positive for vimentin
(anti-vimentin antibody; cat. no. ab52942; 1:500; Abcam, Cambridge,
UK) and S100 (anti-S100 antibody; cat. no. ab66041; 1:500; Abcam),
and only scattered proliferating cells were positive for the
proliferative marker Ki-67 (labeling index, 4.2%; anti-Ki-67
antibody; cat. no. ab833; 1:500; Abcam). Thus, a pathological
diagnosis of classical chondrosarcoma was confirmed. Postoperative
cranial MRI identified no residual tumor (Fig. 2). The patient was discharged on the
10th postoperative day with no neurological deficits.
One month later, the patient underwent adjuvant
γ-knife treatment (total dose, 60 Gy; 2 Gy once a day for 6 weeks).
Follow-up MRI scans were taken at 1, 3 and 6 months after
radiosurgery. A follow-up serial MRI performed 9 months
postoperatively revealed no evidence of recurrence. The patient is
currently alive and shows no obvious symptoms. Informed consent was
obtained from the patient for the publication of this study.
Discussion
Intracranial chondrosarcoma, a subtype of
chondrosarcoma, is a rare malignant cartilaginous tumor that was
first reported by Mott in 1899 (6).
Intracranial chondrosarcoma typically affects patients in the
fourth and fifth decades of life, with no gender preference
(7). The clinical presentation of
chondrosarcomas has been extensively reported in the literature
(8,9).
Generally, patients present with an extensive history of headaches
and symptoms associated with increased intracranial pressure.
Histologically, intracranial chondrosarcomas are classified into
three subtypes: Well-differentiated (classical type), intermediate
(myxoid type) and undifferentiated (mesenchymal type) (10,11). In a
review of 192 chondrosarcoma cases by Chandler et al
(12), 62% were of the classical
subtype, while the mesenchymal and myxoid types accounted for 30
and 8% of cases, respectively. Korten et al (8) reviewed 192 cases of chondrosarcoma and
reported that in general, the mesenchymal type is malignant and
occasionally spreads to distant areas, while the classical subtype
is the most benign of the three subtypes. In this study, a case of
classical type intracranial chondrosarcoma that occurred in the
left frontal region of the skull was presented, which has rarely
been reported in the literature to date. Generally, classical
chondrosarcomas occur in the base of the cranium and affect
patients between the fourth and sixth decades of life (3). In the review of chondrosarcomas by
Korten et al (8), 37% of
tumors were located in the petrous bone, while 23% occurred in the
occipital bone and clivus, 20% in the sphenoid bone and 14% in the
frontal, ethmoidal and parietal bones; the remaining 6% were in
dural tissue, which does not typically contain cartilage.
CT scans usually reveal an isodense/hyperdense mass
with heterogeneous enhancement and varying degrees of calcification
in patients with chondrosarcomas (3,13). MRI
usually reveals a hypointense mass on T1-weighted images and an
extremely hyperintense mass on T2-weighted images (1,14).
On immunological examination, chondrosarcomas
usually exhibit significant positivity for S100 and vimentin, while
only scattered proliferating cells exhibit positivity for the
proliferative marker, Ki-67. These features differentiate
chondrosarcoma from meningioma, hemangiopericytoma, metastasis and
vascular malformations.
Radical excision is the standard treatment for
intracranial chondrosarcoma. In addition, postoperative adjuvant
radiotherapy has been reported to improve patient outcomes for
intracranial chondrosarcoma (15).
Due to the invasive nature of chondrosarcoma, adjuvant radiotherapy
may be recommended even after successful radical resection
(4). According to a previous study,
the 5-year recurrence rate for chondrosarcoma patients treated with
surgery alone is 44%, which is markedly reduced to 9% following the
addition of adjuvant radiation therapy (16). Combined surgical and postoperative
proton radiation therapy has also demonstrated promising results
with regard to tumor control (17).
In the present case, the tumor was located at the surface of the
left frontal region, and exhibited good dissection margins from the
surrounding tissue. In the present case, the patient was treated
with adjuvant radiotherapy following surgery, and subsequently
exhibited a favorable prognosis after total resection.
In conclusion, intracranial chondrosarcoma is a rare
malignant cartilaginous tumor that generally arises from the base
of the skull. Due to its rarity and similar imaging findings with
meningioma, a differential diagnosis is often challenging.
Pathological diagnosis is the gold standard and neurosurgical
resection is the mainstay of therapy, although, as a result of its
high propensity for recurrence, radiotherapy is often necessary.
According to this rare case of a low-grade, classic intracranial
chondrosarcoma, the present study has provided a more objective
protocol for clinicians managing these patients.
References
|
1
|
Bingaman KD, Alleyne CJ Jr and Olson JJ:
Intracranial extraskeletal mesenchymal chondrosarcoma: Case report.
Neurosurgery. 46:207–212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Güneş M, Günaldi O, Tuğcu B, Tanriverdi O,
Güler AK and Cöllüoğlu B: Intracranial chondrosarcoma: A case
report and review of the literature. Minim Invasive Neurosurg.
52:238–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gay E, Sekhar LN, Rubinstein E, Wright DC,
Sen C, Janecka IP and Snyderman CH: Chordomas and chondrosarcomas
of the cranial base: Results and follow-up of 60 patients.
Neurosurgery. 36:887–897. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bosma JJ, Kirollos RW, Broome J and
Eldridge PR: Primary intradural classic chondrosarcoma: Case report
and literature review. Neurosurgery. 48:420–423. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bloch OG, Jian BJ, Yang I, Han SJ, Aranda
D, Ahn BJ and Parsa AT: A systematic review of intracranial
chondrosarcoma and survival. J Clin Neurosci. 16:1547–1551. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mott FW: Chondrosarcoma springing from the
sella turcica. Arch Neurol Psychiat. 1:432–433. 1899.
|
|
7
|
Parker JR, Zarabi MC and Parker JC Jr:
Intracerebral mesenchymal chondrosarcoma. Ann Clin Lab Sci.
19:401–407. 1989.PubMed/NCBI
|
|
8
|
Korten AG, ter Berg HJ, Spincemaille GH,
van der Laan RT and Van de Wel AM: Intracranial chondrosarcoma:
Review of the literature and report of 15 cases. J Neurol Neurosurg
Psychiatry. 65:88–92. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Currie J, Lubin JH and Lesseil S: Chronic
isolated abducens paresis from tumors at the base of the brain.
Arch Neurol. 40:226–229. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wanebo JE, Bristol RE, Porter RR, Coons SW
and Spetzler RF: Management of cranial base chondrosarcomas.
Neurosurgery. 58:249–255. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Neff B, Sataloff RT, Storey L, Hawkshaw M
and Spiegel JR: Chondrosarcoma of the skull base. Laryngoscope.
112:134–139. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chandler JP, Yashar P, Laskin WB and
Russell EJ: Intracranial chondrosarcoma: A case report and review
of the literature. J Neurooncol. 68:33–39. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim MJ, Cho KJ, Ayala AG and Ro JY:
Chondrosarcoma: With updates on molecular genetics. Sarcoma.
2011:4054372011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Salcman M, Scholtz H, Kristt D and
Numaguchi Y: Extraskeletal myxoid chondrosarcoma of the falx.
Neurosurgery. 31:344–348. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park JH, Kim MJ, Kim CJ and Kim JH:
Intracranial extraskeletal myxoid chondrosarcoma: Case report and
literature review. J Korean Neurosurg Soc. 52:246–249. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bloch OG, Jian BJ, Yang I, Han SJ, Aranda
D, Ahn BJ and Parsa AT: Cranial chondrosarcoma and recurrence.
Skull Base. 20:149–156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amichetti M, Amelio D, Cianchetti M,
Enrici RM and Minniti G: A systematic review of proton therapy in
the treatment of chondrosarcoma of the skull base. Neurosurg Rev.
33:155–165. 2010. View Article : Google Scholar : PubMed/NCBI
|