Introduction
Nuclear factor κB (NF-κB) proteins comprise a family
of transcription factors that includes RelA (also known as p65),
RelB, c-Rel, p50 (which originates from the p105 precursor) and p52
(which originates from the p100 precursor) (1,2). NF-κB
transcription factors have essential roles in numerous
physiological and pathological processes, which have been
extensively reviewed (3–5). NF-κB members form homodimeric and
heterodimeric complexes that are involved in signaling through
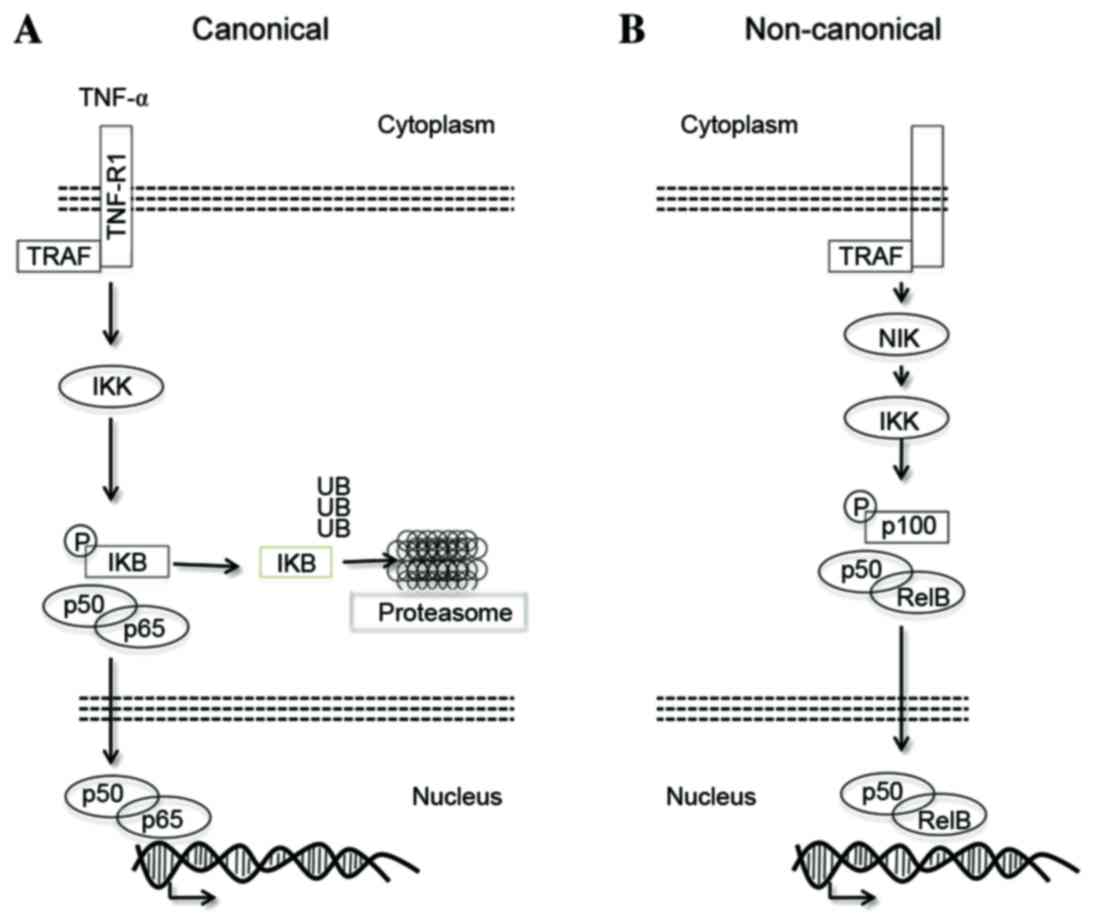
canonical and non-canonical pathways (Fig. 1). In particular, the NF-κB canonical
pathway is based on the p65/p50 dimer, which is physiologically
trapped in the cytoplasm by the inhibitor protein NFKB inhibitor α
(IκB-α) (Fig. 1A) (6). Various stimuli, including inflammatory
cytokines [e.g. tumor necrosis factor α (TNF-α)], radiation, stress
signals and cell surface receptors, induce the activation of the
IκB-kinase (IKK) complex, which is in turn able to promote the
serine phosphorylation of IκB-α (7).
This event primes IκB-α for proteasomal degradation (8) and, as a consequence, free NF-κB dimers
are allowed to migrate into the nucleus, where they regulate gene
expression, leading to cellular proliferation and resistance to
apoptosis (1). Notably, one of the
first genes to be transcribed is NFΚBIA, the gene that
encodes IκB-α, allowing the generation of new IκB-α protein in
order to terminate NF-κB signaling through its removal from the
nucleus (9,10).
The non-canonical NF-κB activation pathway promotes
the activation of the p50 and p52 NF-κB subunits (Fig. 1B). This pathway, also known as the
alternative pathway, requires the activation of NF-κB-inducing
kinase, which is able to promote IKK-α-mediated p100 degradation
(11). Similarly to IκB-α, p100
prevents the translocation of RelB/p50 and RelB/p52 into the
nucleus.
These two pathways are involved in numerous
biological and pathological processes, ranging from immunological
responses to cancer pathogenesis. Due to the key role of NF-κB in
such processes, NF-κB signaling has been extensively investigated
from a therapeutic standpoint (12).
In addition to the development of several selective inhibitors of
the kinases involved in NF-κB signaling (e.g. IKK) (13), the most relevant and clinically used
therapeutic approach is to prevent the degradation of IκB-α through
proteasome inhibitors (14). In
particular, bortezomib is routinely used for the treatment of
multiple myeloma and other cancers, due to its ability to prevent
IκB-α degradation and therefore block NF-κB signaling (15,16).
Clinical experiences with proteasome inhibitors have demonstrated
that IκB proteins act as essential mediators of NF-κB signaling;
however, the true contribution of IκB proteins to cancer
pathogenesis is far from being exhaustively investigated.
IκB proteins
IκB proteins comprise a family of proteins that
includes the typical IκBs expressed in the cytoplasm, which undergo
stimuli-dependent phosphorylation, degradation and re-synthesis;
the atypical IκBs (such as B-cell CLL/lymphoma 3), which are barely
expressed in basal conditions, and are upregulated upon stimulation
and translocated into the nucleus; and the precursor proteins p100
and p105 (7). The typical IκBs
include IκB-α, IκB-β and IκB-ε, and are characterized by the
presence of six ankyrin repeats (7,17). It has
been generally reported that IκB proteins mask the nuclear
localization signal of NF-κB (18,19).
However, a lack of IκBs only partially promotes the translocation
of p65 into the nucleus, due to the compensation of other IκB
proteins, suggesting that IκBs are primarily involved in the
regulation and inhibition of basal NF-κB activation (20,21). IκB-α
is a 37-kDa protein, which is phosphorylated in response to
Toll-like receptor, TNF-α, interleukin 1 and other stimulatory
signals, and is predominantly able to bind to heterodimers
containing p50, p65 and c-Rel. IκB-α is the product of the
NFΚBIA gene at chromosome 14 (22).
IκB-α-knockout murine models
IκB-α-knockout mice are apparently normal at birth,
but inevitably die within few days due to skin defects and
extensive granulopoiesis. The bone marrow cells exhibit increased
NF-κB activity, and the concurrent deletion of p50 partially
rescues the phenotype (20). Similar
data have been observed in a different knockout model (21). In particular, IκB-α homozygous null
mice are normal at birth but die by day 10 due to the development
of dry, flaky skin. In this model, the authors observed an increase
in myelopoiesis in the spleen, which was considered to be of
reactive origin (21).
IκB-α involvement in cancer from a genetic
perspective
The role of IκB-α in murine models suggests that
IκB-α acts as a tumor suppressor. In line with these phenotypes,
IκB-α has been suggested to act as a tumor suppressor in various
types of cancer. In particular, ~25% of glioblastomas harbor
heterozygous NFΚBIA gene deletions at chromosome 14q13
(23,24). Notably, the loss of NFΚBIA has
also been associated with shortened patient survival time; patients
with NFKBIA loss had outcomes similar to those observed in
patients with EGFR amplification, which are poorer than those
observed in patients with normal levels of NFKBIA and EGFR,
suggesting that NFKBIA deletion is a prognostic factor in
glioblastoma (23). In Hodgkin
lymphoma, NFKBIA was found to be mutated in rare cases,
indicating a role of NFΚBIA as a tumor suppressor in this
lymphoid cancer (25). In 16% of lung
cancer specimens, immunohistochemical analysis revealed an absence
of IκB-α protein expression. In particular, NFΚBIA appeared
to be silenced in cases with wild-type epidermal growth factor
receptor/wild-type RAS and in the absence of echinoderm
microtubule-associated protein-like 4/anaplastic lymphoma kinase
gene rearrangement (26). However,
the NFΚBIA gene has not been reported to be mutated/deleted
in lung cancer.
NF-κB/IκB-α pathway in cancer
Aside from the genetic inactivation of IκB-α in some
cases of Hodgkin lymphoma and glioblastoma, IκB-α has not been
found to be recurrently mutated or deleted in other cancer types.
However, NF-κB/IκB-α signaling has been shown to play essential
roles in various types of hematological cancers, which have been
extensively reviewed (27). In
particular, NF-κB was demonstrated to have a key role in acute
myeloid leukemia (AML), via canonical (28,29) and
non-canonical pathways (30), and via
different modalities in chronic myeloid leukemia (CML) (31). In particular, during the chronic phase
of CML, NF-κB was found to be active, although not as much as in
AML blasts; in the blast phase of CML, NF-κB activity appeared to
substantially increase, as reviewed recently (31). More recently, the IκB-α kinase, IKK-α,
was revealed to be essential in CML pathogenesis, suggesting that
IκB-α may also play a key role in this disease (32). The fact that NFKBIA is
occasionally mutated/deleted in various cancer types, and in
particular in AML/CML, while IκB-α has an essential therapeutic
role when stabilized by proteasome inhibitors, suggests that IκB-α
may have NF-κB-independent functions in the context of cancer.
IκB-α expression pattern in CML
While investigating the level of expression of IκB-α
in CML samples, we discovered that, surprisingly, IκB-α is highly
expressed in primary CML cells and that it retained a marked
cytosolic compartmentalization (33).
This observation prompted us to investigate other IκB-α functions
in this particular cancer. CML is a BCR-ABL-driven
myeloproliferative disorder (34,35), which
is effectively treated with BCR-ABL inhibitors, although tyrosine
kinase inhibitors are unable to fully eradicate this disease
(36). In the search for
NF-κB-independent IκB-α functions, previous studies clearly
demonstrated that IκB-α is able to bind with either the p65 subunit
of NF-κB, as is well known, or p53 (37–40).
However, while these findings were described by independent groups,
the IκB-α/p53 interaction has not been observed and analyzed in the
setting of primary cancer cells. As reported in our recent study,
in CML, BCR-ABL is able to interact with IκB-α, which is in turn
able to bind to p53 (33). This
complex forces p53 to become localized in the cytoplasm, with
consequent loss of the nuclear pool; this is responsible for the
majority of p53 tumor-suppressive functions. Notably, inactivation
of BCR-ABL is associated with the re-localization of p53 into the
nucleus. Finally, the study also demonstrated that IκB-α promotes
the inactivation of p53 as a transcriptional factor in the setting
of CML (33).
Discussion
IκB-α is well-established to negatively regulate the
NF-κB canonical pathway through its ability to prevent p50/p65
translocation into the nucleus (1,2). However,
IκB-α is also able to constrain p53 to the cytoplasm, thereby
counteracting the tumor-suppressive functions of p53 (33,37–40). IκB-α
appears to act as a traffic light at the crossroads between
oncogenes and tumor suppressors. Further investigations are
necessary to better understand how, when and in which cellular
context IκB-α is able to modulate these two opposing signals. At
least in CML, where oncogenic signals are constitutively active due
to the presence of BCR-ABL, IκB-α appears to be stably associated
with p53, promoting its inactivation as a transcriptional factor,
due to its re-localization to the cytoplasm (33). In our opinion, the IκB-α-mediated
functional inactivation of p53 is a challenging opportunity for
cancer therapy, particularly for cancers without effective
molecularly targeted therapies. The concept of functional
inactivation of wild-type tumor suppressors has been extensively
studied for PTEN (41,42), and in particular in CML (43,44). While
genetically mutated/deleted tumor suppressors cannot be
specifically targeted, the identification of functionally
inactivated tumor suppressors may offer the potential to design
therapies that will reactivate them, leading to cancer-selective
induction of apoptosis.
In summary, in addition to promoting the loss of p53
tumor-suppressive functions via changes in cellular
compartmentalization, IκB-α may also orchestrate p53 cytoplasmic
functions. Therefore, further analyses must also investigate the
role of the IκB-α/p53 complex on p53 cytoplasmic functions.
Acknowledgements
The authors would like to thank members of the
Professor G. Saglio laboratory for their assistance and
discussions. This review was supported by the Italian Ministry of
Health, Ricerca Finalizzata-Giovani Ricercatori (project code
#GR-2011-02351167), awarded to Alessandro Morotti.
References
|
1
|
Perkins ND: The diverse and complex roles
of NF-κB subunits in cancer. Nat Rev Cancer. 12:121–132.
2012.PubMed/NCBI
|
|
2
|
Aggarwal BB: Nuclear factor-kappaB: The
enemy within. Cancer Cell. 6:203–208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xia Y, Shen S and Verma IM: NF-κB, an
active player in human cancers. Cancer Immunol Res. 2:823–830.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bradford JW and Baldwin AS: IKK/nuclear
factor-kappaB and oncogenesis: Roles in tumor-initiating cells and
in the tumor microenvironment. Adv Cancer Res. 121:125–145. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chaturvedi MM, Sung B, Yadav VR, Kannappan
R and Aggarwal BB: NF-κB addiction and its role in cancer: ‘One
size does not fit all.’. Oncogene. 30:1615–1630. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hinz M and Scheidereit C: The IκB kinase
complex in NF-κB regulation and beyond. EMBO Rep. 15:46–61. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hinz M, Arslan SÇ and Scheidereit C: It
takes two to tango: IκBs, the multifunctional partners of NF-κB.
Immunol Rev. 246:59–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karin M: How NF-kappaB is activated: The
role of the IkappaB kinase (IKK) complex. Oncogene. 18:6867–6874.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Werner SL, Barken D and Hoffmann A:
Stimulus specificity of gene expression programs determined by
temporal control of IKK activity. Science. 309:1857–1861. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O'Dea EL, Barken D, Peralta RQ, Tran KT,
Werner SL, Kearns JD, Levchenko A and Hoffmann A: A homeostatic
model of IkappaB metabolism to control constitutive NF-kappaB
activity. Mol Syst Biol. 3:1112007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Häcker H and Karin M: Is NF-kappaB2/p100 a
direct activator of programmed cell death? Cancer Cell. 2:431–433.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baud V and Karin M: Is NF-kappaB a good
target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov.
8:33–40. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suzuki J, Ogawa M, Muto S, Itai A, Isobe
M, Hirata Y and Nagai R: Novel IkB kinase inhibitors for treatment
of nuclear factor-kB-related diseases. Expert Opin Investig Drugs.
20:395–405. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sánchez-Serrano I: Success in
translational research: Lessons from the development of bortezomib.
Nat Rev Drug Discov. 5:107–114. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li ZW, Chen H, Campbell RA, Bonavida B and
Berenson JR: NF-kappaB in the pathogenesis and treatment of
multiple myeloma. Curr Opin Hematol. 15:391–399. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gilmore TD: Multiple myeloma: Lusting for
NF-kappaB. Cancer Cell. 12:95–97. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Viatour P, Merville MP, Bours V and
Chariot A: Phosphorylation of NF-kappaB and IkappaB proteins:
Implications in cancer and inflammation. Trends Biochem Sci.
30:43–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Beg AA, Ruben SM, Scheinman RI, Haskill S,
Rosen CA and Baldwin AS Jr: I kappa B interacts with the nuclear
localization sequences of the subunits of NF-kappa B: A mechanism
for cytoplasmic retention. Genes Dev. 6:1899–1913. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ganchi PA, Sun SC, Greene WC and Ballard
DW: I kappa B/MAD-3 masks the nuclear localization signal of
NF-kappa B p65 and requires the transactivation domain to inhibit
NF-kappa B p65 DNA binding. Mol Biol Cell. 3:1339–1352. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Beg AA, Sha WC, Bronson RT and Baltimore
D: Constitutive NF-kappa B activation, enhanced granulopoiesis, and
neonatal lethality in I kappa B alpha-deficient mice. Genes Dev.
9:2736–2746. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klement JF, Rice NR, Car BD, Abbondanzo
SJ, Powers GD, Bhatt PH, Chen CH, Rosen CA and Stewart CL:
IkappaBalpha deficiency results in a sustained NF-kappaB response
and severe widespread dermatitis in mice. Mol Cell Biol.
16:2341–2349. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bredel M, Scholtens DM, Yadav AK, Alvarez
AA, Renfrow JJ, Chandler JP, Yu IL, Carro MS, Dai F, Tagge MJ, et
al: NFKBIA deletion in glioblastomas. N Engl J Med. 364:627–637.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rinkenbaugh AL and Baldwin AS: Monoallelic
deletion of NFKBIA in glioblastoma: When less is more. Cancer Cell.
19:163–165. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jungnickel B, Staratschek-Jox A,
Bräuninger A, Spieker T, Wolf J, Diehl V, Hansmann ML, Rajewsky K
and Küppers R: Clonal deleterious mutations in the IkappaBalpha
gene in the malignant cells in Hodgkin's lymphoma. J Exp Med.
191:395–402. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Furukawa M, Soh J, Yamamoto H, Ichimura K,
Shien K, Maki Y, Muraoka T, Tanaka N, Ueno T, Asano H, et al:
Silenced expression of NFKBIA in lung adenocarcinoma patients with
a never-smoking history. Acta Med Okayama. 67:19–24.
2013.PubMed/NCBI
|
|
27
|
Gasparini C, Celeghini C, Monasta L and
Zauli G: NF-κB pathways in hematological malignancies. Cell Mol
Life Sci. 71:2083–2102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guzman ML, Neering SJ, Upchurch D, Grimes
B, Howard DS, Rizzieri DA, Luger SM and Jordan CT: Nuclear
factor-kappaB is constitutively activated in primitive human acute
myelogenous leukemia cells. Blood. 98:2301–2307. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guzman ML, Swiderski CF, Howard DS, Grimes
BA, Rossi RM, Szilvassy SJ and Jordan CT: Preferential induction of
apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci
USA. 99:16220–16225. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morotti A, Parvis G, Cilloni D, Familiari
U, Pautasso M, Bosa M, Messa F, Arruga F, Defilippi I, Catalano R,
et al: CD7/CD56-positive acute myeloid leukemias are characterized
by constitutive phosphorylation of the NF-kB subunit p65 at Ser536.
Leukemia. 21:1305–1306. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carrà G, Torti D, Crivellaro S, Panuzzo C,
Taulli R, Cilloni D, Guerrasio A, Saglio G and Morotti A: The
BCR-ABL/NF-κB signal transduction network: A long lasting
relationship in Philadelphia positive Leukemias. Oncotarget. Aug
22–2016.(Epub ahead of print). View Article : Google Scholar
|
|
32
|
Hsieh MY and Van Etten RA: IKK-dependent
activation of NF-κB contributes to myeloid and lymphoid
leukemogenesis by BCR-ABL1. Blood. 123:2401–2411. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Crivellaro S, Panuzzo C, Carrà G, Volpengo
A, Crasto F, Gottardi E, Familiari U, Papotti M, Torti D, Piazza R,
et al: Non genomic loss of function of tumor suppressors in CML:
BCR-ABL promotes IκBα mediated p53 nuclear exclusion. Oncotarget.
6:25217–25225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Apperley JF: Chronic myeloid leukaemia.
Lancet. 385:1447–1459. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morotti A, Fava C and Saglio G: Milestones
and monitoring. Curr Hematol Malig Rep. 10:167–172. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Morotti A, Panuzzo C, Fava C and Saglio G:
Kinase-inhibitor-insensitive cancer stem cells in chronic myeloid
leukemia. Expert Opin Biol Ther. 14:287–299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chang NS: The non-ankyrin C terminus of
Ikappa Balpha physically interacts with p53 in vivo and dissociates
in response to apoptotic stress, hypoxia, DNA damage, and
transforming growth factor-beta 1-mediated growth suppression. J
Biol Chem. 277:10323–10331. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dreyfus DH, Nagasawa M, Gelfand EW and
Ghoda LY: Modulation of p53 activity by IkappaBalpha: Evidence
suggesting a common phylogeny between NF-kappaB and p53
transcription factors. BMC Immunol. 6:122005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li X, Xing D, Wang J, Zhu DB, Zhang L,
Chen XJ, Sun FY and Hong A: Effects of IkappaBalpha and its mutants
on NF-kappaB and p53 signaling pathways. World J Gastroenterol.
12:6658–6664. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou M, Gu L, Zhu N, Woods WG and Findley
HW: Transfection of a dominant-negative mutant NF-kB inhibitor
(IkBm) represses p53-dependent apoptosis in acute lymphoblastic
leukemia cells: Interaction of IkBm and p53. Oncogene.
22:8137–8144. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Correia NC, Gírio A, Antunes I, Martins LR
and Barata JT: The multiple layers of non-genetic regulation of
PTEN tumour suppressor activity. Eur J Cancer. 50:216–225. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Leslie NR and Foti M: Non-genomic loss of
PTEN function in cancer: Not in my genes. Trends Pharmacol Sci.
32:131–140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Morotti A, Panuzzo C, Crivellaro S,
Pergolizzi B, Familiari U, Berger AH, Saglio G and Pandolfi PP:
BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through
phosphorylation-dependent activation of HAUSP. Leukemia.
28:1326–1333. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Morotti A, Panuzzo C, Crivellaro S, Carrà
G, Fava C, Guerrasio A, Pandolfi PP and Saglio G: BCR-ABL
inactivates cytosolic PTEN through Casein Kinase II mediated tail
phosphorylation. Cell Cycle. 14:973–979. 2015. View Article : Google Scholar : PubMed/NCBI
|