Introduction
Cervical cancer (CC) is one of the most common types
of carcinoma in females worldwide, and is frequently associated
with high-risk human papillomavirus type 16 (HPV 16) infection
(1–3).
HPV 16 infections are common; however, limited progress has been
made towards the elucidation of the mechanisms underlying the
formation of precursor lesions, cervical intraepithelial neoplasia
grade 2/3 (CIN2/3) and CC itself (4,5).
Malignancy is a rare outcome involving complex interactions between
the host, the environment and the virus (6,7).
Initially, cytology-based screening programs were successful in
reducing the incidence and mortality rates of invasive CC in
developed countries (7–9). Due to the limitations of the screening
programs, including suboptimal sensitivity for CIN2/3 and cancer
and limited uptake, it is necessary to develop a novel screening
method. In recent years, screening through the detection of HPV 16
DNA has emerged as a method of identifying cervical lesions
(10). However, this method is unable
to distinguish between those infections that may progress to cancer
and self-limited infections (9,10).
Quantitative evaluation of HPV DNA methylation may, therefore, be a
simple method of triaging the severity levels of cervical lesions
and overcome the limitations of the current screening programs.
DNA methylation is the process of the addition of a
methyl group to the 5′position of cytosine, which often occurs at
the position of cytosine-phosphate-guanine (CpG) dinucleotides
(11). As a major epigenetic
modification mechanism, DNA methylation is able to regulate gene
expression (12). Hypermethylation of
CpG sites in certain genes has been recognized as a frequent early
event in the pathogenesis of numerous forms of cancer, including CC
(13). The quantification of HPV DNA
methylation may be of value in the early detection of precursors of
certain types of cancer, providing reassurance to HPV-infected
patients whom, to avoid unnecessary overtreatment, are candidates
for less-frequent screening.
The L1 gene is an HPV 16 late gene that encodes the
major capsid protein (3). Previous
studies have demonstrated that methylation is highly prevalent in
the L1 gene of HPV 16, the products of which are not required for
neoplastic processes (11,14,15). As
the primary control region of the virus, the long control region
(LCR) contains numerous regulatory elements that are involved in
viral DNA replication and transcription, including promoters and
enhancers (12,13). A previous study revealed an
association between LCR variation and an increased risk of
developing CIN2/3 in an HPV 16-infected population of female
Chinese patients (16). It has been
predicted that variations in the LCR are able to alter specific
transcription factor binding sites (16). To the best of our knowledge, the
methylation status of HPV 16 in the L1 and the LCR has not yet been
investigated in female patients living in northeastern China. The
current study aimed to examine the methylation status of HPV 16 in
a portion of the L1 gene and the LCR in a collection of clinical
tissue samples, and to determine the association between HPV 16
methylation and the cervical disease grade of infected
patients.
Materials and methods
Samples and DNA preparations
Following routine diagnoses using flow-through
hybridization (Hybribio Ltd, Hong Kong, China), HPV 16-positive
clinical cervical swabs between April 2014 and May 2015 were
obtained from 180 female patients (18–70 years old) admitted to The
Affiliated Shengjing Hospital, China Medical University (Shenyang,
China) for use in the present retrospective study. According to the
histological evaluation of fresh tissue specimens from the female
patients, 122 clinical tissue samples were selected between April
2014 and May 2015, including 42 cases of normal and low-grade
squamous intraepithelial lesion (normal tissue/LSIL), 40 cases of
high-grade squamous intraepithelial lesion (HSIL) and 40 cases of
cervical cancer (CC). A total of 58 samples were not included due
to indefinite diagnosis and require clinical examination).
Liquid-based cytology (LBC) samples were processed according to the
manufacturer's instructions and stained using the Papanicolaou
method. The liquid-based specimens were processed with the Cytyc
T2000 Processor (Cytyc, Inc., Marlborough, MA, USA), and final
cytologic diagnoses were issued using the Bethesda System (17). All cytology testing was performed by 2
independent pathologists who were blinded to the HPV detection
results. The present study was approved by the ethics committee of
the Affiliated Shengjing Hospital.
DNA was extracted and purified from a 200 µl aliquot
of cervical swab suspended in specimen transport medium, using a
QIAamp DNA Mini kit (Qiagen, Inc., Valencia, CA, USA), according to
the manufacturer's protocol. The extracted DNA was suspended in 50
µl buffer AE [10 mM Tris-HCl, 0.5 mM EDTA (pH 9.0)].
Bisulfite modification
For each cervical swab sample, the extracted DNA was
treated with sodium bisulfite using the EZ DNA Methylation-Gold™
kit (Zymo Research, Corp, Irvine, CA, USA), according to the
recommended protocol. Briefly, 200–500 ng of each DNA sample was
mixed with 130 µl CT Conversion Reagent (900 µl water, 300 µl
M-Dilution buffer and 50 µl M-dissolving buffer) and incubated at
98°C for 10 min and 64°C for 2.5 h. Subsequently, the mixture and
600 µl M-Binding Buffer were added to a Zymo-Spin™ IC Column (Zymo
Research). Following centrifugation (1,500 × g at 25°C for 1 min),
washing with 100 ml M-wash buffer and desulfonation with 100 ml
M-desulfonation buffer (incubation for 20 min at 25°C,
centrifugation (1,500 × g at 25°C for 1 min) and washing with 100
ml M-wash buffer), the converted DNA was eluted in 10 µl TE buffer
(10 mM Tris-HCl; 1 mM EDTA; pH 8.0) and the recovered DNA was
stored at −20°C.
Amplification of bisulfite-treated
DNA, TA cloning and sequencing
Certain regions of the HPV 16 genome were analyzed
by amplification of bisulfite-treated DNA, TA cloning and
sequencing, including the 3′end of the L1 gene (3′-L1) and the full
length of the LCR. As sections of the DNA may have been degraded
into small fragments during the bisulfite treatment, the analyzed
DNA segment was divided into three amplicons (14), including the 3′-L1/5′-LCR, enhancer
and promoter regions. A total of three pairs of primers were
designed using the MethPrimer Design program (MethPrimer 2.0; Li
Lab, Beijing, China) (18) and are
listed in Table I.
 | Table I.Polymerase chain reaction primer
sequences. |
Table I.
Polymerase chain reaction primer
sequences.
| Genomic region | Primers | CpG nt
position |
|---|
| 3′-L1 and
5′-LCR | F:
5′-TAGGATTGAAGGTTAAATTAAAATT-3′ | 7,089a, 7,134a, 7,143a, 7,268, |
|
| R:
5′-AACACATTTTATACCAAAAAACA-3′ | 7,426, 7,452,
7,458 |
| Enhancer | F: 5′-
TATGTTTTTTGGTATAAAATGTGTTTTT-3′ | 7,532, 7,550,
7,673, 7,679, |
|
| R:
5′-TAAATTAATTAAAACAAACCAAAAATATAT-3′ | 7,691 |
| Promoter | F:
5′-TTTGTAAAATTGTATATGGGTGT-3′ | 7,859b, 31, 37, 43, 52, 58 |
|
| R:
5′-TCCTAAAACATTACAATTCTCTTT-3′ |
|
Polymerase chain reaction was performed with 1 µl
bisulfite-modified DNA, 10 mM dNTPs, 0.5 mM primers, 50 mM MgCl2,
10X PCR buffer (200 mM Tris-HCl (pH 8.4), 500 mM KCl) and
Platinum® Taq DNA polymerase (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for a total reaction volume of
50 µl. The PCR thermocycling conditions were as follows: 94°C for 9
min, followed by 44 cycles of 94°C for 30 sec, 5°C for 30 sec and
72°C for 45 sec, with a final extension at 72°C for 10 min.
Amplicon presence was confirmed using 1% agarose gel
electrophoresis with ethidium bromide and the DNA samples were
purified using the Wizard® SV Gel and PCR Clean-Up
system (Promega Corporation, Madison, WI, USA). The purified PCR
products were cloned into the pCR™ 2.1 vector using the Original TA
Cloning® kit (Invitrogen; Thermo Fisher Scientific,
Inc.), then conversion into E. coli (DH5α; Tiangen Biotech
Co., Ltd., Beijing, China) and cultivated at 37°C. Strains of E.
coli DH5α containing recombinant plasmid were inoculated into 5
ml LB liquid medium with 100 µg/ml Ampicillin and agitated
overnight at 37°C for 12 h. Subsequently, the plasmid was purified
using Wizard Plus SV Minipreps DNA Purification System (Promega
Corporation). A total of 5 clones of each DNA sample were selected
randomly for determination of methylation at CpG sites. In brief,
each clone selected was propagated for PCR and sequencing. The PCR
reaction was amplified with a pair of primers: Forward,
5′-TGTAAAACGACGGCCAGT-3′ and reverse, 5′-CAGGAAACAGCTATGACC-3′. The
reaction was setup in a total volume of 20 µl containing 10 mM
dNTPs, 0.2 mM primers, 10X PCR (Mg2+ plus) buffer and 0.8 unit Taq
DNA polymerase (Takara Taq™; Takara Bio, Inc., Otsu, Japan).
Amplicon presence was confirmed using 1% agarose gel
electrophoresis with ethidium bromide. The clones that were
successful amplified were used for sequencing (Invitrogen, Beijing,
China), and the results were subsequently analyzed by Bioedit
version 4.8.10 (Department of Microbiology, North Carolina State
University). The DNA samples were considered to be methylated at a
certain CpG site if ≥1 clone exhibited a methylated cytosine at
this site (19). The methylation
frequency of each CpG site in a particular DNA sample was
calculated based on the ratio of methylated clones to the five
detected clones.
Statistical analysis
The differences in the proportion of methylation in
DNA samples from the various patient groups were analyzed using the
χ2 test. Receiver operating characteristic (ROC) analysis was
performed in order to examine the sensitivity and specificity of
the methylation frequency of each CpG site for distinction between
the various types of cervical lesion using the optimal threshold
values and, therefore, determine the most informative methylation
frequency for CpG sites. ROC analyses were performed for three
groups, as follows: CC vs. normal tissue/LSIL and HSIL; CC and HSIL
vs. normal tissue/LSIL; CC vs. HSIL. The significance of each
combination of CpG sites was assessed by comparing the obtained
area under the curve (AUC) values. ROC curves and AUC values were
also calculated for the mean methylation frequency of the
combination of CpG sites. All tests were two-tailed and P<0.05
was considered to indicate a statistically significant
difference.
Results
The clinical DNA specimens were
sequenced effectively
A total of 122 clinical DNA specimens were examined
in the present study. The methylation status of CpG sites in the
3′-L1/5′-LCR was detected in cervical swab samples from 30 patients
with normal tissue/LSIL, 35 patients with HSIL and 35 patients with
CC. DNA specimens from the enhancer region were successfully
analyzed in tissue samples from 30, 31 and 30 patients with normal
tissue/LSIL, HSIL and CC, respectively. In the promoter region, the
methylation status of the CpG sites was effectively analyzed in
tissue samples from 37 patients with normal tissue/LSIL, 35
patients with HSIL and 37 patients with CC.
Methylation proportion observed in HPV
16 positive cervical swab samples
A total of 18 CpG sites were scattered in the
section of the genome studied. Among the 18 CpG sites, 3 were
located in the 3′-L1 at nucleotide (nt) 7,089, 7,134 and 7,143, 4
were located in the 5′-LCR at nt 7,268, 7,426, 7,452 and 7,458 and5
were located in the enhancer region at nt 7,532, 7,550, 7,73, 7,679
and 7,691. In addition, 1 CpG site was located in the replication
origin at nt 7,859, and 5 were identified in the promoter region at
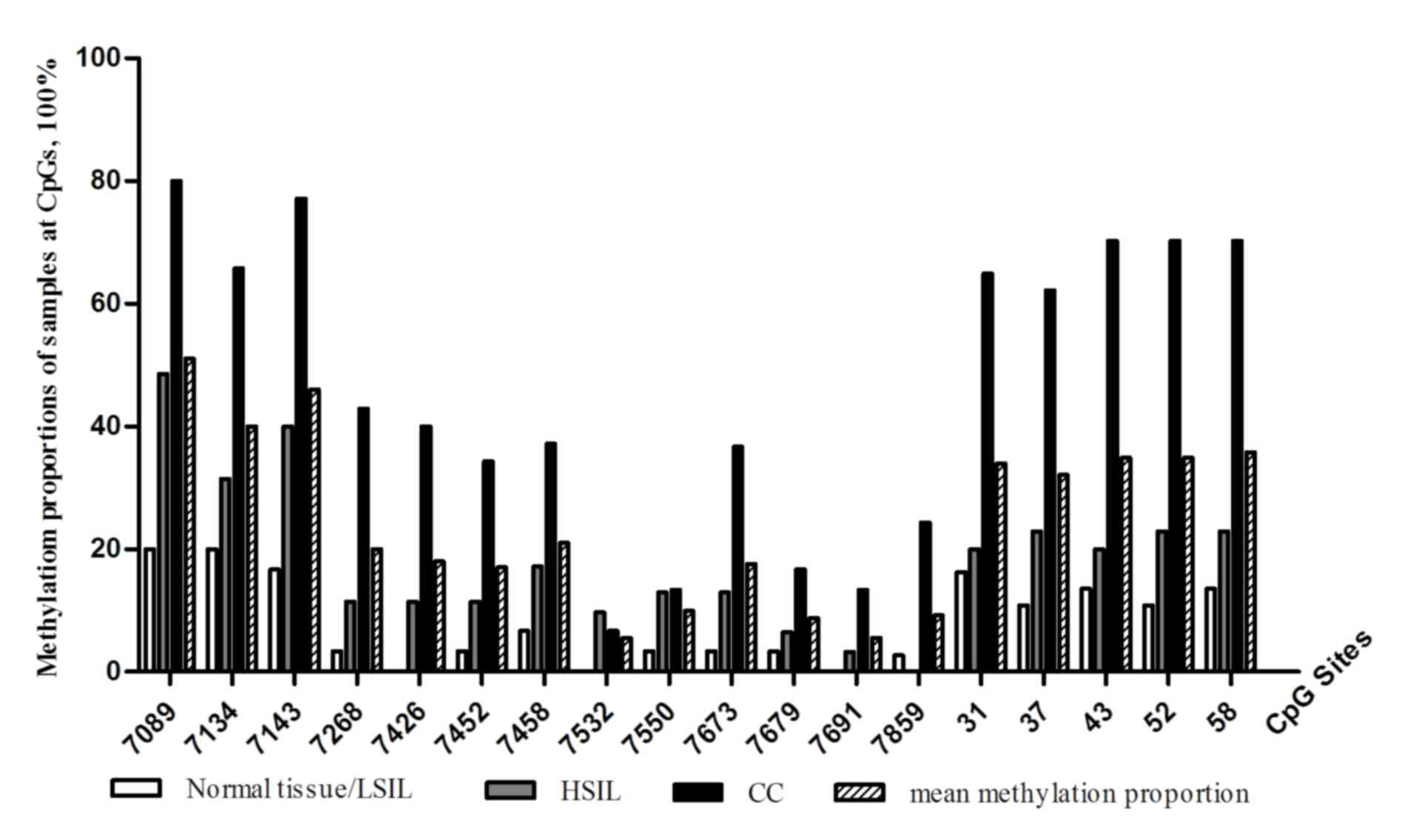
nt 31, 37, 43, 52 and 58. The mean methylation frequency at each
CpG site in DNA samples obtained from the various patient groups is
summarized in Fig. 1. The mean
methylation proportion of the 18 CpG sites of all the analyzed DNA
samples was 25.04% (453 methylated/1,809 analyzed CpG sites). The
mean methylation proportion of the study DNA samples at individual
CpG sites ranged from between 5.49 and 51.00%. Overall, the
methylation proportion of DNA samples at the 3 CpG sites within the
3′-L1 region and the 5 CpG sites in the promoter region was higher,
compared with that observed in the 5′-LCR and enhancer regions.
Association between the methylation
proportion at the studied CpG sites in HPV 16-positive DNA samples
and the severity of cervical lesions in infected female
patients
The mean methylation proportion at the 18 CpG sites
in DNA samples from patients with normal tissue/LSIL, HSI and CC
was 8.42% (49 methylated/582 analyzed CpG sites), 18.36% (112
methylated/610 analyzed CpG sites) and 47.33% (292 methylated/617
analyzed CpG sites), respectively (χ2 test, P<0.001; Table II). The methylation proportion of CpG
sites within the studied regions was highest in cervical swab DNA
samples from patients with CC, and lowest in those from patients
with normal tissue/LSIL (Fig. 1).
| Table II.Number of methylated nucleotides in
DNA samples from patients with various cervical lesions at each CpG
site. |
Table II.
Number of methylated nucleotides in
DNA samples from patients with various cervical lesions at each CpG
site.
| CpGs | 7,089 | 7,134 | 7,143 | 7,268 | 7,426 | 7,452 | 7,458 | 7,532 | 7,550 | 7,673 | 7,679 | 7,691 | 7,859 | 31 | 37 | 43 | 52 | 58 | Total |
|---|
| Normal/LSIL | 6a(30b) | 6 (30) | 5 (30) | 1(30) | 0(30) | 1(30) | 2(30) | 0 (30) | 1 (30) | 1 (30) | 1 (30) | 0 (30) | 1 (37) | 6 (37) | 4 (37) | 5 (37) | 4 (37) | 5 (37) | 49 (582) |
| HSIL | 17 (35) | 11 (35) | 14 (35) | 4 (35) | 4 (35) | 4 (35) | 6 (35) | 3 (31) | 4 (31) | 4 (31) | 2 (31) | 1 (31) | 0 (35) | 7 (35) | 8 (35) | 7 (35) | 8 (35) | 8 (35) | 112 (610) |
| CC | 28 (35) | 23 (35) | 27 (35) | 15 (35) | 14 (35) | 12 (35) | 13 (35) | 2 (30) | 4 (30) | 11 (30) | 5 (30) | 4 (30) | 9 (37) | 24 (37) | 23 (37) | 26 (37) | 26 (37) | 26 (37) | 292 (617) |
| P1 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.002 | 0.009 | 0.238 | 0.339 | 0.002 | 0.161 | 0.061 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| P2 | 0.016 | 0.296 | 0.039 | 0.363 | 0.118 | 0.363 | 0.270 | – | – | 0.354 | – | – | 1.000 | 0.677 | 0.170 | 0.460 | 0.170 | 0.303 | 0.000 |
| P3 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.002 | 0.004 | – | – | 0.001 | – | – | 0.007 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| P4 | 0.006 | 0.004 | 0.002 | 0.003 | 0.006 | 0.023 | 0.060 | – | – | 0.031 | – | – | 0.002 | 0.000 | 0.001 | 0.000 | 0.000 | 0.000 | 0.000 |
The proportion of CpG methylation at nt 7,089,
7,134, 7,143, 7,268, 7,426, 7,452, 7,458, 7,673, 31, 37, 43, 52 and
58 was highest in DNA samples from patients with CC, followed by
those from patients with HSIL, and were lowest in DNA samples from
patients with normal tissue/LSIL (P<0.05; Table II). The proportion of methylation in
DNA samples from patients with normal tissue/LSIL and patients with
CC in the origin of replication, at the CpG site 7,859 was 2.70%
and 24.32%, respectively (P<0.001).
As presented in Table
II, the variation in the methylation proportion between
cervical swab DNA samples from patients with normal tissue/LSIL and
HSIL, normal tissue/LSIL and CC, and HSIL and CC at CpG sites 7,089
and 7,143 was significantly different (P<0.05; Table II). No significant differences in the
methylation proportion were identified between DNA samples from
patients with normal tissue/LSIL and HSIL at the CpG sites 7,134,
7,268, 7,426, 7,452, 7,673, 7,859, 31, 37, 43, 52 and 58; however,
significant differences were observed between patients with normal
tissue/LSIL and patients with CC, HSIL and CC at these sites
(P<0.05; Table II). The
difference in the proportion of methylation at CpG site 7,458 was
significant only between DNA samples from patients with normal
tissue/LSIL and CC (P=0.004; Table
II).
Association between the methylation
frequency of CpG sites 7,089 and 7,143 and the severity of cervical
lesions of infected female patients
In order to elucidate the association between the
methylation frequency of CpG sites 7,089 and 7,143 (individually or
in combination) and the severity of the cervical lesions in
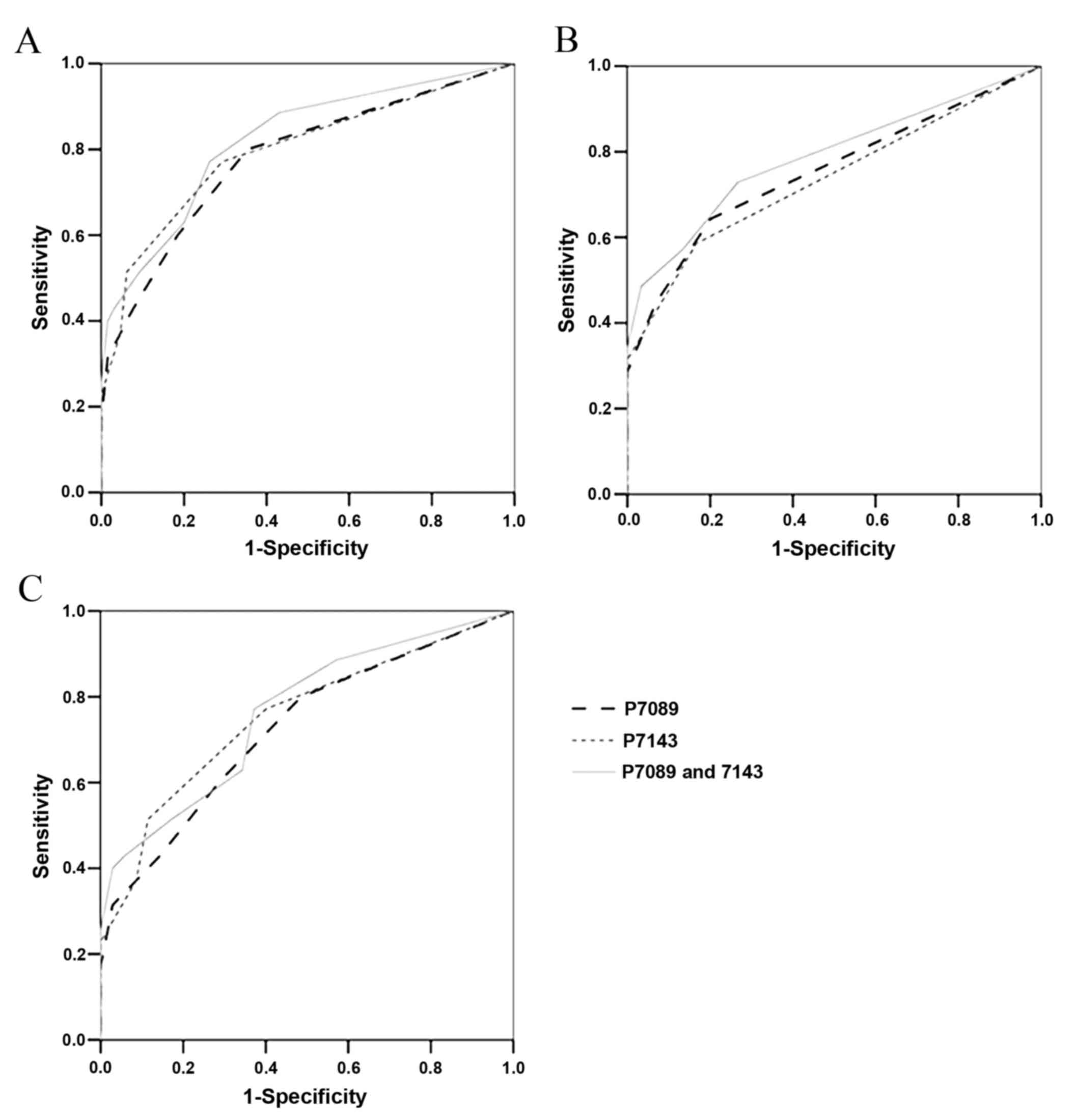
infected patients, ROC analysis was conducted. The results revealed
that the methylation frequencies of the two CpG sites, individually
or in combination, were able to differentiate between cervical swab
DNA patients with CC and samples from patients with normal
tissue/LSIL or HSIL (Fig. 2A). The
AUC values for distinguishing CC from normal tissue/LSIL and HSIL
were 0.782 (95% CI, 0.683–0.881) for the methylation frequency of
CpG site 7,089, and 0.796 (95% CI, 0.697–0.895) for that of CpG
site 7,143 (Table III). The AUCs
values for distinguishing CC and HSIL from normal tissue/LSIL were
0.754 (95% CI, 0.660–0.848) for the methylation frequency of CpG
site 7,089, and 0.736 (95% CI, 0.640–0.832) for that of CpG site
7,143. The AUC values were improved using a combination of the
methylation frequencies of the two CpG sites, with an effective
distinction between CC and normal tissue/LSIL and HSIL (77.1%
sensitivity, 73.8% specificity) obtained using the mean methylation
frequency of CpG sites 7,089 and 7,134 (AUC=0.822; 95% CI,
0.733–0.911) when a 15% methylation frequency was used as a
threshold (Fig. 2A and Table III). CC and HSIL were
distinguishable from normal tissue/LSIL with 72.9% sensitivity and
73.3%specificity, using the mean methylation frequencies of the CpG
sites 7,089 and 7,134 (AUC=0.787; 95% CI, 0.700–0.874) and a 5%
methylation frequency as a critical threshold (Fig. 2B and Table
III). Concordant trends were observed when distinguishing CC
from HSIL using a 15% methylation frequency as the threshold, which
yielded 77.1% sensitivity and 62.9% specificity (AUC=0.763; 95% CI,
0.652–0.874; Fig. 2C; Table III).
| Table III.AUC values obtained from ROC analyses
of individual or combined CpG sites. |
Table III.
AUC values obtained from ROC analyses
of individual or combined CpG sites.
| CpG site | AUC (95% CI) | Sensitivity, % | Specificity, % | Threshold, % |
|---|
| CC vs. normal
tissue/LSIL and HSIL |
|
|
|
|
|
7,089 | 0.782
(0.683–0.881) | 80.0 | 64.6 | 10 |
|
7,143 | 0.796
(0.697–0.895) | 77.1 | 70.8 | 10 |
| 7,089,
7,143 | 0.822
(0.733–0.911) | 77.1 | 73.8 | 15 |
| CC and HSIL vs.
normal tissue/LSIL |
|
|
|
|
|
7,089 | 0.754
(0.660–0.848) | 64.3 | 80.0 | 10 |
|
7,143 | 0.736
(0.640–0.832) | 58.6 | 83.3 | 10 |
| 7,089, 7,143 | 0.787
(0.700–0.874) | 72.9 | 73.3 | 5 |
| CC vs. HSIL |
|
|
|
|
|
7,089 | 0.726
(0.608–0.844) | 80.0 | 51.4 | 10 |
|
7,143 | 0.753
(0.639–0.868) | 51.4 | 88.6 | 30 |
| 7,089,
7,143 | 0.763
(0.652–0.874) | 77.1 | 62.9 | 15 |
Discussion
DNA methylation is an epigenetic mechanism that is
able to inhibit gene expression directly by interfering with
transcription factor binding, or indirectly by recruiting histone
deacetylases through methyl-DNA-binding proteins (20,21).
Previous studies have demonstrated that DNA methylation is also an
important method of regulating the expression of the HPV 16 gene by
blocking certain transcription factor binding sites, and is
associated with the presence of cervical lesions (22,23). At
present, it is acknowledged that HPV 16 exhibits varied methylation
patterns in asymptomatic infection, or in low-grade lesions,
high-grade lesions and CC, and that this mechanism primarily
targets the late genes and the LCR (5). Hypermethylation of the CpG islands
located at the 3′L1 gene and the LCR of the HPV genome may be
associated with cancer progression; however, the methylation status
of the region in HPV 16 DNA has yet to be elucidated in an infected
Chinese population.
The present study examined the methylation status of
18 CpG sites encompassing the 3′ end of the L1 gene and the LCR of
the HPV 16 genome using bisulfite modification, cloning and
sequencing. The 18 CpG sites were located as follows: 3 in the
3′-L1 at nt 7,089, 7,134 and 7,143; 4 in the 5′-LCR at nt 7,268,
7,426, 7,452 and 7,458; 5 in the enhancer at nt 7,532, 7,550,
7,673, 7,679 and 7,691; 1 in the replication origin at nt 7,859; 5
in the promoter at nt 31, 37, 43, 52 and 58. Bisulfite modification
is able to convert all unmethylated cytosines, but not methylated
cytosines, to uracils (5). Whether
bisulfite treatment was completed or not may be determined by the
sequencing results of unmethylated cytosines in the study region.
In the present study, all DNA samples were effectively treated with
sodium bisulfite.
The results of the current study revealed that the
three CpG sites in the 3′-L1 fragment at nt 7,089, 7,134 and 7,143
were hypermethylated, and the methylation proportions of cervical
swab DNA samples from patients with normal tissue/LSIL, HSIL and CC
at these three sites were significantly different. Although
hypermethylation of the L1 region may provide an improved estimate
of the methylation level of the virus (5), to the best of our knowledge, no studies
have suggested that methylated L1 promotes the development of
CC.
Methylation may be a host cell defense against HPV
infection (6). During carcinogenesis,
HPV DNA is integrated into the host genome (24–28), and
the hypermethylation observed in patients with CC may result from a
host defense mechanism that senses the integrated viral genome as
foreign and targets it for epigenetic modification (19). In patients with normal tissue/LSIL,
the L1 gene is unmethylated and expresses the capsid protein of the
episomal form of the virus (5). As
additional HPV DNA integrates into the host genome during the
process of carcinogenesis (29), the
L1 gene is methylated and translation is inhibited, which may
reduce the expression of the capsid protein and trigger viral
evasion of host immune control during HPV reproduction (30). Kalantari et al (5) revealed an increased methylation rate at
the three CpG sites in the HPV 16 L1 region, occurring during the
progression from LSIL/CIN1 to invasive cancer. This result is
concordant with the data from the present study, which revealed
that the methylation proportions at CpG sites 7,268, 7,426 and
7,452 in the 5′-LCR, between DNA samples from patients with normal
tissue/LSIL and HSIL and CC, increased during the growth of
cervical lesions. A similar association was observed in the three
CpG sites in the L1 region, with the differences being
statistically significant.
The promoter is controlled by the enhancer, the
activity of which is retained in the enhancer core (20). Therefore, the promoter and the
enhancer core are the crucial segments of the HPV 16 LCR. For the
CpG sites in the enhancer and promoter regions, the methylation
proportion was lowest in cervical swab DNA samples from patients
with normal tissue/LSIL, and highest in those with CC. Previous
studies have demonstrated that methylation is able to regulate gene
expression as an epigenetic mechanism (31–33).
Numerous transcription factor binding sites in the HPV 16 enhancer
and promoter regions contain CpG sites (19). CpG sites at nt 7,532, 7,550 and 7,679
are in proximity to the activator protein 1 and nuclear factor I
binding sites, the site at nt 31 is within the specificity protein
1 binding site; and those CpG sites at nt 37, 43, 52 and 58 are
within the first twoE2 binding sites (19). Methylation of these CpG sites promotes
the substantial suppression of transcriptional activities by
directly or indirectly blocking the binding sites of certain
transcription factors. The E6 and E7 genes, which are suppressed by
HPV E2 gene products, are able to express oncogenic proteins that
are necessary for transformation and malignant progression
(34). During productive infection,
E6 and E7 are expressed at relatively low levels due to the
transcriptional suppression of E2 gene products (35). The methylation of E2 binding sites
prevents the E2 protein from binding directly to DNA, leading to
reduced regulation of E6 and E7 gene expression levels and the
expression of oncoproteins (36). In
the present study, the proportion of methylation was highest in
cervical swab DNA samples from patients with CC, and was lowest at
CpG sites in the promoter region in DNA samples from patients with
normal tissue/LSIL. The methylation proportions of DNA samples
between patients with normal tissue/LSIL, HSIL and CC were
significantly different. The methylation proportions of cervical
swab DNA samples at the 5 CpG sites (nt 31, 37, 43, 52 and 58) may
aid evaluation of the incidence of cervical cancer. Among the DNA
samples studied in the present study, an increased number of
methylated CpG sites were identified in the HPV 16 promoter region,
compared with the enhancer core, suggesting that the methylation of
the HPV 16 promoter may be of increased importance, compared with
that of the enhancer (21). CpG site
7,859, which is located in the origin of replication, exhibited no
methylation in the DNA samples from patients with HSIL,
hypomethylation in DNA samples from patients with LSIL (2.70%) and
hypermethylation in DNA samples from patients with CC (24.32%). As
a component of the HPV replication origin, the low methylation
proportion at CpG site 7,859 in DNA samples from patients with LSIL
and HSIL may increase viral replication and aid the establishment
of infection (37).
Previous studies have reported that the proportion
of methylation is higher in DNA specimens from patients without
CIN2/3, and lower in those from patients with CIN2/3 at the 11 CpG
sites within the LCR (19). A study
examining the methylation patterns of 19 HPV 16 CpG site
dinucleotides within the L1 gene and the long control region
observed the frequency of methylation to be highest in CC and
lowest in CINs (low and high grade) (37). In another study, the methylation
status of 8 CpG sites in the HPV 16 promoter region and enhancer
core were examined in clinical DNA samples using pyrosequencing,
demonstrating that the proportion of methylated DNA samples was
highest in CC, followed by asymptomatic infection and CIN3, and was
lowest in CIN1/2 (21). The
discrepancy of the methylation status between these results may be
due to the methods utilized for methylation evaluation and the
classification of cervical disease grade and differences between
ethnic groups. The present study observed that the proportion of
methylation in DNA samples at HPV 16 3′-L1 and the LCR increased as
cervical lesions progressed. The increased methylation proportion
in CC samples appears paradoxical, as the DNA methylation mechanism
generally leads to transcriptional suppression (21). However, certain HPV genomes exist in
the episomal, unmethylated form in cancer cells, which maintains
the transformation of viral genomes (38).
The present study revealed that CpG sites in the L1
gene provided effective distinction of DNA samples from patients
with CC. Among them, CpG 7,134 was able to distinguish between
normal tissue/LSIL and CC, as well as HSIL and CC; however, was not
appropriate for detecting normal tissue/LSIL vs. HSIL. ROC curves
were constructed to assess the diagnostic utility of the methylated
CpG sites 7,089 and 7,143 for various cervical lesions. The results
demonstrated that the AUC values were improved using combinations
of these two CpG sites, yielding 77.1% sensitivity and 73.8%
specificity for the detection of CC with a critical threshold value
of 15% methylation frequency. A similar study conducted by Bryant
et al (11) demonstrated that
the optimum separation between normal and dyskaryotic samples was
achieved by assessment of the CpG sites nt 5,600 and 5,609 in the
L1/L2 region, based on ROC curve analysis. Mirabello et al
(39) quantified the methylation
frequency of CpG sites in the HPV 16 L1 region (nt 6,367 and
6,389), revealing that the methylation level of CpG 6,367 was a
predictor of the development of CIN2 (AUC values>0.7), and that
CpG 6,389 was able to identify various cervical lesions. All the
aforementioned results demonstrate that the L1 gene may be the
primary target of methylation by host defense mechanisms during the
development of cervical lesions, and may be of potential use in
identifying cervical disease.
In conclusion, an association between HPV 16
3′-L1/LCR methylation and CC development was observed in female
patients from northeastern China. Of the 18 CpG sites investigated,
the most informative were at nt 7,089 and 7,143 within the L1
region. Cervical lesions in normal tissue/LSIL, HSIL or CC may be
distinguished according to the methylation frequencies of these two
CpG sites. The mean methylation frequency of the combination of CpG
sites 7,089 and 7,143 was particularly effective for determining
sensitivity and specificity. The methylation status of the CpG
sites at nt 31, 37, 43, 52 and 58 was valuable for evaluating the
incidence of CC. These data indicate that the quantification of HPV
DNA methylation in the L1 gene and the promoter region may provide
a candidate biomarker to aid the distinction between benign HPV
infections and those that may progress to precancerous
infections.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81171580 and
81171581) and the Outstanding Scientific Fund of Shengjing Hospital
(grant no. 201105).
References
|
1
|
Murakami I, Fujii T, Dan K, Saito M, Ohno
A, Iwata T and Aoki D: Methylation of human papillomavirus-52 and
−58 is a candidate biomarker in cervical neoplasia. J Clin Virol.
58:149–154. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marongiu L, Godi A, Parry JV and Beddows
S: Human Papillomavirus 16, 18, 31 and 45 viral load, integration
and methylation status stratified by cervical disease stage. BMC
Cancer. 14:3842014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schiffman M, Castle PE, Jeronimo J,
Rodriguez AC and Wacholder S: Human papillomavirus and cervical
cancer. Lancet. 370:890–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clarke MA, Wentzensen N, Mirabello L,
Ghosh A, Wacholder S, Harari A, Lorincz A, Schiffman M and Burk RD:
Human papillomavirus DNA methylation as a potential biomarker for
cervical cancer. Cancer Epidemiol Biomarkers Prev. 21:2125–2137.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kalantari M, Osann K, Calleja-Macias IE,
Kim S, Yan B, Jordan S, Chase DM, Tewari KS and Bernard HU:
Methylation of human papillomavirus 16, 18, 31, and 45 L2 and L1
genes and the cellular DAPK gene: Considerations for use as
biomarkers of the progression of cervical neoplasia. Virology.
448:314–321. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Whiteside MA, Siegel EM and Unger ER:
Human papillomavirus and molecular considerations for cancer risk.
Cancer. 113:(10 Suppl). S2981–S2994. 2008. View Article : Google Scholar
|
|
7
|
Kim SS, Suh DS, Kim KH, Yoon MS and Choi
KU: Clinicopathological significance of atypical glandular cells on
Pap smear. Obstet Gynecol Sci. 56:76–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou J, Tomashefski JF Jr, Sawady J,
Ferrer H and Khiyami A: The diagnostic value of the ThinPrep pap
test in endometrial carcinoma: A prospective study with
histological follow-up. Diagn Cytopathol. 41:408–412. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Strooper LM, van Zummeren M,
Steenbergen RD, Bleeker MC, Hesselink AT, Wisman GB, Snijders PJ,
Heideman DA and Meijer CJ: CADM1, MAL and miR124-2 methylation
analysis in cervical scrapes to detect cervical and endometrial
cancer. J Clin Pathol. 67:1067–1071. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arbyn M, Ronco G, Anttila A, Meijer CJ,
Poljak M, Ogilvie G, Koliopoulos G, Naucler P, Sankaranarayanan R
and Peto J: Evidence regarding human papillomavirus testing in
secondary prevention of cervical cancer. Vaccine. 30:(Suppl 5).
F88–F99. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bryant D, Tristram A, Liloglou T, Hibbitts
S, Fiander A and Powell N: Quantitative measurement of human
papillomavirus type 16 L1/L2 DNA methylation correlates with
cervical disease grade. J Clin Virol. 59:24–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baylin SB and Herman JG: DNA
hypermethylation in tumorigenesis: Epigenetics joins genetics.
Trends Genet. 16:168–174. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shivapurkar N, Sherman ME, Stastny V,
Echebiri C, Rader JS, Nayar R, Bonfiglio TA, Gazdar AF and Wang SS:
Evaluation of candidate methylation markers to detect cervical
neoplasia. Gynecol Oncol. 107:549–553. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kalantari M, Villa LL, Calleja-Macias IE
and Bernard HU: Human papillomavirus-16 and −18 in penile
carcinomas: DNA methylation, chromosomal recombination and genomic
variation. Int J Cancer. 123:1832–1840. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wentzensen N, Sun C, Ghosh A, Kinney W,
Mirabello L, Wacholder S, Shaber R, LaMere B, Clarke M, Lorincz AT,
et al: Methylation of HPV18, HPV31, and HPV45 genomes and cervical
intraepithelial neoplasia grade III. J Natl Cancer Inst.
104:1738–1749. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun Z, Lu Z, Liu J, Wang G, Zhou W, Yang
L, Liu C, Wang B and Ruan Q: Genetic variations of E6 and long
control region of human papillomavirus type 16 from patients with
cervical lesion in Liaoning, China. BMC Cancer. 13:4592013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Verma I, Jain V and Kaur T: Application of
bethesda system for cervical cytology in unhealthy cervix. J Clin
Diagn Res. 8:OC26–OC30. 2014.PubMed/NCBI
|
|
18
|
Li LC and Dahiya R: MethPrimer: Designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xi LF, Jiang M, Shen Z, Hulbert A, Zhou
XH, Lin YY, Kiviat NB and Koutsky LA: Inverse association between
methylation of human papillomavirus type 16 DNA and risk of
cervical intraepithelial neoplasia grades II or III. PLoS One.
6:e238972011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu WG, Srinivasan K, Dai Z, Duan W,
Druhan LJ, Ding H, Yee L, Villalona-Calero MA, Plass C and Otterson
GA: Methylation of adjacent CpG sites affects Sp1/Sp3 binding and
activity in the p21 (Cip1) promoter. Mol Cell Biol. 23:4056–4065.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hong D, Ye F, Lu W, Hu Y, Wan X, Chen Y
and Xie X: Methylation status of the long control region of HPV 16
in clinical cervical specimens. Mol Med Rep. 1:555–560.
2008.PubMed/NCBI
|
|
22
|
Bryant D, Onions T, Raybould R, Jones S,
Tristram A, Hibbitts S, Fiander A and Powell N: Increased
methylation of human papillomavirus type 16 DNA correlates with
viral integration in Vulval Intraepithelial Neoplasia. J Clin
Virol. 61:393–399. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding DC, Chiang MH, Lai HC, Hsiung CA,
Hsieh CY and Chu TY: Methylation of the long control region of
HPV16 is related to the severity of cervical neoplasia. Eur J
Obstet Gynecol Reprod Biol. 147:215–220. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Doerfler W, Remus R, Müller K, Heller H,
Hohlweg U and Schubbert R: The fate of foreign DNA in mammalian
cells and organisms. Dev Biol (Basel). 106:89–97; discussion
143–160. 2001.PubMed/NCBI
|
|
25
|
Daniel B, Mukherjee G, Seshadri L,
Vallikad E and Krishna S: Changes in the physical state and
expression of human papillomavirus type 16 in the progression of
cervical intraepithelial neoplasia lesions analysed by PCR. J Gen
Virol. 76:2589–2593. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Daniel B, Rangarajan A, Mukherjee G,
Vallikad E and Krishna S: The link between integration and
expression of human papillomavirus type 16 genomes and cellular
changes in the evolution of cervical intraepithelial neoplastic
lesions. J Gen Virol. 78:1095–1101. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schwarz E, Freese UK, Gissmann L, Mayer W,
Roggenbuck B, Stremlau A and zur Hausen H: Structure and
transcription of human papillomavirus sequences in cervical
carcinoma cells. Nature. 314:111–114. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vinokurova S, Wentzensen N, Kraus I, Klaes
R, Driesch C, Melsheimer P, Kisseljov F, Dürst M, Schneider A and
von Knebel Doeberitz M: Type-dependent integration frequency of
human papillomavirus genomes in cervical lesions. Cancer Res.
68:307–313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dutta S, Chakraborty C, Dutta AK, Mandal
RK, Roychoudhury S, Basu P and Panda C: Physical and methylation
status of human papillomavirus 16 in asymptomatic cervical
infections changes with malignant transformation. J Clin Pathol.
68:206–211. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qiu C, Zhi Y, Shen Y, Gong J, Li Y and Li
X: High-resolution melting analysis of HPV-16L1 gene methylation: A
promising method for prognosing cervical cancer. Clin Biochem.
48:855–859. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fuks F: DNA methylation and histone
modifications: Teaming up to silence genes. Curr Opin Genet Dev.
15:490–495. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mino A, Onoda N, Yashiro M, Aya M,
Fujiwara I, Kubo N, Sawada T, Ohira M, Kato Y and Hirakawa K:
Frequent p16 CpG island hypermethylation in primary remnant gastric
cancer suggesting an independent carcinogenic pathway. Oncol Rep.
15:615–620. 2006.PubMed/NCBI
|
|
34
|
Münger K, Baldwin A, Edwards KM, Hayakawa
H, Nguyen CL, Owens M, Grace M and Huh K: Mechanisms of human
papillomavirus-induced oncogenesis. J Virol. 78:11451–11460. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang HJ: Aberrant DNA methylation in
cervical carcinogenesis. Chin J Cancer. 32:42–48. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tan SH, Leong LE, Walker PA and Bernard
HU: The human papillomavirus type 16 E2 transcription factor binds
with low cooperativity to two flanking sites and represses the E6
promoter through displacement of Sp1 and TFIID. J Virol.
68:6411–6420. 1994.PubMed/NCBI
|
|
37
|
Kalantari M, Calleja-Macias IE, Tewari D,
Hagmar B, Lie K, Barrera-Saldana HA, Wiley DJ and Bernard HU:
Conserved methylation patterns of human papillomavirus type 16 DNA
in asymptomatic infection and cervical neoplasia. J Virol.
78:12762–12772. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rajeevan MS, Swan DC, Nisenbaum R, Lee DR,
Vernon SD, Ruffin MT, Horowitz IR, Flowers LC, Kmak D, Tadros T, et
al: Epidemiologic and viral factors associated with cervical
neoplasia in HPV-16-positive women. Int J Cancer. 115:114–120.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mirabello L, Schiffman M, Ghosh A,
Rodriguez AC, Vasiljevic N, Wentzensen N, Herrero R, Hildesheim A,
Wacholder S, Scibior-Bentkowska D, et al: Elevated methylation of
HPV16 DNA is associated with the development of high grade cervical
intraepithelial neoplasia. Int J Cancer. 132:1412–1422. 2013.
View Article : Google Scholar : PubMed/NCBI
|