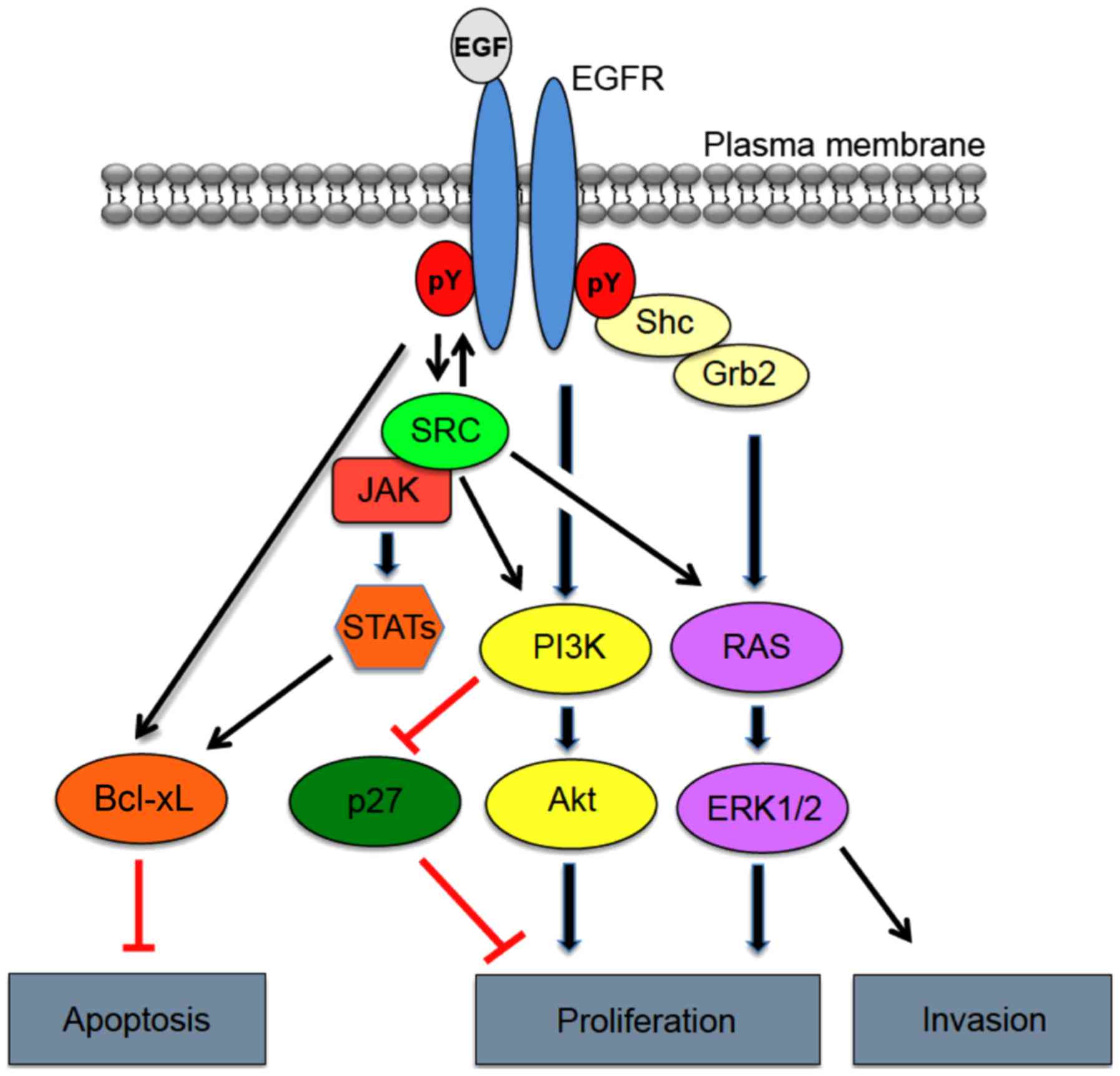
Epidermal growth factor receptor (EGFR), also
referred to as HER1/ErbB1, belongs to a larger family of ErbB
receptors with tyrosine kinase activity (1,2). Other
members of the HER family include ErbB2/HER2, ErbB3/HER3 and
ErbB4/HER4. EGFR is frequently overexpressed and/or hyper-activated
in human malignancies, including glioblastoma, and therefore
EGFR-directed therapeutic strategies are often utilized. Increased
activation of EGFR can occur through a variety of different
mechanisms, both ligand-dependent and ligand-independent (3–5). Among
these mechanisms include: Aberrant enhancement of ligand
production; constitutive receptor activation by multiple exon
deletion or missense mutations (6);
crosstalk with other receptors; increased receptor protein level
via gene amplification; and malfunction in receptor degradation.
EGFR overexpression and activation are known to significantly
impact cancer cell hallmark traits, such as increased cell
survival, proliferation and invasion (7) (Fig.
1).
EGFR is overexpressed in ~60% of primary
glioblastomas versus only 10% of secondary glioblastomas and is
characteristic of more aggressive glioblastoma phenotypes. In
addition to overexpression, several alternative mechanisms account
for aberrant induction of EGFR activation in glioblastoma,
including enhanced autocrine expression of cognate ligands
(8). Gene amplification and mutation
of EGFR also enhance EGFR activation and occur in upwards of 57% of
glioblastomas as determined by the TCGA dataset (9). From a subtype perspective, classical
glioblastoma are synonymous with focal amplification of EGFR
(~95%), whereas mesenchymal, neural and proneural glioblastomas are
associated with reduced rates of EGFR amplification at 29, 67 and
17%, respectively. Mutations of EGFR occur in roughly one-third of
all classical tumors and often in mesenchymal, proneural and neural
glioblastomas as well (10). Of these
mutations, extracellular domain EGFR mutations are most commonly
observed in glioblastoma (11).
The most frequently occurring EGFR mutation in
glioblastoma, EGFRΔIII, arises from an in-frame deletion of 801 bp
in the DNA sequence encoding the extracellular domain, rendering a
truncated yet constitutively active form of the receptor (12) EGFRΔIII is a cancer specific mutation,
as it not detected in normal tissues, making it an attractive
target for therapeutic intervention. Several different studies
(12–14) have indicated that EGFRΔIII is
expressed in ~50% of glioblastomas that amplify wild-type EGFR
(13). Additionally, data mined from
the TCGA indicates that EGFRΔIII is most commonly present in the
classical tumors (23%), where EGFR amplification is most
prevalent.
Despite being constitutively active, EGFRΔIII
sustains a low-level signal capable of evading internalization and
downregulation, which primarily result from inefficient
dimerization (14). In contrast,
wild-type EGFR is rapidly degraded following acute stimulation with
ligand (15). Though low-level in
nature, constitutive signaling downstream of EGFRΔIII leads to
increased glioblastoma cell survival in vivo through
selective augmentation of various mitogenic factors, namely Akt and
repression of apoptosis via enhanced Bcl2 family member expression
(16).
EGFRΔIII has also been associated with
transformative properties, as INK4A/Arf depleted astrocytes and
neural stem cells form high grade tumors in vivo when
expressing EGFRΔIII (17). Given
this, EGFRΔIII may act as a critical initiating event in tumor
development. Not only is EGFRΔIII likely an important factor in
gliomagenesis, but the tumorigenic potential of glioma cells in
vivo are significantly increased by EGFRΔIII expression when
compared to xenografts expressing the wild-type EGFR (18,19).
Studies have also shown that EGFRΔIII-expressing glioblastoma cells
are approvingly resilient towards both chemotherapy as well as
radiation (19–21). Interestingly, recent reports indicate
that co-expression of EGFRΔIII and the GSC marker CD133+
defines a population of GSCs harboring the greatest
tumor-initiating ability, thus further defining its importance in
glioblastoma. Taken together, it is not surprising that EGFRΔIII
expression has been strongly associated with a poor survival
prognosis for patients whose tumors amplify EGFR (21). In addition to EGFRΔIII, sequence
analysis of the EGFR coding region in a cohort of 151 glioblastoma
tumors and cell lines identified a number of novel ectodomain
missense mutations (22).
Approximately 14% of glioblastoma patient samples and 13% of
glioblastoma cell lines displayed this form of mutation. Using
missense mutants encoding R108K, T263P, A289V, G598V, and L861Q it
was determined that these mutations were: i) hyper-phosphorylated
receptor in the absence of ligand; ii) accompanied by an increased
EGFR gene dosage; and iii) exhibited a stronger transforming
phenotype relative to wild-type EGFR as determined by
anchorage-independent growth in NIH-3T3 cells. Importantly, of the
missense mutations evaluated, EGFR-R108K shares the greatest degree
of signaling and behavioral homology to EGFRΔIII, particularly as
it relates to therapeutic resistance (23).
Overexpression of EGFR has been noted in multiple
epithelial tumors, supporting the notion that deregulated EGFR
expression and signaling are pivotal events in the origin of human
cancers (24). This led to the
development of multiple inhibitors of EGFR, including EGFR-targeted
monoclonal antibodies (mAB) such as mAB C225 69 and mAB 528
(25). Mechanistically, EGFR-directed
mAbs compete with cognate ligands for binding, effectively
down-regulating receptor expression and leading to inhibition of
cell growth by induction of cell cycle arrest (26). Initially, mAB C225, dubbed cetuximab,
demonstrated promising antitumor effects in cell cultures and
xenograft models, leading to its implementation as a therapeutic
agent (27). Since, cetuximab has
been approved for use in metastatic colorectal cancer (CRC) as well
as squamous cell carcinoma of the head and neck (HNSCC) (28,29).
Cetuximab has additionally been under evaluation in progressive
non-small cell lung cancer (NSCLC), where activating mutations of
EGFR commonly occur (30). Notably,
preclinical studies in glioblastoma cell cultures and mouse models
have demonstrated the antitumor and radio-sensitizing effects of
cetuximab in this setting (31).
Preclinical data also suggest that cetuximab is active against
EGFRΔIII, where it binds to and engenders receptor internalization,
rendering a reduction in kinase activation (32). Though cetuximab has displayed
promising effects in clinical trials involving CRC, HNSCC and
NSCLC, phase I/II trials in patients with recurrent glioblastoma
have failed to confer any efficacious advantages over standard of
care regimens (33). Insufficient
intratumoral accumulation of cetuximab was cited in the failed
inhibition of EGFR autophosphorylation and degradation in these
studies.
Small molecule tyrosine kinase inhibitors (TKIs)
that competitively target receptor catalytic activity via the EGFR
kinase domain adenosine triphosphate (ATP)-binding pocket, present
another approach to targeting EGFR (34). Despite being low in molecular weight
and more likely to penetrate the BBB, the specificity of these
inhibitors is diminished by the fact that the EGFR ATP-binding
pocket shares homology with that of other RTKs, resulting in
off-target effects (35). Three TKIs
of EGFR (gefitinib, erlotinib and lapatinib) have previously
received regulatory approval for use in NSCLC and breast cancer
(36). In contrast, several phase II
clinical trials evaluating gefitinib, erlotinib or lapatinib in
newly diagnosed or recurrent glioblastoma have yielded minimal
clinical activity as either a monotherapy or in combination
regimens (37–39). The lack of clinical effects were
attributed to insufficient inhibition of Akt activation, which
correlated most strongly with EGFRΔIII expression and loss of PTEN.
Collectively, these findings highlight the need for novel
therapeutic targets capable of improving clinical responses in this
deadly disease.
The EGFR family is a complex system involved in
growth factor cellular signaling. Phosphorylation of EGFR at the
plasma membrane leads to the recruitment of multiple effector
proteins via recognition and binding of Src homology 2 (SH2) and
phosphotyrosine-binding (PTB) domains to phosphotyrosine motifs on
the receptor. Formation of the EGFR signaling complex, in turn,
triggers a variety signaling cascades involved in tumor cell
proliferation, angiogenesis, motility, differentiation, and
survival (Fig. 1) (40). Interestingly, similar substrates are
activated downstream of EGFR and EGFRΔIII, but with differing
levels of intensity. These pathways include the phosphoinositide
3-kinase (PI3K), mitogen-activated protein kinase (MAPK), signal
transducer and activator of transcription 3 (STAT3) pathways and
Src family kinases (41).
The class IA PI3Ks form heterodimers that are
recruited to trigger RTKs and adaptor proteins through regulatory
subunits, including p85a, p55a and p50a, or PIK3R1; p85b or PIKR2;
and p55y or PIKR3. p85a associates with EGFR either through ErbB3
heterodimerization or through phosphorylation of EGFR by the SFK
c-Src (42). p85a association with
EGFR results in a conformational change in p85a, releasing the
inhibition of the catalytic subunit p110 of PI3K. PI3K then
localizes to the plasma membrane, where it functions to catalyze
the formation of phosphatidylinositol 3,4,5-trisphosphate (PIP3)
via the phosphorylation of phosphatidylinositol 4,5-bisphosphate
(PIP2). The resulting PIP3 is a critical activator of Akt, which
consequently phosphorylates, or inhibits, numerous target proteins
involved in regulating cellular metabolism, motility and protein
synthesis (43). Akt activation
additionally results in phosphorylation of Bad, a Bcl family
member, which when phosphorylated fails to inhibit the survival
protein Bcl-xL, thus precluding apoptotic induction (44). Activation of PI3K can also arise from
point mutations, of which ~15% have been catalogued in glioblastoma
tumors. These mutations occur most commonly in the adaptor-binding
domain (ABD) and less frequently in the C2 helical and kinase
domains of the catalytic subunit (PIK3CA) (45). Though mutations in the regulatory
subunit (PI3KR1) are uncommon, prior sequencing analysis from the
TCGA indicated the presence of 9 such mutations occurring among a
cohort of 91 glioblastoma samples. As a result, aberrant PI3K
activation and subsequent activation of Akt is observed in upwards
of 85% of glioblastoma samples (46).
PI3K signaling is negatively regulated by various proteins, most
notably PTEN; PTEN, however, is commonly inactivated (~50%) in
glioblastoma by either epigenetic silencing or deletion mutation
(37). Loss of PTEN, therefore,
disrupts the PI3K:PTEN balance resulting in increased Akt
activation and uncontrolled cell growth. Given the frequency of
PI3K pathway aberrations occurring in glioblastoma, inhibition of
its signaling components is an important contribution for a
therapeutic avenue. Based on this, the rapamycin analogs,
everolimus (Afinitor) and temsirolimus (Torisel), both of which
inhibit mammalian target of rapamycin complex 1 (mTORC1) are
regulatory-approved for treatment of advanced renal cell carcinoma
and have been evaluated in glioblastoma patients. Unfortunately,
the clinical application of rapamycin analogs has yielded
infrequent and short-lived responses in glioblastoma. Additionally,
the PKC/PI3K/AKT inhibitor, enzastaurin, was the first targeted
therapy for glioblastoma evaluated in a phase III clinical trial
(47).
Following EGFR activation, the MAPK signaling
pathway is triggered by the growth factor receptor-bound protein 2
(Grb2) binding directly to EGFR via Y1068 and Y1086 or indirectly
by SHC binding Y1173 and Y1143 (48).
Grb2 also houses 2 SH3 domains, allowing for interactions with
proline-rich sequences, namely those of son of sevenless (SOS)
(49). The Grb2/Shc/EGFR interaction
precedes recruitment of SOS to the plasma membrane. SOS is a
guanine nucleotide exchange factor, which functions to promote the
conversion of Ras-GDP to the active Ras-GTP. Subsequently, Ras
activates Raf, a serine-threonine protein kinase, which then
phosphorylates and activates MEK1/2, resulting in activation of
ERK1/2 (MAPK) (50).
The present study concludes that the concept of EGFR
signaling and related receptors and associated factors is evolving,
however, it needs detailed evaluation for future clinical
applications in cancer patients.
|
1
|
Brosseau S, Viala M, Varga A, Planchard D,
Besse B and Soria JC: 3rd generation's TKI in lung cancer non-small
cell EGFR-mutated having acquired a secondary T790M resistance.
Bull Cancer. 102:749–757. 2015.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ymer SI, Greenall SA, Cvrljevic A, Cao DX,
Donoghue JF, Epa VC, Scott AM, Adams TE and Johns TG: Glioma
specific extracellular missense mutations in the first cysteine
rich region of epidermal growth factor receptor (EGFR) initiate
ligand independent activation. cancers (Basel). 3:2032–2049. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li N, Lorinczi M, Ireton K and Elferink
LA: Specific Grb2-mediated interactions regulate clathrin-dependent
endocytosis of the cMet-tyrosine kinase. J Biol Chem.
282:16764–16775. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shinomiya N, Gao CF, Xie Q, Gustafson M,
Waters DJ, Zhang YW and Woude GF Vande: RNA interference reveals
that ligand-independent met activity is required for tumor cell
signaling and survival. Cancer Res. 64:7962–7970. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pillay V, Allaf L, Wilding AL, Donoghue
JF, Court NW, Greenall SA, Scott AM and Johns TG: The plasticity of
oncogene addiction: implications for targeted therapies directed to
receptor tyrosine kinases. Neoplasia. 11:448–458. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Frederick L, Eley G, Wang XY and James CD:
Analysis of genomic rearrangements associated with EGRFvIII
expression suggests involvement of Alu repeat elements. Neuro
Oncol. 2:159–163. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bertotti A, Burbridge MF, Gastaldi S,
Galimi F, Torti D, Medico E, Giordano S, Corso S, Rolland-Valognes
G, Lockhart BP, et al: Only a subset of Met-activated pathways are
required to sustain oncogene addiction. Sci Signal. 2:ra80. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang PH, Xu AM and White FM: Oncogenic
EGFR signaling networks in glioma. Sci Signal. 2:re62009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brennan CW, Verhaak RG, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al TCGA Research Network, : The somatic genomic
landscape of glioblastoma. Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Verhaak RGW, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al
Cancer genome atlas research network, : Integrated genomic analysis
identifies clinically relevant subtypes of glioblastoma
characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1.
Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohgaki H and Kleihues P: Genetic
alterations and signaling pathways in the evolution of gliomas.
Cancer Sci. 100:2235–2241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wong AJ, Ruppert JM, Bigner SH, Grzeschik
CH, Humphrey PA, Bigner DS and Vogelstein B: Structural alterations
of the epidermal growth factor receptor gene in human gliomas. Proc
Natl Acad Sci USA. 89:pp. 2965–2969. 1992; View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shinojima N, Tada K, Shiraishi S, Kamiryo
T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, et
al: Prognostic value of epidermal growth factor receptor in
patients with glioblastoma multiforme. Cancer Res. 63:6962–6970.
2003.PubMed/NCBI
|
|
14
|
Hwang Y, Chumbalkar V, Latha K and Bogler
O: Forced dimerization increases the activity of ΔEGFR/EGFRvIII and
enhances its oncogenicity. Mol Cancer Res. 9:1199–1208. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schmidt MHH, Furnari FB, Cavenee WK and
Bögler O: Epidermal growth factor receptor signaling intensity
determines intracellular protein interactions, ubiquitination, and
internalization. Proc Natl Acad Sci USA. 100:pp. 6505–6510. 2003;
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nagane M, Coufal F, Lin H, Bögler O,
Cavenee WK and Huang HJ: A common mutant epidermal growth factor
receptor confers enhanced tumorigenicity on human glioblastoma
cells by increasing proliferation and reducing apoptosis. Cancer
Res. 56:5079–5086. 1996.PubMed/NCBI
|
|
17
|
Bachoo RM, Maher EA, Ligon KL, Sharpless
NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R,
et al: Epidermal growth factor receptor and Ink4a/Arf: Convergent
mechanisms governing terminal differentiation and transformation
along the neural stem cell to astrocyte axis. Cancer Cell.
1:269–277. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishikawa R, Ji XD, Harmon RC, Lazar CS,
Gill GN, Cavenee WK and Huang HJ: A mutant epidermal growth factor
receptor common in human glioma confers enhanced tumorigenicity.
Proc Natl Acad Sci USA. 91:pp. 7727–7731. 1994; View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cavenee WK: Genetics and new approaches to
cancer therapy. Carcinogenesis. 23:683–686. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lammering G, Valerie K, Lin PS, Hewit TH
and Schmidt-Ullrich RK: Radiation-induced activation of a common
variant of EGFR confers enhanced radioresistance. Radiother Oncol.
72:267–273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heimberger AB, Hlatky R, Suki D, Yang D,
Weinberg J, Gilbert M, Sawaya R and Aldape K: Prognostic effect of
epidermal growth factor receptor and EGFRvIII in glioblastoma
multiforme patients. Clin Cancer Res. 11:1462–1466. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee JC, Vivanco I, Beroukhim R, Huang JHY,
Feng WL, DeBiasi RM, Yoshimoto K, King JC, Nghiemphu P, Yuza Y, et
al: Epidermal growth factor receptor activation in glioblastoma
through novel missense mutations in the extracellular domain. PLoS
Med. 3:e4852006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Raizer JJ, Abrey LE, Lassman AB, Chang SM,
Lamborn KR, Kuhn JG, Yung WKA, Gilbert MR, Aldape KA, Wen PY, et
al: North American Brain Tumor Consortium: A phase II trial of
erlotinib in patients with recurrent malignant gliomas and
nonprogressive glioblastoma multiforme postradiation therapy.
Neuro-oncol. 12:95–103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Libermann TA, Razon N, Bartal AD, Yarden
Y, Schlessinger J and Soreq H: Expression of epidermal growth
factor receptors in human brain tumors. Cancer Res. 44:753–760.
1984.PubMed/NCBI
|
|
25
|
Kawamoto T, Sato JD, Le A, Polikoff J,
Sato GH and Mendelsohn J: Growth stimulation of A431 cells by
epidermal growth factor: Identification of high-affinity receptors
for epidermal growth factor by an anti-receptor monoclonal
antibody. Proc Natl Acad Sci USA. 80:pp. 1337–1341. 1983;
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Waksal HW: Role of an anti-epidermal
growth factor receptor in treating cancer. Cancer Metastasis Rev.
18:427–436. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goldstein NI, Prewett M, Zuklys K,
Rockwell P and Mendelsohn J: Biological efficacy of a chimeric
antibody to the epidermal growth factor receptor in a human tumor
xenograft model. Clin Cancer Res. 1:1311–1318. 1995.PubMed/NCBI
|
|
28
|
Sobrero AF, Maurel J, Fehrenbacher L,
Scheithauer W, Abubakr YA, Lutz MP, Vega-Villegas ME, Eng C,
Steinhauer EU, Prausova J, et al: EPIC: Phase III trial of
cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin
failure in patients with metastatic colorectal cancer. J Clin
Oncol. 26:2311–2319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bonner JA, Harari PM, Giralt J, Azarnia N,
Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pirker R, Pereira JR, Szczesna A, von
Pawel J, Krzakowski M, Ramlau R, Vynnychenko I, Park K, Yu CT,
Ganul V, et al: FLEX Study Team: Cetuximab plus chemotherapy in
patients with advanced non-small-cell lung cancer (FLEX): An
open-label randomised phase III trial. Lancet. 373:1525–1531. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eller JL, Longo SL, Kyle MM, Bassano D,
Hicklin DJ and Canute GW: Anti-epidermal growth factor receptor
monoclonal antibody cetuximab augments radiation effects in
glioblastoma multiforme in vitro and in vivoNeurosurgery. 56. pp.
155–162. discussion 162; 2005, View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Patel D, Lahiji A, Patel S, Franklin M,
Jimenez X, Hicklin DJ and Kang X: Monoclonal antibody cetuximab
binds to and down-regulates constitutively activated epidermal
growth factor receptor vIII on the cell surface. Anticancer Res.
27(5A): 3355–3366. 2007.PubMed/NCBI
|
|
33
|
Stragliotto G, Vega F, Stasiecki P, Gropp
P, Poisson M and Delattre JY: Multiple infusions of anti-epidermal
growth factor receptor (EGFR) monoclonal antibody (EMD 55,900) in
patients with recurrent malignant gliomas. Eur J Cancer.
32A:636–640. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baselga J: Targeting tyrosine kinases in
cancer: The second wave. Science. 312:1175–1178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vivanco I, Robins HI, Rohle D, Campos C,
Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N,
et al: Differential sensitivity of glioma- versus lung
cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer
Discov. 2:458–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baumann M, Krause M, Dikomey E, Dittmann
K, Dörr W, Kasten-Pisula U and Rodemann HP: EGFR-targeted
anti-cancer drugs in radiotherapy: Preclinical evaluation of
mechanisms. Radiother Oncol. 83:238–248. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rich JN and Bigner DD: Development of
novel targeted therapies in the treatment of malignant glioma. Nat
Rev Drug Discov. 3:430–446. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
van den Bent MJ, Brandes A, Rampling R,
Kouwenhoven M, Kros JM, Carpentier AF, Clement P, Klughammer B,
Gorlia T and Lacombe D: Randomized phase II trial of erlotinib (E)
versus temozolomide (TMZ) or BCNU in recurrent glioblastoma
multiforme (GBM): EORTC 26034. J Clin Oncol. 25:20072005.
|
|
39
|
Thiessen B, Stewart C, Tsao M, Kamel-Reid
S, Schaiquevich P, Mason W, Easaw J, Belanger K, Forsyth P,
McIntosh L, et al: A phase I/II trial of GW572016 (lapatinib) in
recurrent glioblastoma multiforme: Clinical outcomes,
pharmacokinetics and molecular correlation. Cancer Chemother
Pharmacol. 65:353–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jorissen RN, Walker F, Pouliot N, Garrett
TPJ, Ward CW and Burgess AW: Epidermal growth factor receptor:
Mechanisms of activation and signalling. Exp Cell Res. 284:31–53.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mizoguchi M, Betensky RA, Batchelor TT,
Bernay DC, Louis DN and Nutt CL: Activation of STAT3, MAPK, and AKT
in malignant astrocytic gliomas: Correlation with EGFR status,
tumor grade, and survival. J Neuropathol Exp Neurol. 65:1181–1188.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stover DR, Becker M, Liebetanz J and Lydon
NB: Src phosphorylation of the epidermal growth factor receptor at
novel sites mediates receptor interaction with Src and P85 alpha. J
Biol Chem. 270:15591–15597. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Courtney KD, Corcoran RB and Engelman JA:
The PI3K pathway as drug target in human cancer. J Clin Oncol.
28:1075–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao TT, Le Francois BG, Goss G, Ding K,
Bradbury PA and Dimitroulakos J: Lovastatin inhibits EGFR
dimerization and AKT activation in squamous cell carcinoma cells:
Potential regulation by targeting rho proteins. Oncogene.
29:4682–4692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gallia GL, Rand V, Siu I-M, Eberhart CG,
James CD, Marie SKN, Oba-Shinjo SM, Carlotti CG, Caballero OL,
Simpson AJG, et al: PIK3CA gene mutations in pediatric and adult
glioblastoma multiforme. Mol Cancer Res. 4:709–714. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang H, Wang H, Zhang W, Huang HJ, Liao
WSL and Fuller GN: Analysis of the activation status of Akt,
NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest.
84:941–951. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wick W, Puduvalli VK, Chamberlain MC, van
den Bent MJ, Carpentier AF, Cher LM, Mason W, Weller M, Hong S,
Musib L, et al: Phase III study of enzastaurin compared with
lomustine in the treatment of recurrent intracranial glioblastoma.
J Clin Oncol. 28:1168–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Batzer AG, Rotin D, Ureña JM, Skolnik EY
and Schlessinger J: Hierarchy of binding sites for Grb2 and Shc on
the epidermal growth factor receptor. Mol Cell Biol. 14:5192–5201.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pawson T: Protein modules and signalling
networks. Nature. 373:573–580. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Marshall CJ: Cell signalling. Raf gets it
together. Nature. 383:127–128. 1996. View Article : Google Scholar : PubMed/NCBI
|