Introduction
Tumors that comprise a mass of malignant epithelial
cells are also surrounded by multiple non-cancerous cell
populations, including fibroblasts, endothelial cells, pericytes,
immune regulatory cells and cytokines in the extracellular matrix
(ECM) (1). These stromal cells
surrounding the tumor form a distinct microenvironment and were not
considered to possess a role in cancer progression. However, it
became evident that the molecular and biological abnormalities of
cancer cells could not fully explain the complex changes involved
in the regulation of tumor progression (2). Thus, an increasing number of studies
have focused on the functions of the tumor microenvironment in
cancer progression (3–5).
Activated fibroblasts, termed cancer-associated
fibroblasts (CAFs), are one of the major components of stromal
cells. CAFs were first identified as negative factors in tumor
development that had no effect on tumor cells; however, they have
been identified as an essential component in tumor progression
(6). With the reciprocal crosstalk
between cancer cells and fibroblasts, CAFs undergo various
morphological and biological transitions in response to tumor
progression (7). Furthermore, CAFs
have an important role in maintaining an optimal microenvironment
for cancer cell survival and proliferation (6,7). Studies
investigating the role of CAFs have reported that the therapeutic
targeting of cancer cells alone is insufficient for the treatment
of cancer (8). Thus, cancer therapy
should co-target cancer cells and their microenvironment. CAFs are
essential components to the tumor microenvironment and therefore
represent a molecular target for the treatment of cancer (9).
The present study is a review of the recent
developments in CAF research, and is aimed at gaining an improved
understanding of the biological mechanisms underlying CAF
involvement in tumor progression. Furthermore, the association
between cancer cells and the tumor microenvironment was analyzed in
order to identify novel strategies for the treatment of cancer.
General characteristics of CAFs
CAFs are a heterogeneous population of cells with
various origins, the majority of which are derived from resident
fibroblasts. CAFs may also be derived from other cells, including
mesenchymal stem cells (MSCs), epithelial, pericytes, adipocytes
and endothelial cells (10). CAFs in
the tumor stroma can be differentiated according to their
morphology and specific identifiable markers. CAFs are generally
presented as large spindle-shaped cells similar to smooth muscle
cells (myofilaments and electron dense patches) (11). α-smooth muscle actin is regarded as
the most widely used biomarker for identifying CAFs (12). Fibroblast activation protein α (FAPα)
is a cytomembrane protein that is selectively expressed by
activated CAFs in various types of human epithelial cancer
(13). Furthermore, podoplanin-a,
S100A4, vimentin, fibroblast specific protein-1 (FSP-1), and
platelet-derived growth factor (PDGF) receptors α and β are
expressed in CAFs (14). Insulin-like
growth factor-binding protein 7 (IGFBP7), a novel biomarker for
tumor fibroblasts in epithelial cancer, has also been detected in
CAFs through genetic screenings and immunohistochemical studies.
IGFBP7-expressing CAFs have been demonstrated to promote colon
cancer cell proliferation through paracrine tumor-stroma
interactions in vitro (15).
The application of microarray gene-expression
analysis has enabled the comprehensive characterization of CAFs and
has increased awareness on the importance of CAFs in oncological
studies. A total of 46 differentially expressed genes regulated by
the transforming growth factor (TGF)-β signaling pathway were
identified in 15 paired CAF and normal fibroblast (NF) cell lines
(16). All 46 genes were identified
to encode for paracrine factors that are released into the tumor
microenvironment. Of these results, 11 genes
[intercellular-adhesion molecule 1 (ICAM1), THBS2, MME, OXTR,
PDE3B, B3GALT2, EVI2B, COL14A1, GAL and MCTP2] were used to form a
prognostic signature of CAFs in non-small cell lung cancer (NSCLC)
(16). Similar studies have
identified differentially-expressed genes between CAFs and NFs
(17–20). Integrin α11 was identified to be
primarily expressed in CAFs and possess prognostic significance for
NSCLC (17). Furthermore,
cyclooxygenase 2 and TGF-β2 expression in CAFs was confirmed
through immunohistochemical analysis in metastatic colon cancer
(18). In human primary pancreatic
adenocarcinoma, smoothened homolog was identified to be
overexpressed in CAFs compared with the expression in pancreatic
NFs (19). In addition, numerous
altered gene transcripts have been identified in breast CAFs,
including that of ribsosomal protein S6 kinase α3, fibroblastic
growth factor (FGF) receptor 1, nardilysin that enhances shedding
of EGF (NRD1), cyclin-dependent kinase inhibitor 1B, NFY and
prostaglandin E synthase 2 (20).
However, no significant differences in the gene expression pattern
of NFs were reported with the most upregulated gene being chromobox
2, a polycomb homolog repressor of proto oncogenes (20).
Tumors induce fibroblast activation
When cancer cells metastasize to another organ, they
recruit NFs to the tumor mass. The activated phenotype of
fibroblasts in the tumor mass are induced by different genetic and
epigenetic changes that are self-regulated, and regulated by cancer
cells; however, the mechanisms underlying the transformation of NFs
to CAFs remains unclear (21).
The activation of fibroblasts is induced by numerous
cytokines secreted by cancer cells and other stroma cells,
including TGF-β, epidermal growth factor (EGF), PDGF, FGF2 and
C-X-C motif chemokine ligand (CXCL) 12 (22). Cell-cell communication through
adhesion molecules, including ICAM1 and vascular-cell adhesion
molecule 1 also enables fibroblast activation (23).
MicroRNAs (miRNAs/miRs) are an abundant type of
endogenous small RNA molecule that downregulate target gene
expression (24). A previous study
demonstrated that miR-155 is upregulated, whereas miR-31 and
miR-214 are downregulated in ovarian CAFs (25). C-C motif chemokine ligand (CCL) 5 was
identified as a target gene of miR-214. The results demonstrated
that ovarian cancer cells induce the transformation of NFs to CAFs
partially through regulation by miRNAs when NFs are co-cultured
with cancer cells (25). These
findings suggest that miRNAs have a regulatory role in the
transformation of NFs to CAFs. Other miRNAs that have been
identified to be differentially expressed in CAFs are listed in
Table I (26–30).
 | Table I.The regulation of miRNA in cancer
associated fibroblasts. |
Table I.
The regulation of miRNA in cancer
associated fibroblasts.
| A, Upregulated
miRNAs |
|---|
|
|---|
| Author, year | miRNA | Cancer type | Target gene | (Refs.) |
|---|
| Mitra et al,
2012 | miR-155 | Ovarian |
| (25) |
| Zhao et al,
2012 | miR-266,
miR-221-3p, miR-221-5p, miR-31-3p | Breast | ETS2 | (26) |
| Enkelmann et
al, 2011 | miR-16,
miR-320 | Bladder |
| (27) |
| Aprelikova et
al, 2014 | miR-29b, miR-146a
miR-503 | Endometrial |
| (29) |
| Wang et al,
2013 | miR-138, miR-210,
miR-99a | Colorectal |
| (30) |
| Bronisz et
al, 2012 | miR-320 | Breast |
| (56) |
|
| B, Downregulated
miRNAs |
|
| Mitra et al,
2012 | miR-31 | Ovarian | SATB2 | (25) |
| Mitra et al,
2012 | miR-214 | Ovarian | CCL5 | (25) |
| Zhao et al,
2012 | miR-205, miR-200c,
miR-200b, miR-141, miR-101, miR-342-3p, Let-7g | Breast | ZEB1/SIP1 | (26) |
| Enkelmann et
al, 2011 | miR-143,
miR-145 | Bladder |
| (27) |
| Yu et al,
2010 | miR-17/20 | Breast | IL-8, CXCL1, CK8,
α-ENO | (28) |
| Aprelikova et
al, 2014 | miR-31 | Endometrial | SATB2 | (29) |
| Wang et al,
2013 | miR-29b, miR-494,
miR-126 | Colorectal |
| (30) |
| Verghese et
al, 2013 | miR-26b | Breast | TNKS1BP1, CPSF7,
COL12A1 | (55) |
| Mongiat et
al, 2010 | miR-15, miR-16 | Prostate |
| (57) |
CAFs induce tumor growth, angiogenesis,
metastasis and chemoresistance
CAFs induce tumor growth
Tumor growth depends on the abnormal and
uncontrollable proliferation of cancer cells with simultaneous
changes to the microenvironment. Among the stromal cells in the
microenvironment surrounding the tumor, increasing evidence has
reported that CAFs are targets and inducers of tumorigenic
activation signals (31,32).
CAFs produce autocrine and/or paracrine cytokines
that promote the biological characteristics of tumors. In addition
to classical growth factors, including EGF and hepatocyte growth
factor (HGF), novel CAF-secreted proteins [secreted frizzled
related protein 1, and IGF like family member (IGF) 1 and 2], and
membrane molecules (integrin α11 and syndecan-1) have also been
identified to possess cancer cell-supporting roles (33). These factors directly or indirectly
stimulate tumor growth and survival, or enhance their migratory and
invasive properties.
Previous studies have demonstrated that chemokines
secreted by CAFs into the microenvironment allow for the
recruitment of bone marrow-derived cells (BMCs) and immune cells
(34). CXCL12 (35), CXCL14 (36) and CCL5 (37) have been identified as pro-metastatic
factors. In addition, MSC-derived CAFs are recruited to the stroma
of the dysplastic stomach, and express interleukin (IL)-6, Wnt
family member (Wnt) 5α and bone morphogenetic protein 4, all of
which promote tumor growth through DNA hypomethylation (38). Furthermore, MSC-derived CAFs are
recruited to the tumor through TGF-β and CXCL12 signaling (38). In oral squamous cell carcinoma (OCC),
CCL2 expression in CAFs is upregulated, promoting the production of
endogenous reactive oxygen species (ROS) in OC cells (OCCs)
(37). Consequently, ROS induces the
expression of cell cycle regulatory proteins in OCCs, and promotes
OCC proliferation, migration and invasion (39). Together, these chemokines and
cytokines create a suitable microenvironment allowing for the
proliferation and metastasis of cancer cells.
CAFs stimulate tumor angiogenesis
Vascular endothelial growth factor (VEGF) was
originally identified as a multifunctional cytokine in angiogenesis
and lymphangiogenesis (40). The
interaction between tumor and stromal cells can result in increased
VEGF expression, with CAFs being the primary source of VEGF
(41). Furthermore, CAF-derived PDGF
has been demonstrated to be an essential factor in activating VEGF
production. PDGF/PDGF receptor (R) signaling is an important
regulatory pathway primarily involved in angiogenesis (41). PDGFs indirectly promote angiogenesis
by recruiting stromal fibroblasts that secrete VEGF (42). Furthermore, PDGFs are able to recruit
and induce BMCs to form endothelial or smooth muscle cells.
Subsequently, PDGFs promote the proliferation and migration of
endothelial, and smooth muscle cells (42). PDGF subunit B, which is produced by
endothelial cells can induce the migration of pericytes to the
vessel wall and maintain endothelial stability, thus leading to
tumor angiogenesis (43).
Nagasaki et al (44) reported that cancer cells stimulate the
secretion of IL-6 from fibroblasts, subsequently inducing tumor
angiogenesis. IL-6R neutralization antibody inhibited IL-6
signaling and tumor angiogenesis by inhibiting the interaction
between the cancer, and stroma. This finding suggests that IL-6 is
a novel target for anti-angiogenesis therapy (44).
CAFs mediate tumor metastasis
Increasing evidence suggests a metastatic support
role of CAFs in tumors (45,46), whereas data regarding the presence and
role of CAFs in lymph node and distant metastasis is deficient.
Stromal reactions in metastatic lymph nodes, possibly comprising
metastasis-associated fibroblasts, have been described as reactive
and fibrotic tissue with enhanced deposition of vitronectin and
fibronectin, desmoplasia, nodal fibrosis and hyaline stroma
(47). Immunohistochemical
characterization of CAFs was reported in one of these studies,
which assessed metastatic lymph node tissue from a patient with
uterine cervix adenocarcinoma who received preoperative
chemotherapy (47). Certain studies
have suggested that the mesenchymal-like phenotype of CAFs is
involved in enhancing the metastasis of cancer cells, whereas NFs
with the epithelial-like phenotype inhibit the migration of breast
cancer cells (48). Similarly, normal
prostate epithelial cells induce intraepithelial neoplasia in
vivo when co-injected with CAFs, but not when co-injected with
NFs (49).
YAP is a transcription factor that may be a
signature feature of CAFs. YAP has important roles in matrix
stiffening, cancer cell invasion and angiogenesis, which are
induced by CAFs (50). YAP regulates
the expression of specific cytoskeletal proteins, including anillin
actin binding protein, diaphanous related formin 3 and myosin
regulatory light polypeptide 9 (50).
Additionally, CAFs secrete proinflammatory cytokines that stimulate
the nuclear factor-κB (NF-κB) signaling pathway, subsequently
promoting tumorigenesis (51).
Notably, CAFs in the stroma of triple-negative
breast cancer samples have been demonstrated to select for bone
metastatic cells (52). CAFs produce
CXCL12 and IGF1, which are prognostic markers for bone relapse and
activators of the phosphatidylinositol 3-kinase (PI3K)/AKT
serine/threonine kinase (AKT) signaling pathway (52). Cancer cells are primed for metastasis
in the CXCL12-rich microenvironment of the bone marrow, thus
suggesting an important role of CAFs in tumor metastasis. Another
study demonstrated that a reduction in miR-148a expression in CAFs
results in increased Wnt activity through the upregulation of its
target gene WNT10B. Consequently, increased Wnt activity results in
increased migration of endometrial cancer cells (53).
A study reported that the downregulation of miR-26b
in CAFs stimulates the migration of fibroblasts, which is a
dominant characteristic of the CAF phenotype. Furthermore, CAFs
with reduced expression of miR-26b promote the migration and
invasion of human breast cancer cells (54). Additionally, the PTEN/miR-320/ETS2
axis secretes proteins, such as Emilin2, that distinguish between
normal and malignant stroma, and is associated with a higher rate
of relapse in patients with breast cancer (55). This demonstrates that miR-320 is an
essential regulator of the signaling pathway in fibroblasts
involved in the regulation of the tumor microenvironment. Similar
to in breast cancer, in prostate cancer, the downregulation of
miR-15 and −16 in CAFs is mediated through activation of the AKT,
and extracellular signal-regulated kinase signaling pathways,
promoting prostate cancer migration, and angiogenesis (56).
CAFs induce resistance of cancer cells
to therapy
Compared with cancer cells, CAFs are relatively
genetically stable with a reduced probability of developing
drug-resistance, thus representing as a potential therapeutic
target with lower chances for the development of chemoresistance
(57,58). However, an increasing amount of data
has suggested that fibroblasts have a protective role that allows
cancer cells to evade therapy, as described below.
PDGF
The interstitial fluid pressure (IFP) in the center
of solid tumors is increased compared with that in the surrounding
tumor tissue (59). Higher IFP
reduces the efficiency of drug penetration into the tumor tissue,
thus reducing the concentration of the drug reaching the tumor
cells and increasing tumor cell viability (58). Strategies on improving chemotherapy
have focused on reducing tumor IFP in order to increase the
efficiency of drug transport and penetration into tumors (60).
PDGF and other associated tyrosine kinase receptors
are expressed in various types of cancer. STI571, a receptor
tyrosine kinase inhibitor (TKI), reduces tumor IFP and increases
Taxol uptake in subcutaneously injected undifferentiated anaplastic
thyroid carcinoma KAT-4 cell line-induced transplantable tumors in
severe combined immune deficient mice (61,62).
HGF
HGF has been identified as an essential factor in of
CAF-mediated resistance to B-Raf proto-oncogene serine/threonine
kinase (BRAF) inhibitor therapy in melanoma with
BRAFV600E mutation, as well as lapatinib resistance in
HER2+ breast cancer (63,64).
TKIs exhibit strong inhibitory effects against NSCLC
with epidermal growth factor receptor (EGFR)-activating mutations
(65). However, the possibility of
intrinsic or developing acquired resistance is an important
consideration in the management of patients with cancer. The
overexpression of HGF in CAF, a ligand of HGF receptor (MET), has
been reported to contribute to resistance to EGFR-TKIs (66).
EGFR and HGF are coexpressed in colorectal cancer
(CRC) cell lines, and the activation of both receptors
synergistically induces the proliferation of cancer cells (67). Cetuximab suppresses cell growth
through dephosphorylation of EGFR, mitogen-activated protein kinase
(MAPK), and/or the AKT signaling pathway (68). It was demonstrated that CAF-derived
HGF phosphorylates MET, but not EGFR or receptor tyrosine-protein
kinase erbB-3 in cetuximab-treated cells. Subsequently, this was
revealed to restore cell proliferation and rescue cells from
G1 phase arrest, and apoptosis through restimulation of
the MAPK and AKT signaling pathways (68). Notably, this effect is inhibited by
suppressing MET activation with PHA-665752, a highly specific MET
kinase inhibitor, or by knocking down MET expression using RNA
interference (69).
Together, these data demonstrate that the presence
of fibroblasts secreting HGF confers resistance to therapy. In
addition, HGF can activate MET, which is expressed on
cancer-initiating cells (CICs) in colon cancer, through paracrine
signaling (70). This can sustain
typical CIC properties, including long-term self-renewal,
ultimately leading to resistance to anti-EGFR therapy (70).
Chemokines
Increasing evidence supports the presence of stromal
cytokines that are important in the development of tumor
chemoresistance.
CCL2 is an inflammatory chemokine, which is
recruited by immune cells into the tumor microenvironment and has
been demonstrated to confer resistance to paclitaxel, and docetaxel
in prostate cancer (71). A previous
study demonstrated that CCL2 expression is higher in three
different paclitaxel-resistant ovarian cancer cell lines ES-2/TP,
MES-OV/TP and OVCAR-3/TP compared with parental cells (72). Furthermore, treatment with a CCL2
inhibitor enhances the antitumor efficacy of paclitaxel and
carboplatin combination therapy in ovarian cancer (72). CAFs can induce CCL2 production through
signal transducer and activator of transcription 3 (STAT3)
phosphorylation, and in turn, CAF-derived CCL2 promotes cancer
progression by regulating cancer stem cells through activation of
the Notch signaling pathway (73).
The chemokine CXCL12 is the sole ligand of CXCR4.
CAFs are an important source of CXCL12 in the tumor stroma.
Previous studies have indicated that CXCL12/CXCR4 signaling
contributes to chemoresistance by inducing the activation of focal
adhesion kinase, ERK and AKT signaling pathways, enhancing the
transcriptional activities of β-catenin, and NF-κB, and the
expression of survival proteins (74,75).
Disruption of the CXCR4/CXCL12 signaling pathway has been
demonstrated to sensitize prostate cancer cells to docetaxel
(76). Similar results have been
observed in colon (77) and lung
(78) cancer. Therefore, these
studies suggest that chemokines, including CXCL12, may act as
promising targets for cancer therapy, alone and/or in combination
with other cytotoxic drugs.
Interleukin family
Emerging evidence suggests that the dynamic
crosstalk between tumor cells and stromal fibroblasts underlie drug
resistance. In CRC, IL-17A, which is overexpressed by CAFs in
response to chemotherapy, bind to the IL-17A receptor expressed on
CICs (79). Consequently, this
results in the maintenance and development of therapeutic
resistance of CICs through the upregulation of NF-κB (79). In ER-negative and triple-negative
breast cancer, IL-17A protects from docetaxel-induced cell death
through activation of ERK1, and 2, thus participating in
therapy-resistance development (80).
IL-6, an inflammatory cytokine, is primarily
secreted by CAFs. IL-6 promotes the growth and invasion of cancer
cells through activation of STAT3 (81). NSCLC cells expressing persistently
activated mutant EGFR are also associated with the IL-6 signaling
pathway, which promotes the proliferation and survival of cells,
leading to erlotinib resistance (82,83). IL-6
secreted by CAFs induces tamoxifen resistance through activation of
the Janus kinase (JAK)/STAT3 and PI3K/AKT signaling pathways in
breast cancer cells (84). Inhibition
of proteasome activity, IL-6 activity or the JAK/STAT3, or PI3K/AKT
signaling pathways markedly reduced CAF-induced tamoxifen
resistance (84). These results
demonstrate that IL-6 creates a ‘protective niche’ that maintains
the survival of residual tumor cells, consequently inducing tumor
relapse.
Other factors
WNT16B is an important fibroblast-derived protein
and treatment-induced factor that confers chemotherapy resistance.
The chemotherapy resistance effects of fibroblast-derived WNT16B
have been detected in vivo and in vitro, indicating
that WNT16B reduces apoptosis induced by chemotherapy drugs in
prostatic carcinoma (85). This study
guides novel directions for combination therapies, including
targeting fibroblast-derived WNT16B, which may reverse
chemoresistance in breast and prostate cancer (85). Fibroblast-secreted high mobility group
protein B1 is released into the tumor microenvironment and performs
paracrine signaling on neighboring cancer cells, which has been
suggested to induce chemoresistance in breast cancer (86).
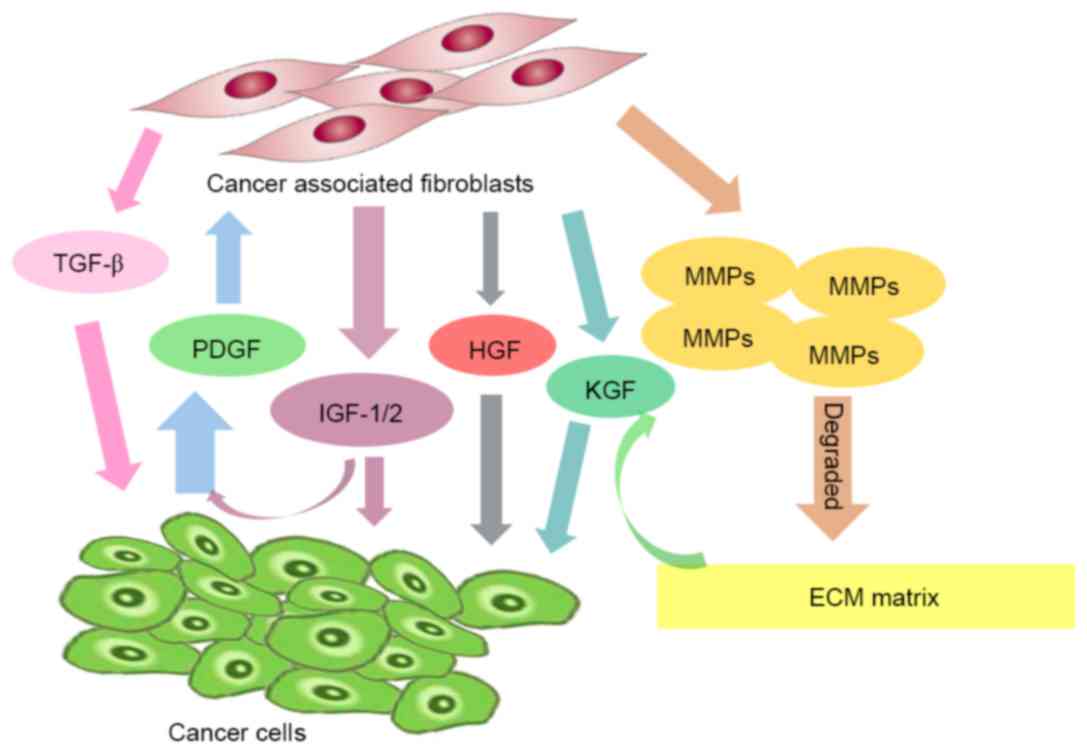
Interaction loop
A bi-directional activation between cancer cells and
fibroblasts has been identified as the leading cause of formation
of the malignant phenotype of cancer. As aforementioned, the
crosstalk between the two is important for tumor progression, and
the interactions between them are induced by the reciprocal
signaling of secreted components, including cytokines, and
regulatory factors in the ECM. Cullen et al (87) reported that cancer cells produce PDGF,
which induces fibroblast proliferation and the expression of IGF I,
and II. Notably, IGFs secreted by fibroblasts in turn induce cancer
cell proliferation and the synthesis of PDGF (87).
Cancer cells induce the production of matrix
metalloproteinases (MMPs) by fibroblasts, which results in
degradation of the extracellular matrix and enhances the
invasiveness of cancer cells (88).
In return, fibroblasts secrete growth factors, including HGF
(89), keratinocyte growth factor
(90), and IGF-1 and −2 (91), which stimulate the proliferation of
cancer cells. Furthermore, a previous study reported that local
cell-cell interactions between breast cancer cells and fibroblasts
exhibit various effects on numerous genes, including the regulation
of the expression of TGF-β-altered genes (92).
These signaling pathways are involved in positive
feedback loops, which result in increased tumor cell numbers and/or
amplification of signaling molecules, and consequently tumor
therapy resistance. Thus, understanding the biological mechanism
underlying CAFs may aid in the development of novel
molecular-targeted therapies to inhibit these signaling feedback
loops (Fig. 1).
Inhibition of the feedback loop as an
approach for anti-cancer therapy
In order to target CAFs, a possible approach is to
inhibit the feedback loop between fibroblasts and cancer cells.
Such therapies have not yet been applied clinically, but based on
the aforementioned evidence, the potential benefits of these
treatments have been demonstrated. Inhibiting the feedback loop may
involve the following approaches: Inhibition of fibroblasts
directly and disruption of CAF-associated paracrine growth factor
signals (Fig. 2) (6).
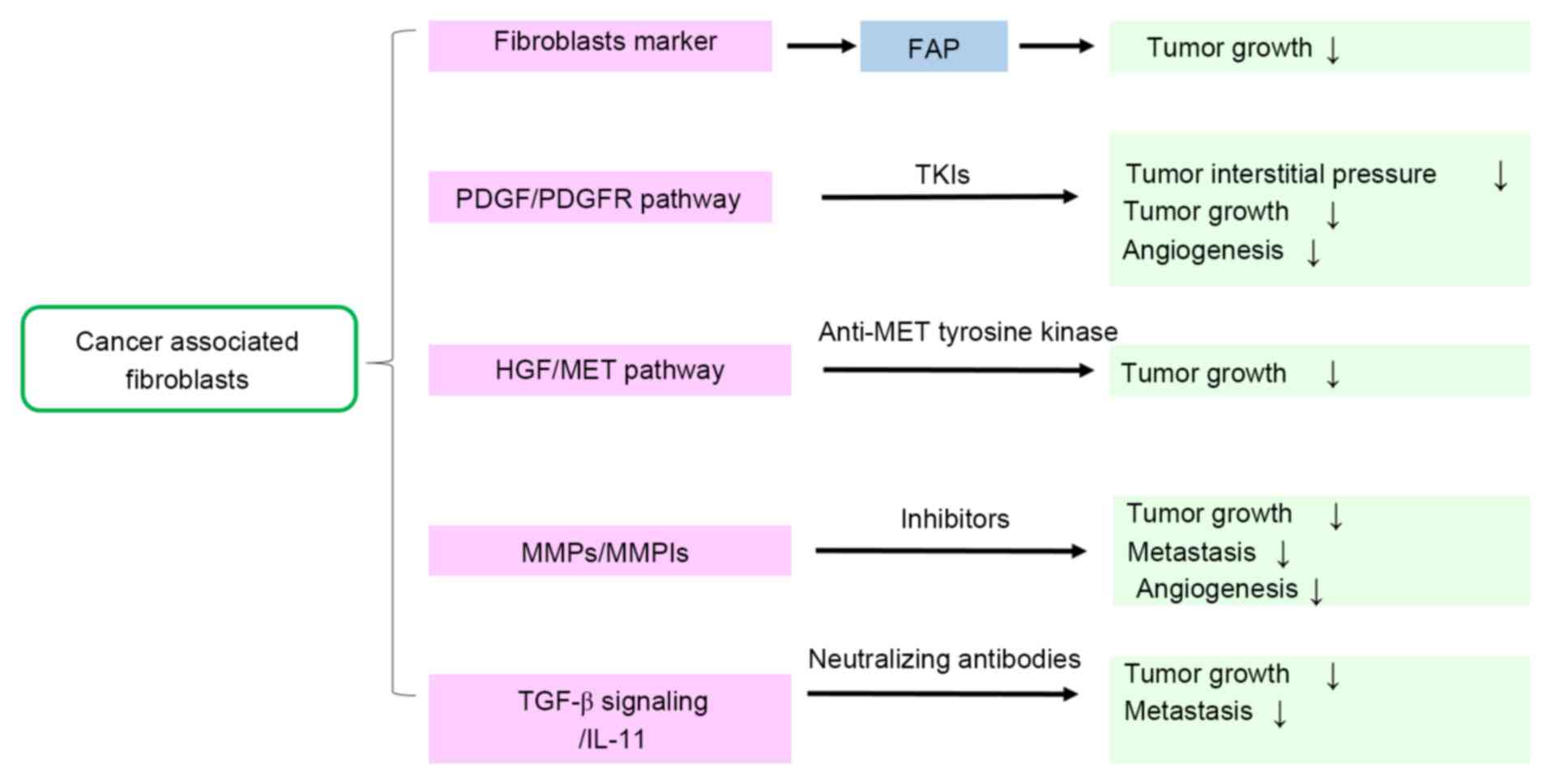 | Figure 2.Therapeutic target markers and
pathways of CAFs. This fig presents the potential strategies of
inhibiting the feedback loop and targeting CAFs during malignant
cancer treatment. CAFs, cancer-associated fibroblasts; TGF-β,
transforming growth factor β; PDGF, platelet-derived growth factor;
PDGFR, PDGF receptor; HGF, hepatocyte growth factor; MET,
hepatocyte growth factor receptor; MMPs, matrix metalloproteinases;
MMPIs, MMP inhibitors; TKIs, tyrosine kinase inhibitors; IL-11,
interleukin-11; FAP, fibroblast activation protein. |
Targeting fibroblast markers
directly
Therapy directed at specific fibroblast markers or
the antigens presented on CAFs make CAFs particularly sensitive to
cancer treatment. FAP is a membrane protein that is exclusively
overexpressed on CAFs (93). FAP has
been shown to support tumor growth and proliferation, making it a
potential target for novel anticancer therapies (94). FAP-specific molecules selectively
target fibroblasts and finally inhibit the growth of surrounding
cancer cells (94,95).
FAPα-specific monoclonal antibodies have
demonstrated therapeutic potential in cancer treatment. FAP5-DM1, a
monoclonal maytansinoid-conjugated antibody, was demonstrated to
inhibit and cause the complete regression of tumor growth in
xenograft models of lung, pancreatic, and head and neck cancer
in vivo (96).
Inhibition of FAPα enzyme activity using specific
inhibitors has also been considered a promising approach to
targeting fibroblasts. Using the peptidase inhibitor, PT-100
(talabostat) was revealed to reduce the tumor growth rate in
numerous types of tumor animal models (97). Knocking down FAPα expression resulted
in distinct tumor growth regression in an
LSL-K-rasG12D genetic mouse model of lung cancer
and in a colon cancer model, suggesting a tumor-supporting role of
endogenous FAPα (98). Furthermore,
treatment with PT-630 was able to inhibit tumor growth in the lung
and colon cancer models (98).
Targeting paracrine signaling of
fibroblasts
PDGF/PDGFR signaling pathway
Cancers stimulate CAFs through the activation of
PDGFR. A previous study demonstrated that following the
overexpression of PDGF in cancer cells, there was an increase in
the fibrotic stroma response, thus suggesting an essential role of
PDGFR signaling in fibroblast activation (99).
Multiple TKIs, including imatinib, sorafenib and
sunitinib, confer anti-PDGFR activity, and the association between
TKIs and PDGFR activity is currently being investigated (100). Imatinib, is a breakpoint cluster
region-ABL proto-oncogene 1 non-receptor tyrosine kinase inhibitor,
which also exhibits anti-PDGFR and anti-c-kit kinase activity,
resulting in decreased proliferation, and protein expression
regulation in human colorectal fibroblasts (101). Furthermore, targeting PDGFRs
increases the uptake and therefore the inhibitory effect of
chemotherapeutics, including paclitaxel, by decreasing the IFP
(62).
The indolinone derivative BIBF1120 is a potent
inhibitor of VEGFR, PDGFR and FGFR family members. It has been
revealed to inhibit MAPK and Akt signaling pathways in endothelial
cells, pericytes, and smooth muscle cells, all of which contribute
to angiogenesis, thus resulting in the inhibition of cancer cell
proliferation and apoptosis. BIBF1120 has been applied clinically
for the treatment of several types of tumor (102). Taken together, these findings
suggest that the inhibition of PDGFR signaling may serve as a novel
treatment approach for cancer.
HGF/MET signaling pathway
HGF is a growth factor that is primarily secreted by
fibroblasts to activate c-Met on cancer cells (103). Genetic and biological studies have
suggested that HGF and its receptor MET are potential targets for
cancer treatment. The progress in understanding the structure and
function of HGF/MET has led to the development of targeting drugs
and numerous small molecule MET kinase inhibitors. Reports from
previous clinical trails demonstrated that inhibiting MET signaling
has great therapeutic value in several types of human cancers,
including NSCLC (104,105).
The use of the anti-HGF monoclonal antibodies
AMG-102 (rilotumumab) and AV-299 (ficlatuzumab) has been
investigated in previous clinical trials (106,107).
Furthermore, the anti-MET agents represent a novel strategy for the
inhibition of the MET signaling pathway. Several phase I and II
clinical trials have investigated the use of novel small molecules
that target MET tyrosine kinase, including tivantinib (108), cabozantinib (109) and crizotinib (110–112).
With the results of these translational and clinical studies,
HGF/MET-targeted therapy is becoming a promising therapeutic choice
for patients with NSCLC.
MMPs/MMP inhibitors (MMPIs)
MMPs are primarily derived from CAFs in various
types of tumor. MMPs have been extensively detected in animal model
experiments, which have demonstrated the importance of these
proteases in inducing tumor growth, metastasis and angiogenesis
(113,114). Inhibitors can be used to
therapeutically target MMPs and lower the enzymatic activity,
providing a prospective for future studies. Even though the
majority of clinical trials on these drugs have reported
insufficient results, research on MMPIs remains ongoing (115,116).
Considering these explanations, one of the major difficulties in
the future is the development of inhibitors or antibodies that bind
to the active site of the enzyme and are highly specific to certain
MMPs (117).
TGF-β signaling
TGF-β stimulates myofibroblast differentiation and
the inhibition of TGF-β signaling in stromal fibroblasts result in
significant regression in tumor growth; however, the antitumor
effects of TGF-β signaling may depend primarily on individual tumor
models (118). The TGF-β signaling
pathway is increasingly considered as a therapeutic target due to
its role in cancer cells and its capacity to instruct a
protumorigenic program in tumor stromal cells (119). Several therapeutic agents that
inhibit the TGF-β signaling pathway have been studied in
preclinical and clinical trials. Neutralizing antibodies, soluble
receptors and antisense oligonucleotides that target the
ligand-receptor interaction, and inhibit the function of TGFBRI or
TGFBRII have been studied in clinical experiments (120). The clinical application of the
TGFBRI kinase inhibitor LY2157299 has been investigated in
glioblastoma (121), hepatocellular
carcinoma (122) and advanced
pancreatic cancer (123); these
studies have provided promising results.
Crosstalk between cancer cells and CAFs through
TGF-β could suggest another therapeutic target. IL-11 has been
recognized for its capacity to promote the maturation of platelets
producing megakaryocyte progenitors in vitro and in the bone
marrow in vivo (124). A
previous study investigated the pro-metastatic effect of IL-11,
which is secreted by TGF-β-stimulated CAFs in CRC (125). It was reported that IL-11 promotes
the survival of tumor cells at the sites of metastatic colonization
(125). This finding suggests that
the clinical use of IL-11 to treat thrombocytopenia caused by
chemotherapy agents should be reconsidered and the use of anti-IL11
therapies against CRC should be evaluated.
Conclusion
CAFs are considered as an essential component of
tumorigenesis. Increasing evidence has suggested that CAFs exhibit
a positive effect on the development of solid tumors. CAFs can
modulate tumor microenvironment through diverse mechanisms, thus
supporting tumor progression. Pre-clinical and clinical trials have
revealed that CAFs are a potential target for the treatment of
solid tumors.
References
|
1
|
Balkwill FR, Capasso M and Hagemann T: The
tumor microenvironment at a glance. J Cell Sci. 125:5591–5596.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Spano D and Zollo M: Tumor
microenvironment: A main actor in the metastasis process. Clin Exp
Metastasis. 29:381–395. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Swartz MA, Iida N, Roberts EW, Sangaletti
S, Wong MH, Yull FE, Coussens LM and DeClerck YA: Tumor
microenvironment complexity: Emerging roles in cancer therapy.
Cancer Res. 72:2473–2480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cirri P and Chiarugi P:
Cancer-associated-fibroblasts and tumour cells: A diabolic liaison
driving cancer progression. Cancer Metast Rev. 31:195–208. 2012.
View Article : Google Scholar
|
|
7
|
Marsh T, Pietras K and McAllister SS:
Fibroblasts as architects of cancer pathogenesis. Biochim Biophys
Acta. 1832:1070–1078. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun Y: Translational horizons in the tumor
microenvironment: Harnessing breakthroughs and targeting cures. Med
Res Rev. 35:408–436. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Slany A, Bileck A, Muqaku B and Gerner C:
Targeting breast cancer-associated fibroblasts to improve
anti-cancer therapy. Breast. 24:532–538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anderberg C and Pietras K: On the origin
of cancer-associated fibroblasts. Cell Cycle. 8:1461–1462. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Y, Hu T, Shen J, Li SF, Lin JW, Zheng
XH, Gao QH and Zhou HM: Separation, cultivation and biological
characteristics of oral carcinoma-associated fibroblasts. Oral Dis.
12:375–380. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sugimoto H, Mundel TM, Kieran MW and
Kalluri R: Identification of fibroblast heterogeneity in the tumor
microenvironment. Cancer Biol Ther. 5:1640–1646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park JE, Lenter MC, Zimmermann RN,
Garin-Chesa P, Old LJ and Rettig WJ: Fibroblast activation protein,
a dual specificity serine protease expressed in reactive human
tumor stromal fibroblasts. J Biol Chem. 274:36505–36512. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim HM, Jung WH and Koo JS: Expression of
cancer-associated fibroblast related proteins in metastatic breast
cancer: An immunohistochemical analysis. J Transl Med. 13:2222015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rupp C, Scherzer M, Rudisch A, Unger C,
Haslinger C, Schweifer N, Artaker M, Nivarthi H, Moriggl R,
Hengstschläger M, et al: IGFBP7, a novel tumor stroma marker, with
growth-promoting effects in colon cancer through a paracrine
tumor-stroma interaction. Oncogene. 34:815–825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Navab R, Strumpf D, Bandarchi B, Zhu CQ,
Pintilie M, Ramnarine VR, Ibrahimov E, Radulovich N, Leung L,
Barczyk M, et al: Prognostic gene-expression signature of
carcinoma-associated fibroblasts in non-small cell lung cancer.
Proc Natl Acad Sci USA. 108:pp. 7160–7165. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu CQ, Popova SN, Brown ER,
Barsyte-Lovejoy D, Navab R, Shih W, Li M, Lu M, Jurisica I, Penn
LZ, et al: Integrin alpha11 regulates IGF2 expression in
fibroblasts to enhance tumorigenicity of human non-small-cell lung
cancer cells. Proc Natl Acad Sci USA. 104:pp. 11754–11759. 2007;
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakagawa H, Liyanarachchi S, Davuluri RV,
Auer H, Martin EW Jr, de la Chapelle A and Frankel WL: Role of
cancer-associated stromal fibroblasts in metastatic colon cancer to
the liver and their expression profiles. Oncogene. 23:7366–7377.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Walter K, Omura N, Hong SM, Griffith M,
Vincent A, Borges M and Goggins M: Overexpression of smoothened
activates the sonic hedgehog signaling pathway in pancreatic
cancer-associated fibroblasts. Clin Cancer Res. 16:1781–1789. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rozenchan PB, Carraro DM, Brentani H, de
Carvalho Mota LD, Bastos EP, e Ferreira EN, Torres CH, Katayama ML,
Roela RA, Lyra EC, et al: Reciprocal changes in gene expression
profiles of cocultured breast epithelial cells and primary
fibroblasts. Int J Cancer. 125:2767–2777. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Räsänen K and Vaheri A: Activation of
fibroblasts in cancer stroma. Exp Cell Res. 316:2713–2722. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Clayton A, Evans RA, Pettit E, Hallett M,
Williams JD and Steadman R: Cellular activation through the
ligation of intercellular adhesion molecule-1. J Cell Sci.
111:443–453. 1998.PubMed/NCBI
|
|
24
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mitra AK, Zillhardt M, Hua Y, Tiwari P,
Murmann AE, Peter ME and Lengyel E: MicroRNAs reprogram normal
fibroblasts into cancer-associated fibroblasts in ovarian cancer.
Cancer Discov. 2:1100–1108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao L, Sun Y, Hou Y, Peng Q, Wang L, Luo
H, Tang X, Zeng Z and Liu M: MiRNA expression analysis of
cancer-associated fibroblasts and normal fibroblasts in breast
cancer. Int J Biochem Cell Biol. 44:2051–2059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Enkelmann A, Heinzelmann J, von Eggeling
F, Walter M, Berndt A, Wunderlich H and Junker K: Specific protein
and miRNA patterns characterise tumour-associated fibroblasts in
bladder cancer. J Cancer Res Clin Oncol. 137:751–759. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu Z, Willmarth NE, Zhou J, Katiyar S,
Wang M, Liu Y, McCue PA, Quong AA, Lisanti MP and Pestell RG:
microRNA 17/20 inhibits cellular invasion and tumor metastasis in
breast cancer by heterotypic signaling. Proc Natl Acad Sci USA.
107:pp. 8231–8236. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aprelikova O, Yu X, Palla J, Wei BR, John
S, Yi M, Stephens R, Simpson RM, Risinger JI, Jazaeri A and
Niederhuber J: The role of miR-31 and its target gene SATB2 in
cancer-associated fibroblasts. Cell Cycle. 9:4387–4398. 2014.
View Article : Google Scholar
|
|
30
|
Wang S, Wang Z, Xu K, Ruan Z and Chen L:
miRNA expression analysis of cancer-associated fibroblasts and
normal fibroblasts in colorectal cancer. J Mod Oncol. 09:1918–1922.
2013.
|
|
31
|
Bhowmick NA, Neilson E G and Moses HL:
Stromal fibroblasts in cancer initiation and progression. Nature.
432:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gonda TA, Varro A, Wang TC and Tycko B:
Molecular biology of cancer-associated fibroblasts: Can these cells
be targeted in anti-cancer therapy? Seminars Cell Dev Biol. 21:pp.
2–10. 2009; View Article : Google Scholar
|
|
33
|
Ostman A and Augsten M: Cancer-associated
fibroblasts and tumor growth-bystanders turning into key players.
Curr Opin Genet Dev. 19:67–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Servais C and Erez N: From sentinel cells
to inflammatory culprits: Cancer-associated fibroblasts in
tumour-related inflammation. J Pathol. 229:198–207. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luker KE, Lewin SA, Mihalko LA, Schmidt
BT, Winkler JS, Coggins NL, Thomas DG and Luker GD: Scavenging of
CXCL12 by CXCR7 promotes tumor growth and metastasis of
CXCR4-positive breast cancer cells. Oncogene. 31:4750–4758. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Augsten M, Sjöberg E, Frings O, Vorrink
SU, Frijhoff J, Olsson E, Borg Å and Östman A: Cancer-associated
fibroblasts expressing CXCL14 rely upon NOS1-derived nitric oxide
signaling for their tumor-supporting properties. Cancer Res.
74:2999–3010. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mi Z, Bhattacharya SD, Kim VM, Guo H,
Talbot LJ and Kuo PC: Osteopontin promotes CCL5-mesenchymal stromal
cell-mediated breast cancer metastasis. Carcinogenesis. 32:477–487.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Quante M, Tu SP, Tomita H, Gonda T, Wang
SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, et al: Bone
marrow-derived myofibroblasts contribute to the mesenchymal stem
cell niche and promote tumor growth. Cancer Cell. 19:257–272. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li X, Xu Q, Wu Y, Li J, Tang D, Han L and
Fan Q: A CCL2/ROS autoregulation loop is critical for
cancer-associated fibroblasts-enhanced tumor growth of oral
squamous cell carcinoma. Carcinogenesis. 35:1362–1370. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shibuya M and Claesson-Welsh L: Signal
transduction by VEGF receptors in regulation of angiogenesis and
lymphangiogenesis. Exp Cell Res. 312:549–560. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gomes FG, Nedel F, Alves AM, Nör JE and
Tarquinio SB: Tumor angiogenesis and lymphangiogenesis:
Tumor/endothelial crosstalk and cellular/microenvironmental
signaling mechanisms. Life Sci. 92:101–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ferrara N: Pathways mediating
VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev.
21:21–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang J and Liu J: Tumor stroma as targets
for cancer therapy. Pharmacol Ther. 137:200–215. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nagasaki T, Hara M, Nakanishi H, Takahashi
H, Sato M and Takeyama H: Interleukin-6 released by colon
cancer-associated fibroblasts is critical for tumour angiogenesis:
Anti-interleukin-6 receptor antibody suppressed angiogenesis and
inhibited tumour-stroma interaction. Br J Cancer. 110:469–478.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Karagiannis GS, Poutahidis T, Erdman SE,
Kirsch R, Riddell RH and Diamandis EP: Cancer-associated
fibroblasts drive the progression of metastasis through both
paracrine and mechanical pressure on cancer tissue. Mol Cancer Res.
10:1403–1418. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vpavlides S, Vera I, Gandara R, Sneddon S,
Pestell RG, Mercier I, Martinez-Outschoorn UE, Whitaker-Menezes D,
Howell A, Sotgia F and Lisanti MP: Warburg meets autophagy:
Cancer-associated fibroblasts accelerate tumor growth and
metastasis via oxidative stress, mitophagy, and aerobic glycolysis.
Antioxid Redox Signal. 16:1264–1284. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
De Wever O, Van Bockstal M, Mareel M,
Hendrix A and Bracke M: Carcinoma-associated fibroblasts provide
operational flexibility in metastasis. Semin Cancer Biol. 25:33–46.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dumont N, Liu B, DeFilippis RA, Chang H,
Rabban JT, Karnezis AN, Tjoe JA, Marx J, Parvin B and Tlsty TD:
Breast fibroblasts modulate early dissemination, tumorigenesis, and
metastasis through alteration of extracellular matrix
characteristics. Neoplasia. 15:249–262. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Olumi AF, Grossfeld GD, Hayward SW,
Carroll PR, Tlsty TD and Cunha GR: Carcinoma-associated fibroblasts
direct tumor progression of initiated human prostatic epithelium.
Cancer Res. 59:5002–5011. 1999.PubMed/NCBI
|
|
50
|
Calvo F, Ege N, Grande-Garcia A, Hooper S,
Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary
E, Charras G and Sahai E: Mechanotransduction and YAP-dependent
matrix remodelling is required for the generation and maintenance
of cancer-associated fibroblasts. Nat Cell Biol. 15:637–646. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Erez N, Truitt M, Olson P, Arron ST and
Hanahan D: Cancer-associated fibroblasts are activated in incipient
neoplasia to orchestrate tumor-promoting inflammation in an
NF-kappaB-dependent manner. Cancer cell. 17:135–147. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang XH, Jin X, Malladi S, Zou Y, Wen YH,
Brogi E, Smid M, Foekens JA and Massagué J: Selection of bone
metastasis seeds by mesenchymal signals in the primary tumor
stroma. Cell. 154:1060–1073. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Aprelikova O, Palla J, Hibler B, Yu X,
Greer YE, Yi M, Stephens R, Maxwell GL, Jazaeri A, Risinger JI, et
al: Silencing of miR-148a in cancer-associated fibroblasts results
in WNT10B-mediated stimulation of tumor cell motility. Oncogene.
32:3246–3253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Verghese ET, Drury R, Green CA, Holliday
DL, Lu X, Nash C, Speirs V, Thorne JL, Thygesen HH, Zougman A, et
al: MiR-26b is down-regulated in carcinoma-associated fibroblasts
from ER-positive breast cancers leading to enhanced cell migration
and invasion. J Pathol. 231:388–399. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bronisz A, Godlewski J, Wallace JA,
Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ,
Martin CK, Li F, et al: Reprogramming of the tumour
microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol.
14:159–167. 2012. View Article : Google Scholar
|
|
56
|
Mongiat M, Marastoni S, Ligresti G,
Lorenzon E, Schiappacassi M, Perris R, Frustaci S and Colombatti A:
The extracellular matrix glycoprotein elastin microfibril interface
located protein 2: A dual role in the tumor microenvironment.
Neoplasia. 12:294–304. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Correia AL and Bissell MJ: The tumor
microenvironment is a dominant force in multidrug resistance. Drug
Resist Updat. 15:39–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kerbel RS: A cancer therapy resistant to
resistance. Nature. 390:335–336. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
59
|
Swartz MA and Lund AW: Lymphatic and
interstitial flow in the tumour microenvironment: Linking
mechanobiology with immunity. Nat Rev Cancer. 12:210–219. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Khawar IA, Kim JH and Kuh HJ: Improving
drug delivery to solid tumors: Priming the tumor microenvironment.
J Control Release. 201:78–89. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pietras K, Östman A, Sjöquist M,
Buchdunger E, Reed RK, Heldin CH and Rubin K: Inhibition of
Platelet-derived growth factor receptors reduces interstitial
hypertension and increases transcapillary transport in tumors.
Cancer Res. 61:2929–2934. 2001.PubMed/NCBI
|
|
62
|
Pietras K, Rubin K, Sjöblom T, Buchdunger
E, Sjöquist M, Heldin CH and Ostman A: Inhibition of PDGF receptor
signaling in tumor stroma enhances antitumor effect of
chemotherapy. Cancer Res. 62:5476–5484. 2002.PubMed/NCBI
|
|
63
|
Wilson TR, Fridlyand J, Yan Y, Penuel E,
Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al:
Widespread potential for growth-factor-driven resistance to
anticancer kinase inhibitors. Nature. 487:505–509. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Straussman R, Morikawa T, Shee K,
Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J,
Frederick DT, et al: Tumour micro-environment elicits innate
resistance to RAF inhibitors through HGF secretion. Nature.
487:500–504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yano S, Wang W, Li Q, Yamada T, Takeuchi
S, Matsumoto K, Nishioka Y and Sone S: HGF-MET in resistance to
EGFR tyrosine kinase inhibitors in lung cancer. Curr Signal Trans
Ther. 6:228–233. 2011. View Article : Google Scholar
|
|
67
|
Liska D, Chen CT, Bachleitner-Hofmann T,
Christensen JG and Weiser MR: HGF rescues colorectal cancer cells
from EGFR inhibition via MET activation. Clin Cancer Res.
17:472–482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yamatodani T, Ekblad L, Kjellén E,
Johnsson A, Mineta H and Wennerberg J: Epidermal growth factor
receptor status and persistent activation of Akt and p44/42 MAPK
pathways correlate with the effect of cetuximab in head and neck
and colon cancer cell lines. J Cancer Res Clin Oncol.
135:3954022009. View Article : Google Scholar
|
|
69
|
Liska D, Chen CT, Bachleitner-Hofmann T,
Christensen JG and Weiser MR: HGF rescues colorectal cancer cells
from EGFR inhibition via MET activation. Clin Cancer Res.
17:472–482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Luraghi P, Reato G, Cipriano E, Sassi F,
Orzan F, Bigatto V, De Bacco F, Menietti E, Han M, Rideout WM III,
et al: MET signaling in colon cancer stem-like cells blunts the
therapeutic response to EGFR inhibitors. Cancer Res. 74:1857–1869.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Qian DZ, Rademacher BL, Pittsenbarger J,
Huang CY, Myrthue A, Higano CS, Garzotto M, Nelson PS and Beer TM:
CCL2 is induced by chemotherapy and protects prostate cancer cells
from docetaxel-induced cytotoxicity. Prostate. 70:433–442.
2010.PubMed/NCBI
|
|
72
|
Moisan F, Francisco EB, Brozovic A, Duran
GE, Wang YC, Chaturvedi S, Seetharam S, Snyder LA, Doshi P and
Sikic BI: Enhancement of paclitaxel and carboplatin therapies by
CCL2 blockade in ovarian cancers. Mol Oncol. 8:1231–1239. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tsuyada A, Chow A, Wu J, Somlo G, Chu P,
Loera S, Luu T, Li AX, Wu X, Ye W, et al: CCL2 mediates cross-talk
between cancer cells and stromal fibroblasts that regulates breast
cancer stem cells. Cancer Res. 72:2768–2779. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Weekes CD, Song D, Arcaroli J, Wilson LA,
Rubio-Viqueira B, Cusatis G, Garrett-Mayer E, Messersmith WA, Winn
RA and Hidalgo M: Stromal cell-derived factor 1α mediates
resistance to mTOR-directed therapy in pancreatic cancer.
Neoplasia. 14:690–701. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Singh S, Srivastava SK, Bhardwaj A, Owen
LB and Singh AP: CXCL12-CXCR4 signalling axis confers gemcitabine
resistance to pancreatic cancer cells: A novel target for therapy.
Br J Cancer. 103:1671–1679. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Domanska UM, Timmer-Bosscha H, Nagengast
WB, Munnink TH Oude, Kruizinga RC, Ananias HJ, Kliphuis NM, Huls G,
De Vries EG, de Jong IJ and Walenkamp AM: CXCR4 inhibition with
AMD3100 sensitizes prostate cancer to docetaxel chemotherapy.
Neoplasia. 14:709–718. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Heckmann D, Maier P, Laufs S, Wenz F,
Zeller WJ, Fruehauf S and Allgayer H: CXCR4 expression and
treatment with SDF-1α or plerixafor modulate proliferation and
chemosensitivity of colon cancer cells. Transl Oncol. 6:124–132.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Burger JA, Stewart DJ, Wald O and Peled A:
Potential of CXCR4 antagonists for the treatment of metastatic lung
cancer. Expert Rev Anticancer Ther. 11:621–630. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lotti F, Jarrar AM, Pai RK, Hitomi M,
Lathia J, Mace A, Gantt GA Jr, Sukhdeo K, DeVecchio J, Vasanji A,
et al: Chemotherapy activates cancer-associated fibroblasts to
maintain colorectal cancer-initiating cells by IL-17A. J Exp Med.
210:2851–2872. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Cochaud S, Giustiniani J, Thomas C,
Laprevotte E, Garbar C, Savoye AM, Curé H, Mascaux C, Alberici G,
Bonnefoy N, et al: IL-17A is produced by breast cancer TILs and
promotes chemoresistance and proliferation through ERK1/2. Sci Rep.
3:34562013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Studebaker AW, Storci G, Werbeck JL,
Sansone P, Sasser AK, Tavolari S, Huang T, Chan MW, Marini FC,
Rosol TJ, et al: Fibroblasts isolated from common sites of breast
cancer metastasis enhance cancer cell growth rates and invasiveness
in an interleukin-6-dependent manner. Cancer Res. 68:9087–9095.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Gao SP, Mark KG, Leslie K, Pao W, Motoi N,
Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B and Bromberg
JF: Mutations in the EGFR kinase domain mediate STAT3 activation
via IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yao Z, Fenoglio S, Gao DC, Camiolo M,
Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V,
et al: TGF-beta IL-6 axis mediates selective and adaptive
mechanisms of resistance to molecular targeted therapy in lung
cancer. Proc Natl Acad Sci USA. 107:pp. 15535–15540. 2010;
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sun X, Mao Y, Wang J, Zu L, Hao M, Cheng
G, Qu Q, Cui D, Keller ET, Chen X, et al: IL-6 secreted by
cancer-associated fibroblasts induces tamoxifen resistance in
luminal breast cancer. Oncogene. doi: 10.1038/onc.2014.158.
|
|
85
|
Sun Y, Campisi J, Higano C, Beer TM,
Porter P, Coleman I, True L and Nelson PS: Treatment-induced damage
to the tumor microenvironment promotes prostate cancer therapy
resistance through WNT16B. Nat Med. 18:1359–1368. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Amornsupuk K, Insawang T, Thuwajit P,
O-Charoenrat P, Eccles SA and Thuwajit C: Cancer-associated
fibroblasts induce high mobility group box 1 and contribute to
resistance to doxorubicin in breast cancer cells. BMC Cancer.
14:9552014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cullen KJ, Smith HS, Hill S, Rosen N and
Lippman ME: Growth factor messenger RNA expression by human breast
fibroblasts from benign and malignant lesions. Cancer Res.
51:4978–4985. 1991.PubMed/NCBI
|
|
88
|
Shay G, Lynch CC and Fingleton B: Moving
targets: Emerging roles for MMPs in cancer progression and
metastasis. Matrix Biol 44–46. 1–206. 2015.
|
|
89
|
Jia CC, Wang TT, Liu W, Fu BS, Hua X, Wang
GY, Li TJ, Li X, Wu XY, Tai Y, et al: Cancer-associated fibroblasts
from hepatocellular carcinoma promote malignant cell proliferation
by HGF secretion. PLoS One. 8:e632432013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lin J, Liu C, Ge L, Gao Q, He X, Liu Y, Li
S, Zhou M, Chen Q and Zhou H: Carcinoma-associated fibroblasts
promotes the proliferation of a lingual carcinoma cell line by
secreting keratinocyte growth factor. Tumor Biol. 32:597–602. 2011.
View Article : Google Scholar
|
|
91
|
Weroha SJ and Haluska P: The insulin-like
growth factor system in cancer. Endocrinol Metab Clin North Am.
41:335–350.vi. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Hawinkels LJ, Paauwe M, Verspaget HW,
Wiercinska E, van der Zon JM, van der Ploeg K, Koelink PJ, Lindeman
JH, Mesker W, ten Dijke P and Sier CF: Interaction with colon
cancer cells hyperactivates TGF-β signaling in cancer-associated
fibroblasts. Oncogene. 33:97–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mueller MM and Fusenig NE: Friends or
foes-bipolar effects of the tumour stroma in cancer. Nat Rev
Cancer. 4:839–849. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Liu R, Li H, Liu L, Yu J and Ren X:
Fibroblast activation protein: A potential therapeutic target in
cancer. Cancer Biol Ther. 13:123–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
LeBeau AM, Brennen WN, Aggarwal S and
Denmeade SR: Targeting the cancer stroma with a fibroblast
activation protein-activated promelittin protoxin. Mol Cancer Ther.
8:1378–1386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ostermann E, Garin-Chesa P, Heider KH,
Kalat M, Lamche H, Puri C, Kerjaschki D, Rettig WJ and Adolf GR:
Effective immunoconjugate therapy in cancer models targeting a
serine protease of tumor fibroblasts. Clin Cancer Res.
14:4584–4592. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Adams S, Miller GT, Jesson MI, Watanabe T,
Jones B and Wallner BP: PT-100, a small molecule dipeptidyl
peptidase inhibitor, has potent antitumor effects and augments
antibody-mediated cytotoxicity via a novel immune mechanism. Cancer
Res. 64:5471–5480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Santos AM, Jung J, Aziz N, Kissil JL and
Puré E: Targeting fibroblast activation protein inhibits tumor
stromagenesis and growth in mice. J Clin Invest. 119:3613–3625.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Heldin CH: Targeting the PDGF signaling
pathway in tumor treatment. Cell Commun Signal. 11:972013.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Steeghs N, Nortier JW and Gelderblom H:
Small molecule tyrosine kinase inhibitors in the treatment of solid
tumors: An update of recent developments. Ann Surg Oncol.
14:942–953. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Pietras K, Pahler J, Bergers G and Hanahan
D: Functions of paracrine PDGF signaling in the proangiogenic tumor
stroma revealed by pharmacological targeting. PLos Med. 5:e192008.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Hilberg F, Roth GJ, Krssak M, Kautschitsch
S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel
A, Quant J, et al: BIBF 1120: Triple angiokinase inhibitor with
sustained receptor blockade and good antitumor efficacy. Cancer
Res. 68:4774–4782. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Cecchi F, Rabe DC and Bottaro DP:
Targeting the HGF/Met signaling pathway in cancer therapy. Expert
Opin Ther Targets. 16:553–572. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Gherardi E, Birchmeier W, Birchmeier C and
Woude G Vande: Targeting MET in cancer: Rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Sadiq AA and Salgia R: MET as a possible
target for non-small-cell lung cancer. J Clin Oncol. 31:1089–1096.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Oliner KS, Tang R, Anderson A, et al:
Evaluation of MET pathway biomarkers in a phase II study of
rilotumumab (R, AMG 102) or placebo (P) in combination with
epirubicin, cisplatin and capecitabine (ECX) in patients (pts) with
locally advanced or metastatic gastric (G) or esophagogastric
junction (EGJ) cancerJ Clin Oncol Amer Soc Clin Oncol. 2318 Mill
Road, Ste 800, Alexandria, Va 22314 USA: 2012
|
|
107
|
Tan E, Park K, Lim WT, et al: Phase 1b
study of ficlatuzumab (AV-299), an anti-hepatocyte growth factor
monoclonal antibody, in combination with gefitinib in Asian
patients with NSCLC. J Clin Oncol. 29 Suppl:493S2011. View Article : Google Scholar
|
|
108
|
Katayama R, Aoyama A, Yamori T, Qi J,
Oh-hara T, Song Y, Engelman JA and Fujita N: Cytotoxic activity of
tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer
Res. 73:3087–3096. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wakelee H, Gettinger S, Engelman J, et al:
A phase Ib/II study of XL184 (BMS 907351) with and without
erlotinib (E) in patients (pts) with non-small cell lung cancer
(NSCLC). ASCO Annual Meeting Proceedings. pp. 30172010;
|
|
110
|
Tanizaki J, Okamoto I, Okamoto K, Takezawa
K, Kuwata K, Yamaguchi H and Nakagawa K: MET tyrosine kinase
inhibitor crizotinib (PF-02341066) shows differential antitumor
effects in non-small cell lung cancer according to MET alterations.
J Thorac Oncol. 6:1624–1631. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Feng Y, Thiagarajan PS and Ma PC: MET
signaling: Novel targeted inhibition and its clinical development
in lung cancer. J Thorac Oncol. 7:459–467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Blumenschein GR Jr, Mills GB and
Gonzalez-Angulo AM: Targeting the hepatocyte growth factor-cMET
axis in cancer therapy. J Clin Oncol. 30:3287–3296. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
114
|
Rundhaug JE: Matrix metalloproteinases and
angiogenesis. J Cell Mol Med. 9:267–285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Pavlaki M and Zucker S: Matrix
metalloproteinase inhibitors (MMPIs): The beginning of phase I or
the termination of phase III clinical trials. Cancer Metastasis
Rev. 22:177–203. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Shepherd FA, Giaccone G, Seymour L,
Debruyne C, Bezjak A, Hirsh V, Smylie M, Rubin S, Martins H, Lamont
A, et al: Prospective, randomized, double-blind, placebo-controlled
trial of marimastat after response to first-line chemotherapy in
patients with small-cell lung cancer: A trial of the national
cancer institute of Canada-clinical trials group and the European
organization for research and treatment of cancer. J Clin Oncol.
20:4434–4439. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Konstantinopoulos PA, Karamouzis MV,
Papatsoris AG and Papavassiliou AG: Matrix metalloproteinase
inhibitors as anticancer agents. Int J Biochem Cell Biol.
40:1156–1168. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Bierie B and Moses HL: TGF-β and cancer.
Cytokine Growth Factor Rev. 17:29–40. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lonning S, Mannick J and McPherson J:
Antibody targeting of TGF-β in cancer patients. Curr Pharm
Biotechnol. 12:2176–2189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Hawinkels LJ and Ten Dijke P: Exploring
anti-TGF-β therapies in cancer and fibrosis. Growth Factors.
29:140–152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Rodon J, Carducci MA, Sepulveda-Sánchez
JM, Azaro A, Calvo E, Seoane J, Braña I, Sicart E, Gueorguieva I,
Cleverly AL, et al: First-in-human dose study of the novel
transforming growth factor-β receptor I kinase inhibitor LY2157299
monohydrate in patients with advanced cancer and glioma. Clin
Cancer Res. 21:553–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Rani B, Dituri F, Cao Y, Engström U, Lupo
L, Dooley S, Moustakas A and Giannelli G: P0320: Targeting TGF-beta
I with the transforming growth factor receptor type I kinase
inhibitor, LY2157299, modulates stemness-related biomarkers in
hepatocellular carcinoma. J Hepatol. 62:S4292015. View Article : Google Scholar
|
|
123
|
Whatcott CJ, Dumas SN, Watanabe A, LoBello
J, Von Hoff DD and Han H: Abstract 2135: TGFβRI inhibition results
in reduced collagen expression in pancreatic ductal adenocarcinoma.
Cancer Res. DOI: 10.1158/1538-7445.
|
|
124
|
Johnstone CN, Chand A, Putoczki TL and
Ernst M: Emerging roles for IL-11 signaling in cancer development
and progression: Focus on breast cancer. Cytokine Growth Factor
Rev. 26:489–498. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Calon A, Espinet E, Palomo-Ponce S,
Tauriello DV, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung
P, Zhang XH, et al: Dependency of colorectal cancer on a
TGF-β-driven program in stromal cells for metastasis initiation.
Cancer Cell. 22:571–584. 2012. View Article : Google Scholar : PubMed/NCBI
|