Introduction
Malignant glioblastoma is an aggressive and
incurable tumor, with an annual incidence of 5.26 per 100,000
population or 17,000 new diagnoses per year (1), which represents nearly 80% of diagnosed
primary brain tumors. In children, glioblastoma accounts for about
one-fifth of all childhood cancers (2). Glioblastoma is among the most feared
types of cancers which are usually associated with poor prognosis
and profoundly impaired life quality. Glioblastoma originates from
glial cells in central nervous system, and previous work
demonstrated that chromosome 10 loss, p16INK4a deletion, p14ARF,
PTEN and p53 mutation, RB1 and MGMT methylation, EGFR amplification
contributed to the pathogenesis of glioblastoma (3,4). The
current standard cure for newly diagnosed glioblastoma patients is
surgical removal combined with radiotherapy and then chemotherapy
with the temozolomide if the tumor is high-grade. However, the
exact molecular cause of glioblastoma is hard to decipher. In
addition, many glioblastoma patients show high resistances to these
therapeutic treatments, especially for the standard chemo
drugs-temozolomide and carmustine (BCNU), and thus tumor
recurrences are frequent. For example, intensive studies found that
the overexpression of MGMT (O6-methylguanine methyl transferase)
and inactivation mutations in the mismatch repair gene MSH6
(mutS homolog 6) were closely related with glioblastoma recurrent
post-temozolomide treatment (5,6), and the
resistance mechanisms should have equal effects for carmustine in
that they shared the same alkylating effect of DNA (7). Therefore, clearly revealing the
underlying mechanisms of chemo-drug tolerance is the most urgent
issue of improving the therapies of glioblastoma.
As is known to all, rapidly-proliferated and
metastatic tumor cells consume lots of nutrients through adequate
blood supply, so anti-angiogenic therapy has become an important
method in the treatment of many solid tumors. Glioblastomas is
highly vascularized (8) and
overexpresses vascular endothelial growth factor A (VEGF-A) that is
responsible for the angiogenesis (9).
As the first available anti-angiogenic drug, bevacizumab was
granted accelerated approval by FDA in 2009 for the treatment of
recurrent multiform glioblastoma. Bevacizumab is one kind of
recombinant humanized monoclonal antibody that targets for VEGF-A
and blocks its binding to VEGF receptor, which thus inhibits the
angiogenesis in a variety of diseases, especially for cancers, such
as colorectal cancer, lung cancer, cervical cancer, ovarian cancer
and renal cell carcinoma (Avastin Prescribing Information;
Genentech, Inc., December 2016). In preclinical experiments and
early clinical trials, bevacizumab had some efficacies on
prolonging progression-free survival, possibly improving quality of
life and decreasing steroid usage. However, it did not show an
overall-survival benefit in a late clinical trial of patients with
glioblastoma (10,11). Some studies were performed to explore
the reason of low efficacy of bevacizumab for glioblastoma
patients. Several mechanisms, including receptor tyrosine kinase
c-Met upregulation, myeloid cell infiltration and stem cell
accumulation, were identified to be associated with the resistance
of glioblastomas to anti-angiogenic therapy (12,13). In
colorectal cancer cells, people found that the prolonged activation
of autocrine VEGF signaling might contribute to the bevacizumab
resistance (14). To improve the
efficacy of bevacizumab, additional researches are still required
to explore the mechanisms of resistance, other pro-angiogenic
pathways and new combination strategies.
Autophagy is a highly conserved system responsible
for the removal of damaged organelles or misfolded proteins by
lysosomal degradation, which contributes to maintain intercellular
homeostasis. Previous studies demonstrated that autophagy could
play significant roles in antigen presentation, cell death,
bacterial and viral infection (15,16).
Dysfunction of autophagy is associated with the pathogenesis of
metabolic and neurodegenerative diseases, viral infection, muscle
diseases, cancer, and hepatic inflammation (17–19).
Autophagy process consists of a series of steps: i) The initiation
of the isolated membrane; ii) cargo recognition and nucleation;
iii) elongation of the isolated membrane; iv) enclosure of membrane
structures and formation of autophagosome; and v) maturation and
degradation of engulfed proteins (20). During autophagy, microtubule
associated protein 1-light chain 3 (LC3, one homolog of ATG8) is
firstly loaded onto the membrane by conjugating with
phosphatidylethanolamine (POPE) in the membrane, which will modify
the curvature of membrane and promote the maturation of
autophagosome. Then, the cargo is loaded into the autophagosome by
the interaction between the specific receptors on cargo proteins
and LC3 on the autophagosome membrane, in which the first
identified selective receptor is SQSTM1(p62) (21). After formation, autophagosome will
fuse with the lysosome to digest the loaded cargo proteins
(22). Previous studies found that
autophagy could either support or suppress the tumor cell growth
depending on the cell context (23).
In normal tissues and cells, autophagy serves as a
tumor-suppressive process (24).
However, once the malignant phenotype has been established,
autophagy is often harnessed to facilitate tumor cell survival
under metabolic stresses caused by antitumor agents (25). It was also reported that autophagy
could be induced in response to chemotherapeutics, promoting the
formation of drug-tolerance and the impairment of tumor therapy
(26–28). Therefore, targeting autophagy is an
attractive and promising therapeutic strategy to potentiate the
effects of chemotherapy and improve clinical outputs in the
treatment of cancer patients (29).
Until now, there are no available reports about the
autophagy involved in the tolerance of glioblastomas to
bevacizumab. Here, we used a glioblastoma cell line, U87-MG cells,
to systematically study the anti-proliferation and pro-apoptosis
effects of bevacizumab on glioblastoma cells. We found that
bevacizumab could induce the downregulate the anti-apoptotic
proteins and upregulate the pro-apoptotic proteins in glioblastomas
cells to promote their apoptosis. However, glioblastomas cells were
able to enhance their autophagy to tolerant bevacizumab through
attenuating Akt-mTOR signaling pathway, while blockade of the
autophagy process by its inhibitor could significantly increase the
tumor-suppressive effect of bevacizumab on glioblastomas.
Materials and methods
Cell culture and reagents
The human glioblastoma cell line, U87-MG was bought
from ATCC and maintained in Dulbecco's modified Eagle's medium
(DMEM) (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
and supplemented with 10% fetal bovine serum at 37°C in a
humidified 5% CO2 incubator. Although one research
published in Science Translational Medicine revealed that glioma
cell line U87-MG from ATCC was likely to be a bona fide human
glioblastoma cell line of unknown origin (30), there was a research also declared that
studies of U87 still reflected brain-cancer biology and didn't need
to be tossed out (31). So, we still
used the U87-MG cell line to study the glioblastoma just like this
research (32) Chloroquine (CQ) was
obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
Bevacizumab was obtained from Roche Diagnostics (Basel,
Switzerland). Anti-Bim, anti-Bcl-2, anti-Bax, anti-survivin,
anti-cleaved caspase-3, anti-cleaved caspase-8, anti-cleaved
caspase-9, anti-PARP, anti-LC3B-I, anti-LC3B-II, anti-SQSTM1 (p62),
anti-Akt, anti-p70S6K, anti-mTOR, anti-GAPDH, anti-p-Akt (T308),
anti-p-Akt (S473), anti-p-p70S6K (T389) and anti-p-mTOR (S2448)
antibodies were from Cell Signaling Technology, Inc. (Danvers, MA,
USA). MTT kit was from Thermo Fisher Scientific, Inc. Annexin V/PI
kit was from Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China).
Cell proliferation measurements by
MTT
Before experiments, U87-MG cells growing in
logarithmic phase were digested with 0.25% Trypsin-EDTA and
pipetted into single cells. Cells were carefully counted by TC20™
Automated Cell Counter (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) and 5×103 cells in 100 µl medium per well were
seeded into 96-well plate supplemented with different
concentrations (0, 0.5, 1, 2, 4, 8, 16, 32 mg/ml) of bevacizumab.
For each concentration, five repeated wells were prepared and a
blank control group with culture medium only was also set, and then
they were cultured in the incubator for 24 or 48 h, respectively.
After that, the cell viability was measured with MTT kit following
the manufacturer's instructions. Briefly, the medium was removed
and replaced by 100 µl of fresh phenol red-free culture medium. 10
µl (10% of the volume of the culture medium) MTT reagent was gently
loaded into the medium in each well, and then cultured in the
incubator at 37°C for 4 h. 75 µl of medium was removed from each
well and then 50 µl DMSO was added into each well and mixed
thoroughly with the pipette. The 96-well plate was then incubated
at 37°C for 10 min. Then the samples were mixed again and the
optical density (OD) was measured at 540 nm for each well by a
plate reader (EON; BioTek Instruments, Inc., Winooski, VT,
USA).
Cell apoptosis measurements by Annexin
V/PI
Cells for Annexin V-FITC/PI staining were harvested
at the same time points and with the same methods mentioned above.
However, to avoid the cell damage due to trypsinization, trypsin
without EDTA was used to digest the cells. Then the cells were
stained with Annexin V-FITC/PI following the manufacturer's
instruction. Briefly, 2×105 U87-MG cells were pooled and
washed twice with cold PBS, and then re-suspended in 500 µl binding
buffer. After that, 5 µl Annexin V-FITC and 5 µl propidium iodide
(PI) were added into the cell suspension and mixed equally by
gently pipetting. Then the cell samples were incubated at room
temperature for 5–15 min and protected from light during this
process. Subsequently, the cell samples were analyzed by flow
cytometry (FACSCalibur; BD Biosciences) to check the apoptosis
within 1 h. All experiments were performed in triplicate and
repeated at least 3 times.
Protein extraction and western blot
analysis
5×105 U87-MG cells were seeded into
6-well plate supplemented with 5 ml fresh DMEM medium and different
concentrations of bevacizumab (0, 1, 2, 4 mg/ml) for 48 h. In the
autophagy blocking experiment, 4 mg/ml bevacizumab with or without
10 µM chloroquine were added into the medium for 48 h. Then the
cells were pooled and washed with PBS twice, then lysed by RIPA
buffer. The same volume of cell lysates was mixed with 4X reducing
loading buffer and then the mixtures were boiled for 10 min. And
the proteins were subjected for SDS-PAGE electrophoresis, and then
the separated proteins in gel were transferred to PVDF membrane,
which was subsequently blocked by 10% BSA and incubated with the
indicated primary antibodies for the target proteins. After TBST
washing for three times, the membrane was then incubated with the
corresponding HRP-conjugated secondary antibodies. After TBST
washing, the PVDF membrane was then incubated with ECL substrate
and used for film exposure in dark room.
Data analysis
All experiments were performed at least for three
times in triplicate, data were expressed as mean ± standard
deviation (SD). Statistical analyses were performed using GraphPad
Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical
significance was determined as indicated in the figure legends.
P<0.05 was considered significant. One-way analysis of variance
(ANOVA) followed by Tukeys post hoc test was used to test for
multiple comparison.
Results
Bevacizumab suppressed the
proliferation of glioblastoma cells
In clinic, bevacizumab is used as an angiogenesis
inhibitor. However, whether it can directly suppress the
proliferation of tumor cells is not clear. To study the effects of
bevacizumab on glioblastoma cells, we firstly administered U87-MG
cells with various concentrations of bevacizumab for 24 and 48 h
respectively, and then checked the cell proliferation and viability
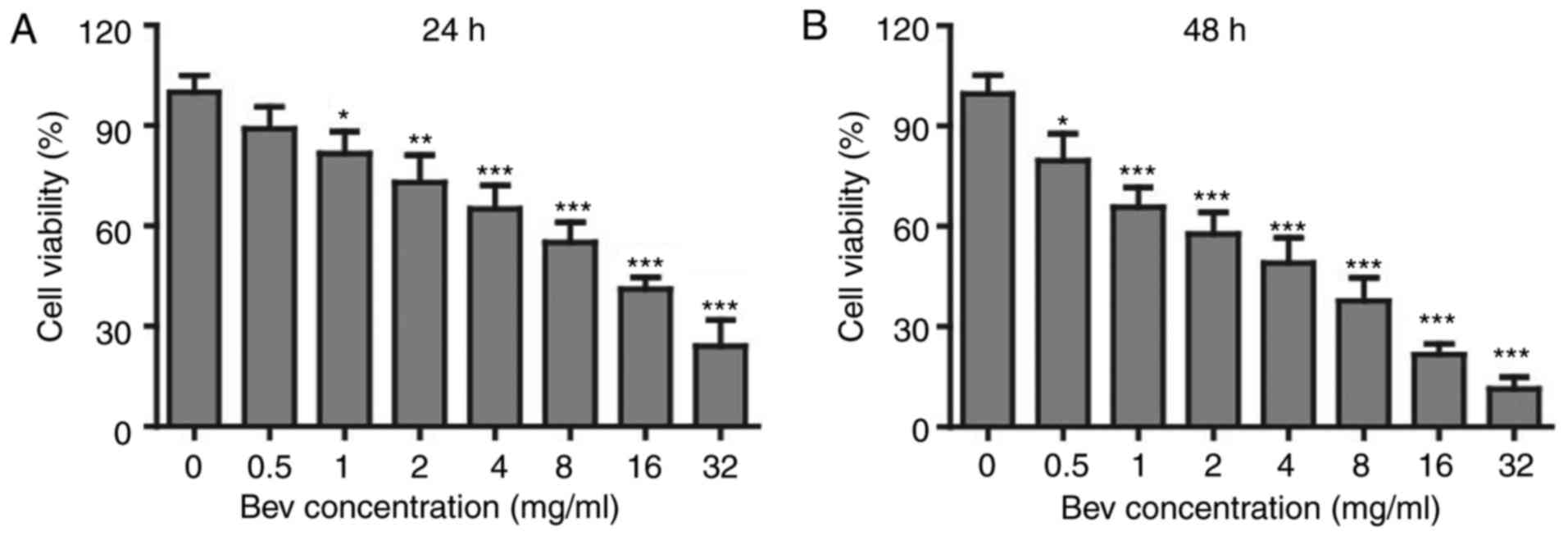
by MTT kit. To our surprise, the cell viability was getting lower
down with the increasing concentration of bevacizumab (Fig. 1A), and the proliferation suppression
was further enhanced if the treatment time was expanded for 48 h
(Fig. 1B). When treated with 4 mg/ml
bevacizumab for 48 h, glioblastoma cells showed only about 50% of
proliferation compared to those without bevacizumab treatment.
According to this result, we mainly used 0, 1, 2, 4 mg/ml doses of
bevacizumab for 48 h in later experiments. Taken together, this
result demonstrated that bevacizumab could directly suppress the
proliferation of glioblastoma cells in a dose and time dependent
manner.
Bevacizumab promoted the apoptosis of
glioblastoma cells
Besides the anti-proliferation effect, we also want
to know whether bevacizumab can promote the apoptosis of
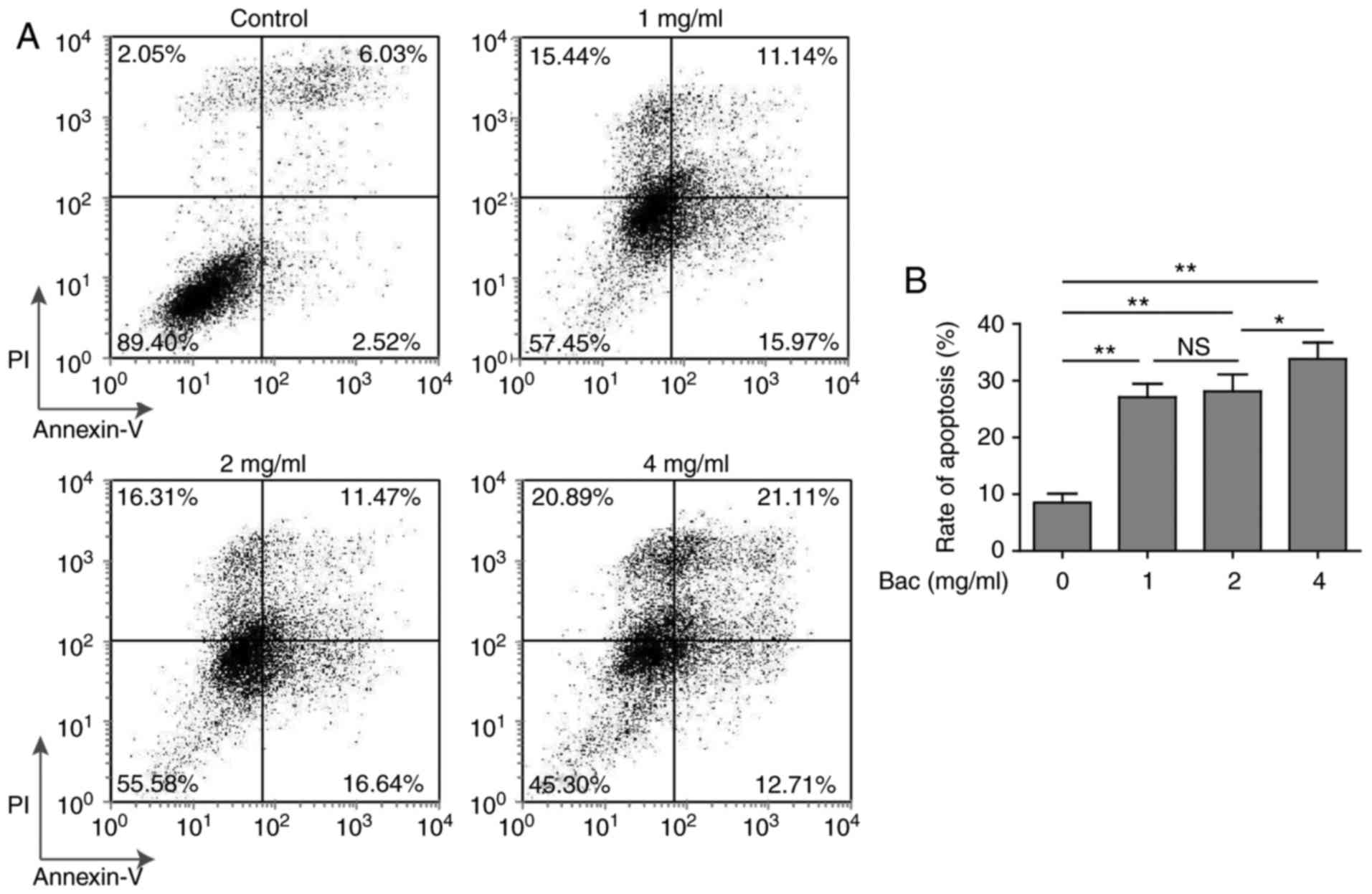
glioblastoma cells. To address this question, we cultured U87-MG
cells with various concentrations of bevacizumab for 48 h, and then
performed the cell apoptosis measurements using Annexin V/PI
method. Without bevacizumab, U87-MG cells showed very low
percentage of apoptosis (2.52 and 6.03% for early and late stages
of apoptosis, respectively). However, even low dose (1 mg/ml) of
bevacizumab was able to induce significantly high level of
apoptosis, both the early and late stages of apoptosis (15.97 and
11.14%, respectively) (Fig. 2). More
importantly, glioblastoma cells showed similar (~30%) apoptosis
with 1 or 2 mg/ml concentrations of bevacizumab (Fig. 2B), that meant some glioblastoma cells
could still survive under high dose of bevacizumab, which thus
reflected that the tolerance of glioblastoma cells to bevacizumab
occurred.
Bevacizumab downregulated
anti-apoptotic protein level and augmented pro-apoptotic protein
level of glioblastoma cells
To uncover the mechanism of bevacizumab on directly
inhibiting proliferation and promoting apoptosis of tumor cells, we
firstly treated U87-MG cells with different concentrations of
bevacizumab for 48 h, and then performed new biochemical
experiments to assess the protein contents of Bcl-2 and survivin,
which were typical anti-apoptotic markers. At the same time, we
measured the expression levels of Bim, Bax and cleaved caspase −3,
−8, and −9 which belonged to pro-apoptotic markers. PARP, which is
involved in DNA damage repair in its full-length form, will be
cleaved into two parts by caspase-3, and the cleavage of PARP will
hinder the DNA repair and therefore serves as an apoptosis marker.
Herein we blotted the full length of PARP to reflect its cleavage
status. The results clearly demonstrated that bevacizumab could
significantly reduce the expression levels of anti-apoptotic
proteins, including Bcl-2, survivin and full length of PARP
(Fig. 3). Meanwhile, bevacizumab
upregulated the expression levels of pro-apoptotic proteins, such
as Bim, Bax and cleaved caspase-3, −8 and −9, which also promoted
the cleavage of PARP (Fig. 3). Both
mechanisms synergistically led to the decreased proliferation and
increased apoptosis of glioblastoma cells with the treatment of
bevacizumab.
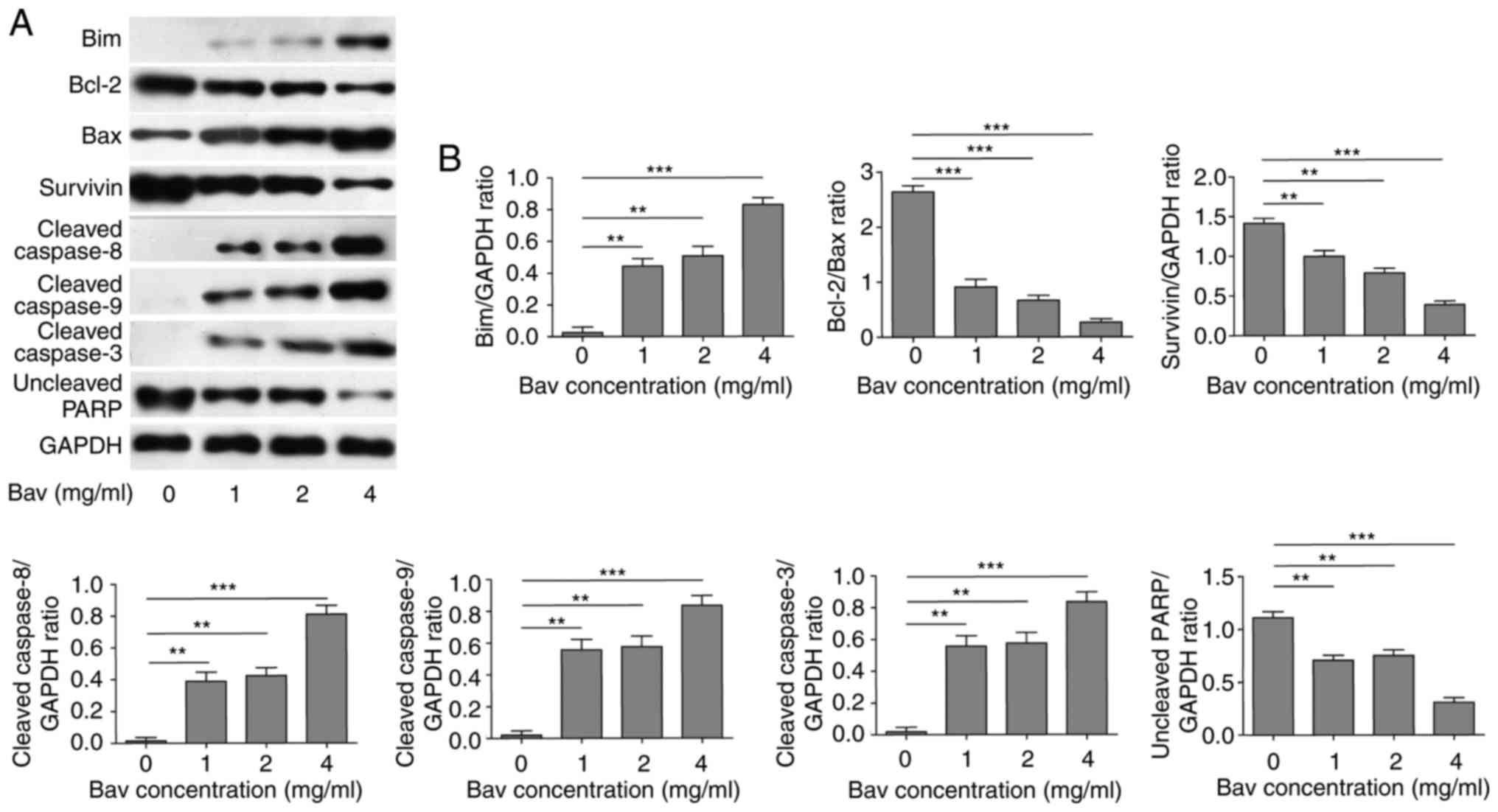 | Figure 3.The attenuated anti-apoptotic protein
level and augmented pro-apoptotic protein level of glioblastoma
cells by bevacizumab treatment. (A) U87-MG cells were treated with
different concentrations of bevacizumab for 48 h, and then the
anti-apoptotic protein contents of Bcl-2 and surviving, and the
pro-apoptotic protein contents of Bim, Bax and cleaved caspase-3,
−8, and −9, as well as the uncleaved PARP, were assessed by western
blot analysis, respectively. GAPDH was used as the loading control.
The result was a representative of three independent experiments.
(B) The quantification of Bcl-2/Bax ratio and the expression levels
of other proteins normalized to GAPDH for result in A. Error bars
represented mean ± SD. P-values were determined by one-way ANOVA
followed by Tukeys post hoc test. ***P<0.001, **P<0.01.
ANOVA, analysis of variance. |
Bevacizumab enhanced the autophagy of
glioblastoma cells
In previous clinical application, there were obvious
chemo-tolerance developed during glioblastoma treatment with
bevacizumab, but the underlying mechanism was still elusive. As is
mentioned before, the autophagy process was found to be involved in
the chemo-drug tolerance. To explore the relationship between
bevacizumab-tolerance and autophagy, we used varying doses of
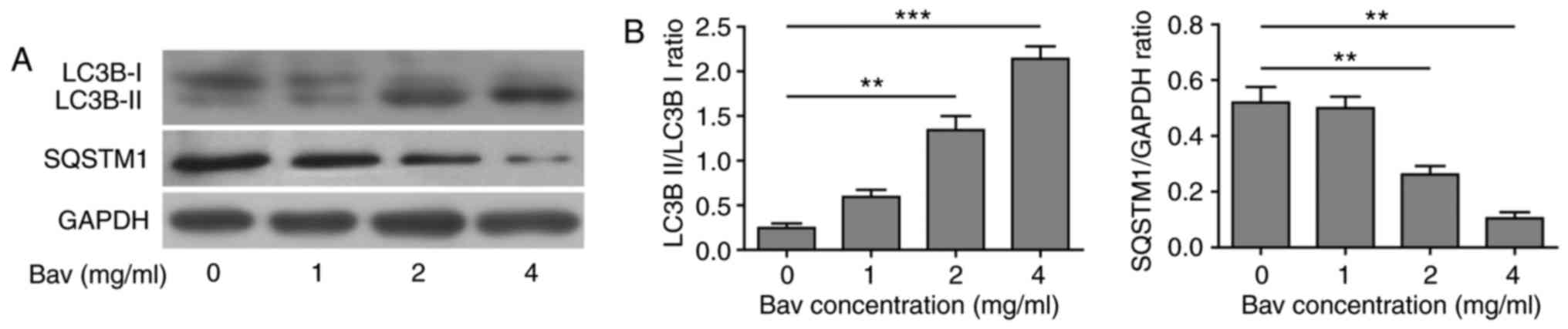
bevacizumab to treat glioblastoma cells for 48 h, and then assessed
two hallmarks of autophagy, the LC3B-II/LC3B-I ratio and
SQSTM1(p62) degradation by biochemical approaches. During
autophagy, a cytosolic form of LC3B-I can be converted to a
membrane-associated form LC3B-II by conjugating LC3B-I to
phosphatidylethanolamine in the pre-autophagosomal and
autophagosomal membranes through a ubiquitin like system (33). Thus, the relative contents of LC3B-II
and the ratio of LC3B-II/LC3B-I were reliable indicators for
monitoring the autophagy and autophagy-related processes in tumor
cells. In addition, the level of SQSTM1 (p62) has also been used
for monitoring autophagy, which is down-regulated when autophagy
occurs (34). Meanwhile, SQSTM1
directly binds to LC3-II and mediates the targeted degradation of
ubiquitinated protein aggregates (35). As is expected, glioblastoma cells
showed gradually decreased LC3B-I, increased LC3B-II and thus
upward LC3B-II/LC3B-I ratio as well as remarkable degradation of
SQSTM1 (p62) upon bevacizumab treatment (Fig. 4). These results clearly proved that
bevacizumab could enhance the autophagy of glioblastoma cells on a
dose dependent manner, for which probably contributed to the
formation of chemo-tolerance of glioblastoma cells.
Bevacizumab induced autophagy by
suppressing Akt-mTOR signaling pathway
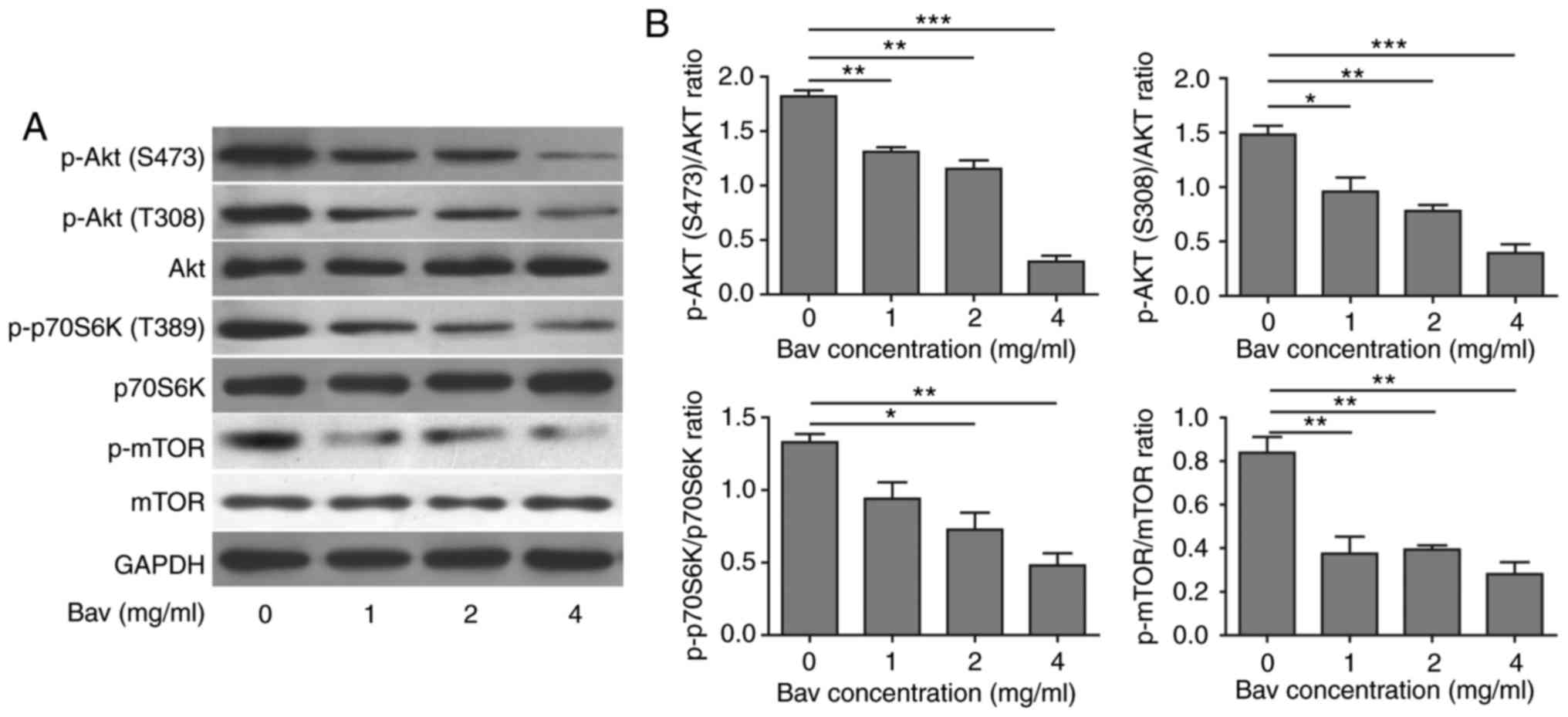
Previous studies have revealed that tumor cells
could suppress Akt-mTOR signaling pathway to induce autophagy
(36). In order to understand the
detailed mechanism of enhanced autophagy in glioblastoma cells
after bevacizumab treatment, we further performed some biochemical
experiments to assess the Akt-mTOR signaling pathway by measuring
the phosphorylation levels of Akt (T308 and S473), mTOR (S2448) and
p70 ribosomal protein S6 kinase (p70S6K, Thr389). p70S6K is a
direct substrate of mTOR and an established marker for mTOR
signaling. Previous studies showed that mTOR was critical for
autophagy induction, and the upstream PI3K-Akt signaling could
activate mTOR thus suppressed autophagy, while repressed mTOR in
the absence of growth factors could active autophagy (37). After treated with varying doses of
bevacizumab for 48 h, as expected, glioblastoma cells showed
dramatically decreased phosphorylation levels of Akt, mTOR and
p70S6K in a dose dependent manner (Fig.
5), which indicated that the Akt-mTOR signaling pathway was
impaired. It has been proved that attenuated Akt-mTOR signaling
pathway can result in autophagy (38). Based on our results, we concluded that
bevacizumab could indeed suppress Akt-mTOR signaling pathway to
induce high level of autophagy in glioblastoma cells, which was
also consistent with previous results reported in other tumor cells
(39).
Bevacizumab induced autophagy can be
blocked by chloroquine in glioblastoma cells
In the later stage of autophagy, autophagosomes fuse
with lysosomes to form autolysosomes, and the sequestered
intra-autophagosomal components will be degraded by lysosomal
hydrolases. Chloroquine (CQ), a well-known lysosome inhibitor, is
therefore often used to inhibit autophagy process as it can
accumulate in lysosome and raise the lysosomal pH, which results in
robust inhibition of lysosomal proteases that require an acidic pH,
the fusion of autophagosome with lysosome and lysosomal protein
degradation (40). Here, we also
treated glioblastoma cells with bevacizumab in the presence of CQ,
western blotting showed that sole bevacizumab treatment could
upregulate the LC3B-II/LC3B-I ratio and downregulate SQSTM1 (p62)
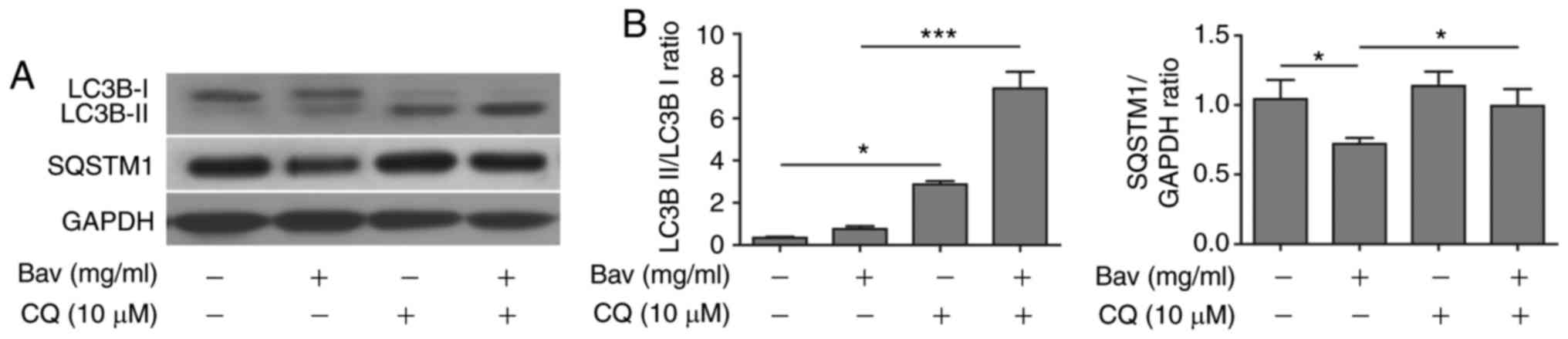
when compared with those without bevacizumab treatment (Fig. 6), which confirmed that autophagy was
indeed induced by bevacizumab. Moreover, bevacizumab plus CQ
further increased the levels of LC3-II and SQSTM1 when compared
with those treated by bevacizumab alone (Fig. 6). The significantly accumulated LC3-II
and SQSTM1 (p62) caused by inhibiting lysosomal protein degradation
indicated that bevacizumab induced autophagy was severely blocked
by CQ in glioblastoma cells.
Blocking the autophagy process
enhanced the cytotoxicity of bevacizumab on glioblastoma cells
To verify whether bevacizumab resistance of
glioblastoma cells was autophagy dependent, we further utilized CQ
to inhibit the autophagy process to examine the apoptosis of
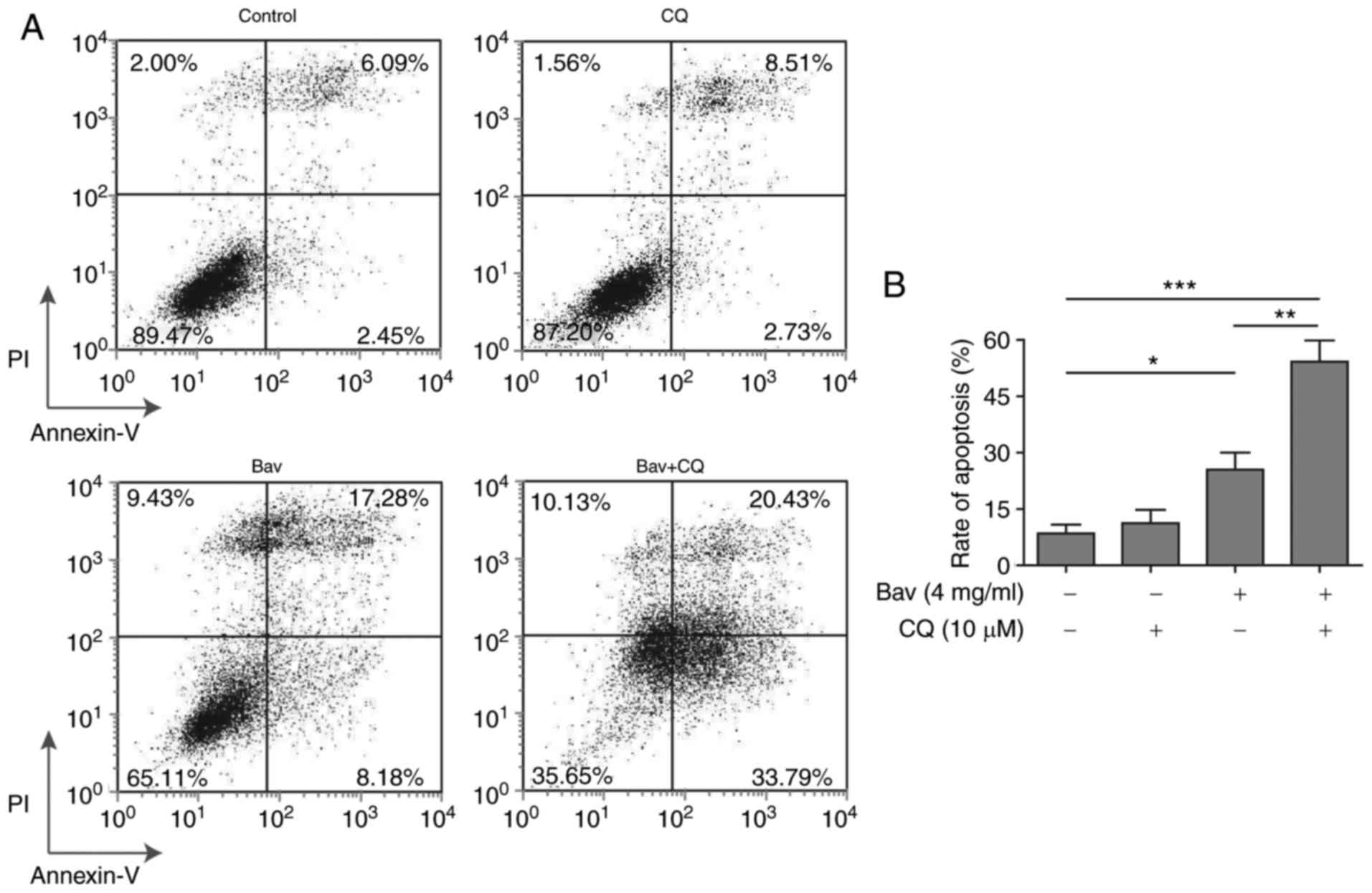
glioblastoma cells under bevacizumab administration. As expected,
the FACS results clearly demonstrated that combined treatment with
bevacizumab and CQ displayed remarkably increased cytotoxicity on
glioblastoma cells when compared to the sole treatment with either
drug alone, from almost 54.22 to 25.46% (bevacizumab alone) or
11.24% (CQ alone) of cell apoptosis (Fig.
7). And the apoptosis result of bevacizumab treatment alone was
consistent with that shown in Fig. 2.
Meanwhile, the data in Fig. 2
demonstrated that 2 mg/ml and 4 mg/ml bevacizumab could cause
distinct tolerance of glioblastoma cells, therefore, the augmented
cell apoptosis resulted from the combination of CQ and the same
dose of bevacizumab further proved that autophagy was a leading
cause of drug-tolerance. Moreover, the tolerance of glioblastoma
cells was relieved by blocking their autophagy process. As stated
above, the tolerance occurred during bevacizumab administration was
principally caused by autophagy induction in glioblastoma cells,
and thus blocking the autophagy process was able to break the
tolerance and enhance the cytotoxicity of bevacizumab.
Discussion
In recent clinical treatments, there are ever
growing obstacles occurred in the therapy of glioblastoma patients,
such as low efficacy, various side effects and knotty
chemo-tolerance. All these problems lead to low cure rate, high
mortality and high recurrence of glioblastoma. Despite we have made
great progresses in developing new chemotherapeutic agents,
chemo-tolerance is still the crucial issue for both clinicians and
drug developers, which could severely dampen the efficacy of
anticancer drugs in clinical application. To overcome this
difficulty, many efforts have been put into the related study,
however, the complexities of mechanisms of resistance that caused
by tumor heterogeneity and microenvironment have seemingly hindered
our steps to solve this problem.
In this study, we explored the effect of
bevacizumab, one anti-angiogenic reagent, not a traditional
chemo-drug, on the tolerance induction of glioblastoma from
different angles. We firstly found that bevacizumab could directly
suppress the proliferation of glioblastoma cells with a dose and
time dependent manner. Meanwhile, bevacizumab was able to promote
the apoptosis of tumor cells. The downregulated anti-apoptotic
protein level and upregulated pro-apoptotic protein level
synergistically lead to this striking effect of bevacizumab.
Nevertheless, the apoptosis of glioblastoma cells reached a plateau
under high dose of bevacizumab, which reflected that some
glioblastoma cells could tolerate bevacizumab. The reasons behind
this phenomenon were complex, and multiple changes were utilized by
tumor cells to survive under metabolic stress in the
microenvironment, which include the elevated autophagy.
Interestingly, we also revealed that high dose of bevacizumab would
induce the autophagy in glioblastoma cells, indicated by the
increased LC3B-II/LC3B-I ratio and remarkable degradation of
SQSTM1, which therefore counteracted the cytotoxicity of
bevacizumab on tumor cells. In other related reports,
hepatocellular carcinoma (HCC) cells treated with bevacizumab also
showed inhibited cell growth, reduced new vessel developments and
upregulated autophagy (41), which
suggested that bevacizumab induced autophagy might be a general
phenomenon in solid tumors.
Previous work has shown that autophagy can constrain
tumor initiation in normal tissues by regulating DNA damage and
oxidative stress. However, some established tumors also rely on
autophagy for tumor promotion and maintenance (42). Mechanistic study further confirmed
that bevacizumab treatment could induce autophagy of glioblastoma
cells by suppressing the Akt-mTOR signaling pathway via reducing
the phosphorylation of Akt, mTOR and mTOR's direct substrates
p70S6K. Besides, AMPK signaling pathway was also shown to be
involved in the autophagy induction (39). In other work, people also found that
chemo-tolerance of non-small cell lung cancer could be contributed
by autophagy in hypoxic conditions (43). Therefore, blocking autophagy process
became an appealing tumor therapy and extensive biomedical studies
were carried out to test the antitumor effect of various autophagy
inhibitors (44–46). Furthermore, CQ was administered in
combination with trastuzumab (Herceptin) in clinic to solve the
drug tolerance in Her2 positive breast cancer patients, which
successfully improved the efficacy of trastuzumab (27). In our study, we used CQ to block
autophagy in glioblastoma cells. Significantly accumulated LC3B-II
and SQSTM1 proteins clearly demonstrated that the autophagy process
was severely blocked. Thus, we found the tumor-suppressive effect
of bevacizumab was significantly enhanced, indicated by the
remarkably increased tumor cell apoptosis after bevacizumab and CQ
combination treatment. In some related reports, people also found
that other autophagy inhibitors also had similar functions in
different cancers (44,45). Nevertheless, these encouraging
improvements need further confirmation in human glioblastoma
treatment when combined autophagy inhibitors with radiotherapy and
chemotherapy. Moreover, the detailed mechanism of CQ on tumor cells
may be complicated, not solely depending on the blocking of
autophagy.
In summary, our study revealed that bevacizumab
could induce the chemo-tolerance of glioblastoma cells by
upregulating their autophagy level through inhibiting the Akt-mTOR
signaling pathway. This novel mechanism will help us better
understand the functional relevance of autophagy within the tumor
microenvironment. Therefore, pharmacological or genetic inhibition
of autophagy is a reasonable and promising way to enhance the
efficacy of chemotherapy for glioblastoma and to improve clinical
treatment of cancer patients. In future, more cooperation between
laboratory and clinical research is still needed to design other
therapeutic strategies to overcome the chemo-tolerance and to
enhance the outcomes of anticancer therapies for cancer
patients.
References
|
1
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Madany M, Thomas TM, Edwards L and Yu JS:
Immunobiology and immunotherapeutic targeting of glioma stem cells.
Adv Exp Med Biol. 853:139–166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kanu OO, Hughes B, Di C, Lin N, Fu J,
Bigner DD, Yan H and Adamson C: Glioblastoma multiforme
oncogenomics and signaling pathways. Clin Med Oncol. 3:39–52.
2009.PubMed/NCBI
|
|
4
|
Tanaka S, Louis DN, Curry WT, Batchelor TT
and Dietrich J: Diagnostic and therapeutic avenues for
glioblastoma: No longer a dead end? Nat Rev Clin Oncol. 10:14–26.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang J, Stevens MF, Laughton CA,
Madhusudan S and Bradshaw TD: Acquired resistance to temozolomide
in glioma cell lines: Molecular mechanisms and potential
translational applications. Oncology. 78:103–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cahill DP, Levine KK, Betensky RA, Codd
PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR,
Curry WT, et al: Loss of the mismatch repair protein MSH6 in human
glioblastomas is associated with tumor progression during
temozolomide treatment. Clin Cancer Res. 13:2038–2045. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ramirez YP, Weatherbee JL, Wheelhouse RT
and Ross AH: Glioblastoma multiforme therapy and mechanisms of
resistance. Pharmaceuticals (Basel). 6:1475–1506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chi AS, Sorensen AG, Jain RK and Batchelor
TT: Angiogenesis as a therapeutic target in malignant gliomas.
Oncologist. 14:621–636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Das S and Marsden PA: Angiogenesis in
Glioblastoma. N Engl J Med. 369:1561–1563. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wick W, Chinot OL, Bendszus M, Mason W,
Henriksson R, Saran F, Nishikawa R, Revil C, Kerloeguen Y and
Cloughesy T: Evaluation of pseudoprogression rates and tumor
progression patterns in a phase III trial of bevacizumab plus
radiotherapy/temozolomide for newly diagnosed glioblastoma. Neuro
Oncol. 18:1434–1441. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chinot OL, Wick W, Mason W, Henriksson R,
Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea
D, et al: Bevacizumab plus radiotherapy-temozolomide for newly
diagnosed glioblastoma. N Engl J Med. 370:709–722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jahangiri A, De Lay M, Miller LM,
Carbonell WS, Hu YL, Lu K, Tom MW, Paquette J, Tokuyasu TA, Tsao S,
et al: Gene expression profile identifies tyrosine kinase c-Met as
a targetable mediator of antiangiogenic therapy resistance. Clin
Cancer Res. 19:1773–1783. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Piao Y, Liang J, Holmes L, Zurita AJ,
Henry V, Heymach JV and de Groot JF: Glioblastoma resistance to
anti-VEGF therapy is associated with myeloid cell infiltration,
stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol.
14:1379–1392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mesange P, Poindessous V, Sabbah M,
Escargueil AE, de Gramont A and Larsen AK: Intrinsic bevacizumab
resistance is associated with prolonged activation of autocrine
VEGF signaling and hypoxia tolerance in colorectal cancer cells and
can be overcome by nintedanib, a small molecule angiokinase
inhibitor. Oncotarget. 5:4709–4721. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wileman T: Aggresomes and autophagy
generate sites for virus replication. Science. 312:875–878. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Münz C: Autophagy and antigen
presentation. Cell Microbiol. 8:891–898. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murrow L and Debnath J: Autophagy as a
stress-response and quality-control mechanism: Implications for
cell injury and human disease. Annu Rev Pathol. 8:105–137. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gugnoni M, Sancisi V, Manzotti G, Gandolfi
G and Ciarrocchi A: Autophagy and epithelial-mesenchymal
transition: An intricate interplay in cancer. Cell Death Dis.
7:e25202016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhong Z, Sanchez-Lopez E and Karin M:
Autophagy, inflammation and immunity: A troika governing cancer and
its treatment. Cell. 166:288–298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dou Z, Xu C, Donahue G, Shimi T, Pan JA,
Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al: Autophagy
mediates degradation of nuclear lamina. Nature. 527:105–109. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Perera RM, Stoykova S, Nicolay BN, Ross
KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK,
Ferrone CR, et al: Transcriptional control of autophagy-lysosome
function drives pancreatic cancer metabolism. Nature. 524:361–365.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cufi S, Vazquez-Martin A,
Oliveras-Ferraros C, Corominas-Faja B, Cuyàs E, López-Bonet E,
Martin-Castillo B, Joven J and Menendez JA: The anti-malarial
chloroquine overcomes primary resistance and restores sensitivity
to trastuzumab in HER2-positive breast cancer. Sci Rep. 3:24692013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang MC, Wu MY, Hwang MH, Chang YT, Huang
HJ, Lin AM and Yang JC: Chloroquine enhances gefitinib cytotoxicity
in gefitinib-resistant nonsmall cell lung cancer cells. PLoS One.
10:e01191352015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aveic S and Tonini GP: Resistance to
receptor tyrosine kinase inhibitors in solid tumors: Can we improve
the cancer fighting strategy by blocking autophagy? Cancer Cell
Int. 16:622016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:354re32016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dolgin E: Venerable brain-cancer cell line
faces identity crisis. Nature. 537:149–150. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han J, Jun Y, Kim SH, Hoang HH, Jung Y,
Kim S, Kim J, Austin RH, Lee S and Park S: Rapid emergence and
mechanisms of resistance by U87 glioblastoma cells to doxorubicin
in an in vitro tumor microfluidic ecology. Proc Natl Acad Sci USA.
113:pp. 14283–14288. 2016; View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ichimura Y, Kirisako T, Takao T, Satomi Y,
Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi
M, et al: A ubiquitin-like system mediates protein lipidation.
Nature. 408:488–492. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Larsen KB, Lamark T, Øvervatn A,
Harneshaug I, Johansen T and Bjørkøy G: A reporter cell system to
monitor autophagy based on p62/SQSTM1. Autophagy. 6:784–793. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Johansen T and Lamark T: Selective
autophagy mediated by autophagic adapter proteins. Autophagy.
7:279–296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schmelzle T and Hall MN: TOR, a central
controller of cell growth. Cell. 103:253–262. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shao X, Lai D, Zhang L and Xu H: Induction
of autophagy and apoptosis via PI3K/AKT/TOR pathways by
azadirachtin a in Spodoptera litura cells. Sci Rep. 6:354822016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zha QB, Zhang XY, Lin QR, Xu LH, Zhao GX,
Pan H, Zhou D, Ouyang DY, Liu ZH and He XH: Cucurbitacin E induces
autophagy via downregulating mTORC1 signaling and upregulating AMPK
activity. PLoS One. 10:e01243552015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guo XL, Li D, Sun K, Wang J, Liu Y, Song
JR, Zhao QD, Zhang SS, Deng WJ, Zhao X, et al: Inhibition of
autophagy enhances anticancer effects of bevacizumab in
hepatocarcinoma. J Mol Med (Berl). 91:473–483. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee JG, Shin JH, Shim HS, Lee CY, Kim DJ,
Kim YS and Chung KY: Autophagy contributes to the chemo-resistance
of non-small cell lung cancer in hypoxic conditions. Respir Res.
16:1382015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dragowska WH, Weppler SA, Wang JC, Wong
LY, Kapanen AI, Rawji JS, Warburton C, Qadir MA, Donohue E, Roberge
M, et al: Induction of autophagy is an early response to gefitinib
and a potential therapeutic target in breast cancer. PLoS One.
8:e765032013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang Z, Liu Y, Wei X, Zhou X, Gong C,
Zhang T, Jin P, Xu S, Ma D and Gao Q: Co-targeting EGFR and
autophagy impairs ovarian cancer cell survival during detachment
from the ECM. Curr Cancer Drug Targets. 15:215–226. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nihira K, Miki Y, Iida S, Narumi S, Ono K,
Iwabuchi E, Ise K, Mori K, Saito M, Ebina M, et al: An activation
of LC3A-mediated autophagy contributes to de novo and acquired
resistance to EGFR tyrosine kinase inhibitors in lung
adenocarcinoma. J Pathol. 234:277–288. 2014.PubMed/NCBI
|