Introduction
Colorectal cancer is one of the most common
malignancies worldwide; imbalanced dietary patterns, such as the
large consumption of red and processed meat, have been identified
as a contributor to the increasing incidence of colorectal cancer
(1,2).
This dietary habit causes increased levels of cholesterol and
low-density lipoproteins (LDL) in the blood, which can eventually
develop into hyperlipidemia. According to recent data,
hyperlipidemia is one factor contributing to the increased
incidence of colorectal cancer (3,4) as high
levels of cholesterol and LDL can enhance cancer growth and
metastasis (5,6). Statin compounds are the most commonly
used drugs to reduce the levels of cholesterol and LDL; therefore,
using statins to prevent cancer is a recommended treatment for
patients with hyperlipidemia. Nielsen, Nordestgaard and Bojesen
(7) have reported that the use of
statins may decrease cancer mortality in Danish population.
Moreover, numerous clinical studies revealed an association between
statin consumption and cancer risk; however, the data regarding
colorectal cancer were inconsistent (8,9).
Consequently, it is worth evaluating the anti-cancer effect of
statins for the purpose of applying them in colorectal cancer
prevention. Atorvastatin (ATST) is one of the main statins in
clinical circulation. Prior studies have indicated the synergistic
antitumor effects of ATST when combined with non-steroidal
anti-inflammatory drugs (10,11), γ-tocotrienol (12) or green tea polyphenols (13). Therefore, exploring a combinational
strategy between statins and other dietary components may be an
effective way to prevent colorectal cancer in patients with
hyperlipidemia.
Previous studies revealed that apples may reduce the
risk of cancer, and that polyphenol and flavonoid compounds can
also contribute to this chemopreventive effect (14–17).
Phloretin (PT) is one of the most abundant phenolic phytochemicals
in apples and apple products. Numerous studies have reported on the
antitumor activities of PT, including its ability to suppress cell
growth and induce apoptosis in human hepatoma cells, HL-60 human
leukemia cells, B16 mouse 4A5 melanoma cells and HT29 human colon
cancer cells (18–21). Both in vitro and in vivo
studies have revealed that PT could potentiate the antitumor
effects of paclitaxel via the induction of cell apoptosis (22). Another study showed that cytochalasin
B could enhance the PT-induced apoptosis of HepG2 cells (21). Although, according to these reports,
the combination of PT with other compounds may enhance its
antitumor effect, little evidence is currently available to support
a synergistic effect between PT and statins.
In this study, the potential synergistic inhibitory
effect between PT and ATST was evaluated in human colon cancer
cells. The synergistic mechanisms involving the cell cycle and
apoptosis were also investigated. The results of the present study
have provided a potential novel chemoprevention strategy for the
hyperlipidemia population, specifically via the combination of
dietary functional components and statin compounds.
Materials and methods
Cell lines and reagents
The human colon cancer cells SW620 and HCT116 were
purchased from the Institute of Basic Medical Cell Center, Chinese
Academy of Medical Sciences. ATST and PT were purchased from the
National Institutes for Food and Drug Control (Beijing, China).
MTT, propidium iodine (PI) and RNase were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The Annexin V
conjugate was purchased from Invitrogen (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Lysis buffer and a BCA assay kit were
purchased from Beyotime Co., (Haimen, China). Antibodies for
poly-ADP-ribose polymerase (PARP), cleaved-PARP, caspase-3, cyclin
B1, phospho-cdc2 (Tyr15) and Myt1 were purchased from Cell
Signaling Technology, Inc., (Danvers, MA, USA).
Cell viability assay
Human SW620 and HCT116 colon cancer cells were
seeded into 96-well plates (2,000 cells/well). After 24 h, the
cells in each well were treated with a series of concentrations of
PT, ATST or a combination (ratio of 10:1, PT and ATST,
respectively). After 24 and 48 h, cell viability was determined
using an MTT assay.
Analysis of synergy
The synergy analysis was conducted according to the
median-effect equation (23). It was
assumed that the dose-response model follows the median-effect
equation,
fa/fu=(D/Dm)m;
the dose-effect curve,
log(fa/fu)=mlogD-mlogDm, is
the logarithmic form of the median-effect equation, which is a
linear regression model with the independent variable logD
and the dependent variable log
(fa/fu). D is the dose;
Dm is the dose required for a 50% effect
(IC50); fa is the fraction affected by
D, which is the same as the ratio of surviving cells;
fu is the unaffected fraction, which is the same
as the ratio of non-surviving cells, m is the slope. This
equation is applied for calculating the effective doses of agent 1
and agent 2, and of a fixed ratio of combination agents, using data
from an MTT assay. Suppose that the combination
(D1, D2) exert the same effect
x as agent 1 alone at dose level D×1, and
agent 2 alone at dose D×2. D×1
and D×2, were calculated from the dose-effect
curve. The interaction index=D1/D×1 +
D2/D×2 +
α+I1·+2)/(D×1·1×2),
If the agents are mutually exclusive, α=0; if the agents are
mutually nonexclusive, α=1. The interaction index was used
to determine whether the combinational dose (D1,
D2) was additive, synergistic or antagonistic,
depending on an interaction index of 1, <1 or >1,
respectively.
Measurement of cell apoptosis
Cell apoptosis was assessed using flow cytometry
combined with an Annexin V/PI double-staining assay. After being
treated with PT (100 µM), ATST (10 µM) or a combination of the two
(100 µM + 10 µM, respectively) for 48 h, cells were harvested,
washed with ice-cold PBS and resuspended in 100 µl Annexin-binding
buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH
7.4), which contained 5 µl Annexin V conjugate and 0.1 µg PI. After
incubation at room temperature for 15 min, the cell suspension was
gently mixed with 400 µl Annexin-binding buffer and analyzed by
FACSCalibur flow cytometry (BD Biosciences, Franklin Lakes, NJ,
USA) at 488 nm.
Detection of the cell cycle
Flow cytometry was performed to analyze the cell
cycle distribution. After being treated with PT (100 µM), ATST (10
µM) or a combination of the two (100 µM + 10 µM, respectively) for
48 h, cells were harvested and washed with ice-cold PBS, then
resuspended in 70% ethanol and stored at 4°C for 24 h. After 24 h,
cells were pelleted by centrifugation, incubated with RNase (50
µM/ml in PBS) and stained with PI (1 mg/ml in PBS) in the dark at
37°C for 30 min. Cells were then evaluated at a wavelength of 550
nm using a FACSCalibur flow cytometer (BD Biosciences), and the
data were analyzed with Summit v5.0 software.
Western blot analysis
After being exposed to PT (100 µM), ATST (10 µM) or
a combination of the two (100 µM + 10 µM, respectively) for 48 h,
the cells were harvested into tubes. The cell pellet from each tube
was incubated on ice for 30 min with 300 µl lysis buffer containing
phenylmethanesulfonyl fluoride (PMSF, 1 mM) and cocktails (1:10).
The cell pellets were resuspended and centrifuged at 12,000 rpm for
20 min to collect the supernatants. Proteins were subjected to
quantification using a BCA assay kit and then resolved via
SDS-PAGE. After electrophoresis, the proteins were transferred to a
polyvinylidene fluoride membrane, which was then blocked. The
membranes were incubated with different primary antibodies at the
concentrations recommended by the manufacturer at 4°C overnight.
Subsequently, the membranes were incubated with secondary
antibodies at room temperature for 1 h, prior to visualization
using an enhanced chemiluminescence reagent. Antibodies for PARP,
cleaved-PARP, caspase-3, cyclin B1, Tyr15 and Myt1 were purchased
from Cell Signaling Technologies, Inc.
Statistical analysis
All data were obtained from at least three
independent experiments and are presented as the mean ± standard
deviation. A Student's t-test was used to assess differences
between two groups. One-way analysis of variance and the Dunnett's
post hoc test were used to compare differences among multiple
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
A synergistic anti-proliferative effect was observed
in SW620 and HCT116 cells treated with a combination of PT and
ATST. The growth inhibitory effect of PT, ATST and their
combination was evaluated in SW620 and HCT116 cells at 24 and 48 h.
The tested concentrations of PT were 50, 75, 100, 125, 150 and 200
µM; the tested concentrations of ATST were 5, 7.5, 10, 12.5, 15 and
20 µM; the combination ratio of PT to ATST was 10:1. After exposure
to these treatments for 24 and 48 h, cell survival was measured
using an MTT assay. The results (Fig.
1) showed that PT and ATST exhibited time- and dose-dependent
growth inhibitory effects on the two cell lines. The
IC50 values for single PT and ATST treatments in HCT116
cells were 137.48±2.14 and 19.52±1.29 µM, respectively. However,
for SW620 cells, the IC50 value of PT was 191.70±2.28
µM, and ATST did not show a significant inhibitory effect on cell
growth.
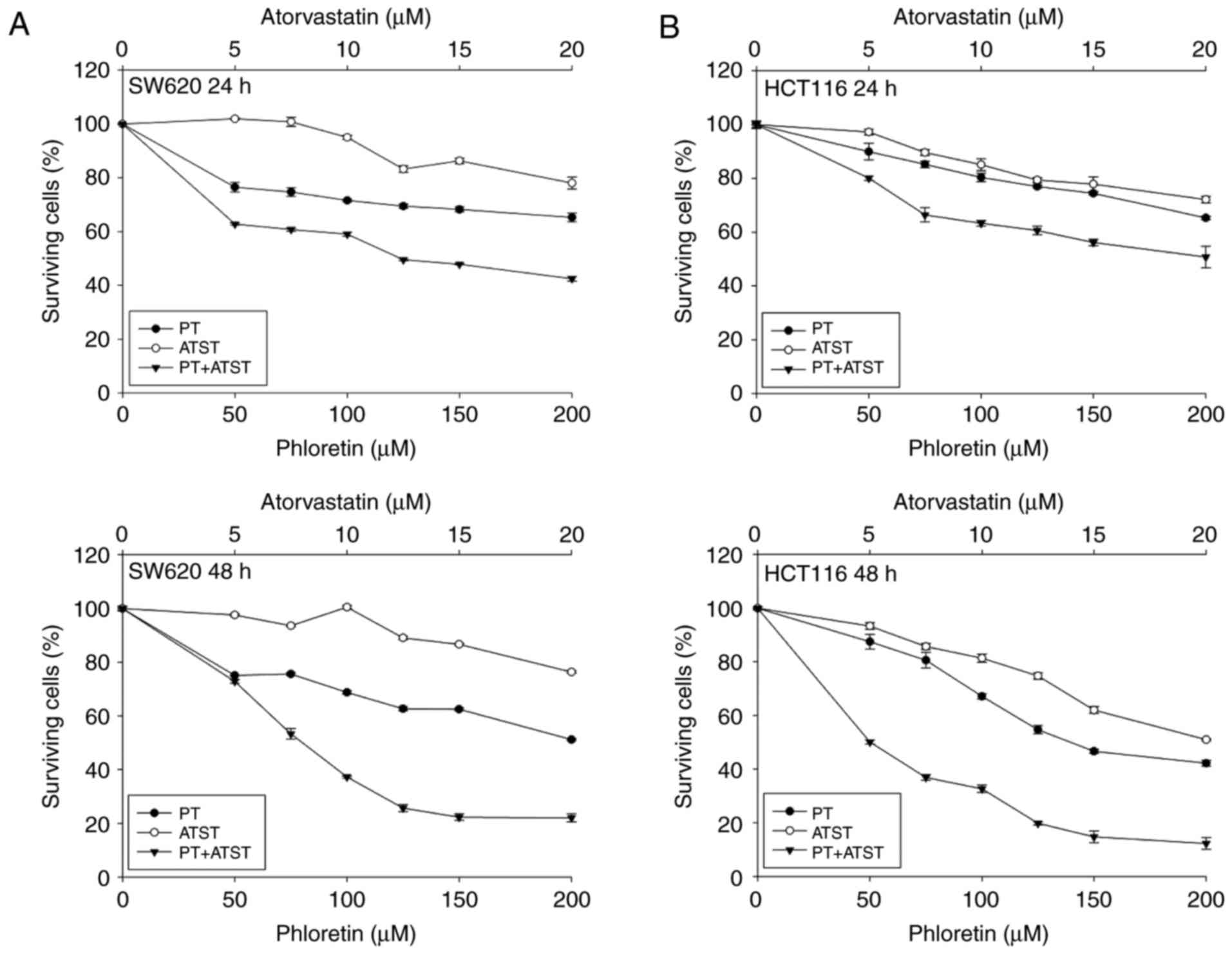 | Figure 1.The viability of SW620 (A) and HCT116
cells (B) after treatment with PT, ATST and their combinations.
Cells were treated with a series of dosages of PT (0, 50, 100, 150
and 200 µM), ATST (0, 5, 10, 15 and 20 µM), or a combination at a
fixed ratio of 10:1 for 24 and 48 h, and then cell viability was
measured using an MTT assay. Data are shown as the mean ± SD
(n=5), and the surviving cell percentage of the respective
controls were set as 100%. |
Compared with individual PT and ATST treatments, the
combination markedly decreased cell viability. The cell viability
rates at 24 h and 48 h following treatment with PT (100 µM)
combined with ATST (10 µM) were 59.08±0.73% and 37.27±0.39%,
respectively, for SW620 cells, and 63.35±1.08% and 32.65±1.34% for
HCT116 cells, respectively. Furthermore, to determine whether the
enhanced inhibitory effect observed with combined PT/ATST was
additive or synergistic, the combination indexes were computed
using the aforementioned Chou and Talalayla method.
The median-effect plots of these two cells are shown
in Fig. 2A and B, and indicate that
the linear regression model fit better with the dose-dependent
manner of PT, ATST and their combinations. According to the
median-effect equation, the effect doses of PT, ATST and their
combination at a dosage ratio of 10:1 were computed, and then used
to compute the interaction indexes. As presented in Fig. 2C and D, the interaction indexes of
each PT and ATST concentration pair were <1.0; notably, a
proportion was <0.5. Consequently, it was determined the
combination of PT and ATST generated a strong synergistic
inhibitory effect on SW620 and HCT116 cell growth. As the results
showed that the inhibitory effect of combination treatment at 48 h
were significantly stronger than the ones at 24 h, we therefore
selected the time point of 48 h to carry out the following
experiments.
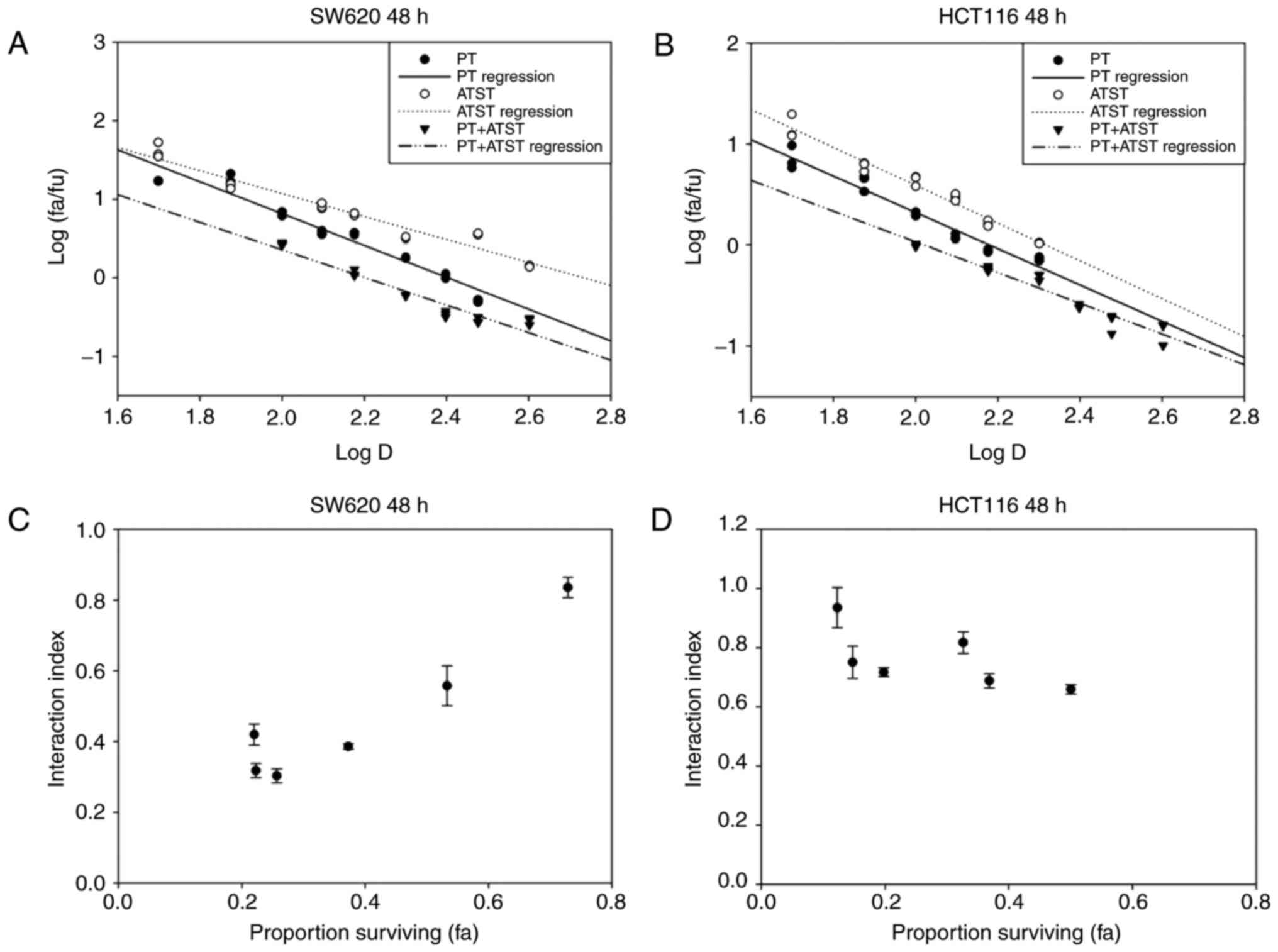 | Figure 2.Median-effect and interaction index
plots of PT, ATST or a combination in SW620 (A and C) and HCT116 (B
and D) cells. Cells were treated with a series of dosages of PT (0,
50, 100, 150 and 200 µM), ATST (0, 5, 10, 15 and 20 µM) or a
combination at a fixed ratio of 10:1 for 48 h, and then cell
viability was measured using an MTT assay. Median-effect plots (A,
B) and interaction index plots (C and D) were computed with the
median-effect equation. Synergy was defined as an interaction index
<1.0. The data of interaction plots are shown as the mean ± SD
(n=5). |
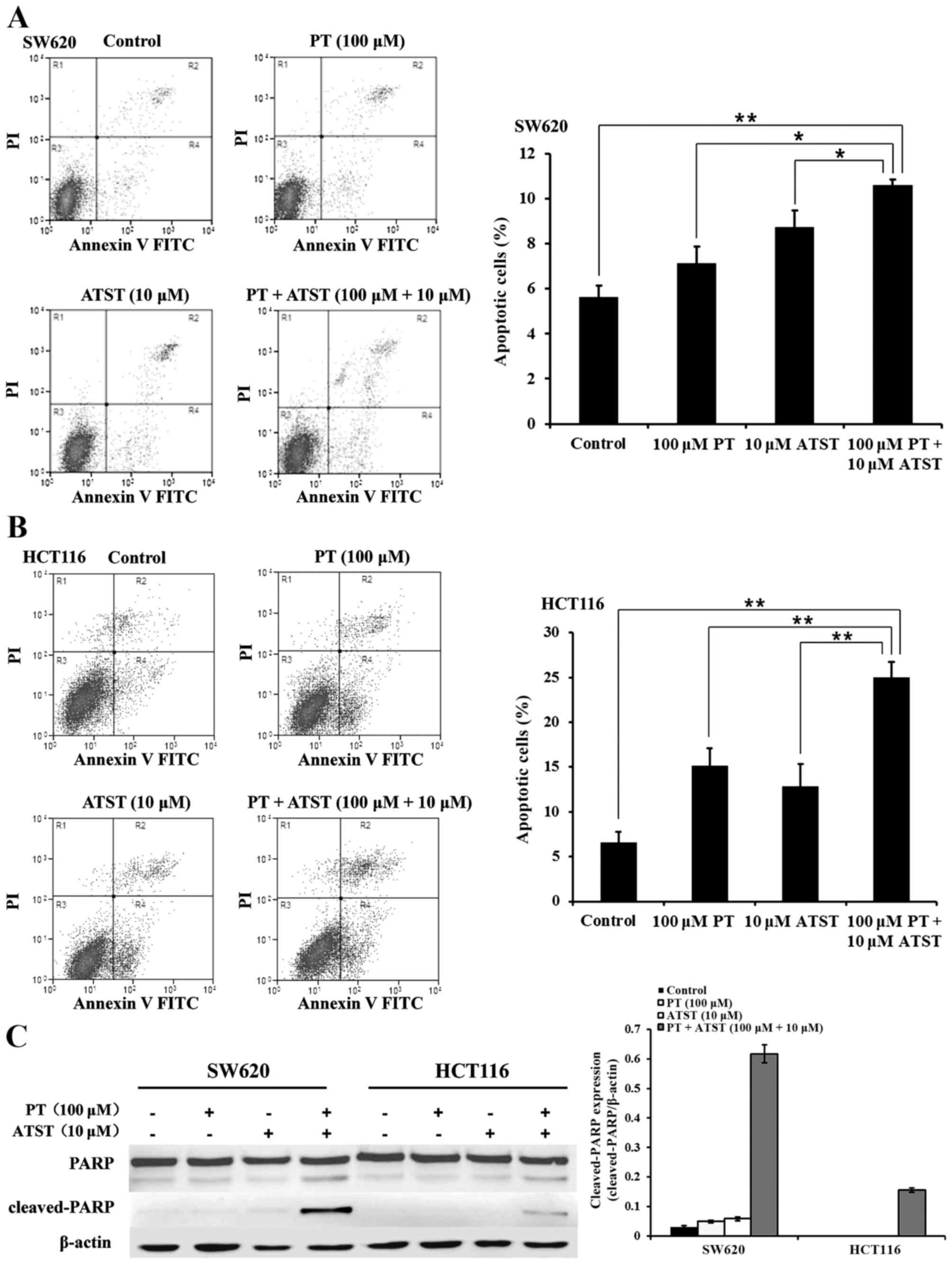
Combined PT and ATST treatment induces apoptosis.
The results revealed a strong synergistic anti-proliferative effect
exerted by the combined PT and ATST treatment of SW620 and HCT116
cells. This observation prompted us to determine whether the
decreased cell survival was related to apoptosis. An Annexin V/PI
double staining assay was used to determine the proportion of
apoptotic cells. The results are presented in Fig. 3A and B. In both SW620 and HCT116
cells, the percentage of apoptotic cells in the group treated with
combined PT and ATST was significantly higher than for those cells
treated with each agent alone. Furthermore, the expression levels
of PARP and cleaved-PARP were determined (Fig. 3C). The PARP fragments were significant
in SW620 and HCT116 cells following the combination treatments. By
contrast, there were no significant levels of cleaved-PARP detected
in either cell line following treatment with the single agents.
These observations, together with the results of the MTT assay,
indicated that combined treatment with PT and ATST could induce
colon cancer cell apoptosis.
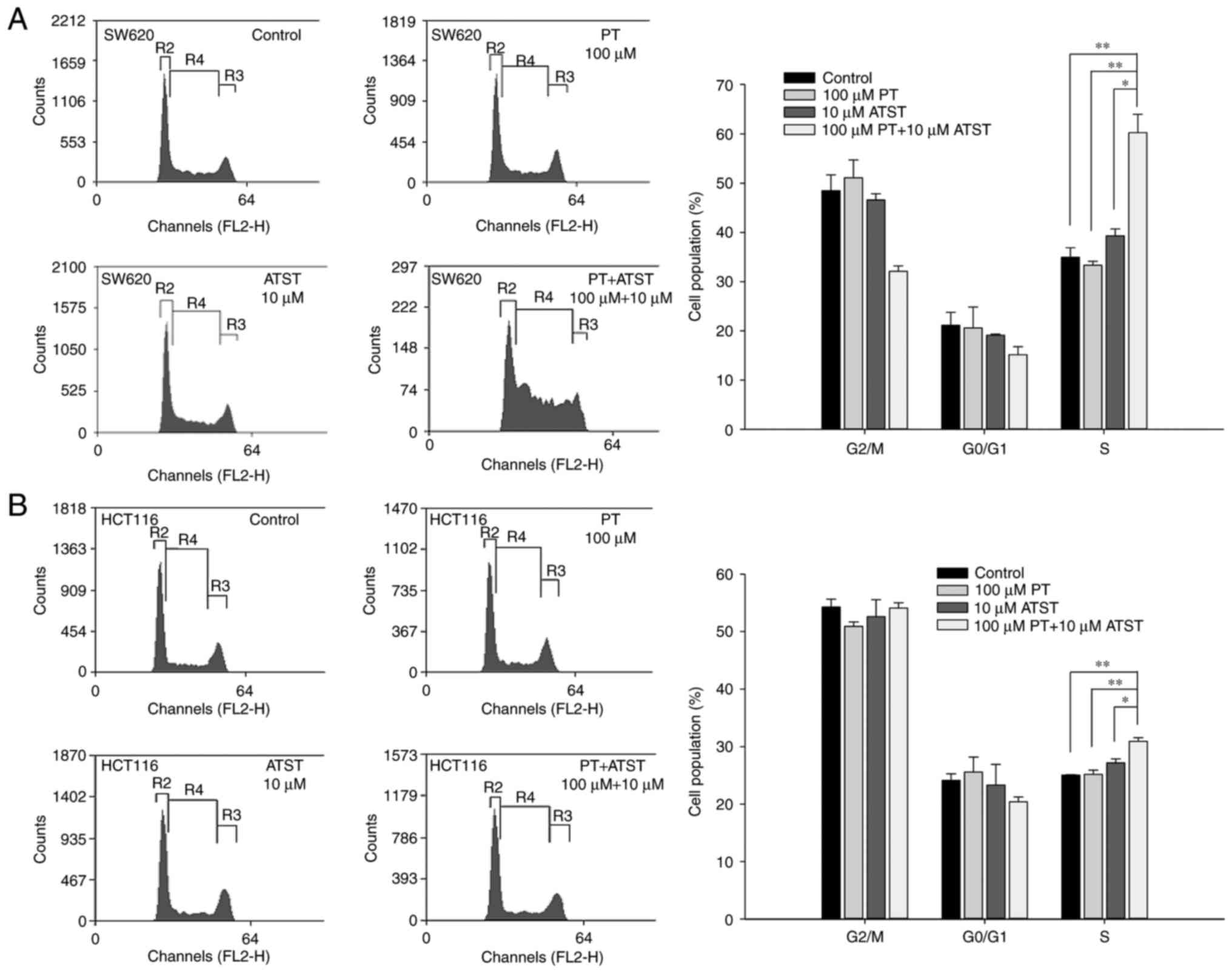
Combined PT and ATST treatment causes cell cycle
arrest at the S phase. The results warranted us to hypothesize that
the synergistic effect of PT and ATST on apoptosis may involve cell
cycle arrest. Flow cytometry was performed to analyze the cell
cycle distribution for both SW620 and HCT116 cells following
treatment with PT, ATST or a combination for 48 h. The results
(Fig. 4) indicated that individual PT
or ATST treatment did not significantly change the cell cycle
distribution for the two cell lines. By contrast, combined PT and
ATST could alter the distribution of cell cycle. For SW620 cells, a
significant increase in the S phase cell proportion was observed
following combined treatment with PT and ATST, which was
correspondingly accompanied by a decrease in the G2/M
phase cell proportion. For HCT116 cells, an increase in the S phase
proportion was also observed with combined treatment; however, this
increase was slighter when compared with the SW620 cells, and no
decrease in the G2/M phase proportion was noted.
Therefore, the results indicated that combined PT and ATST
treatment could arrest SW620 and HCT116 cells in the S phase.
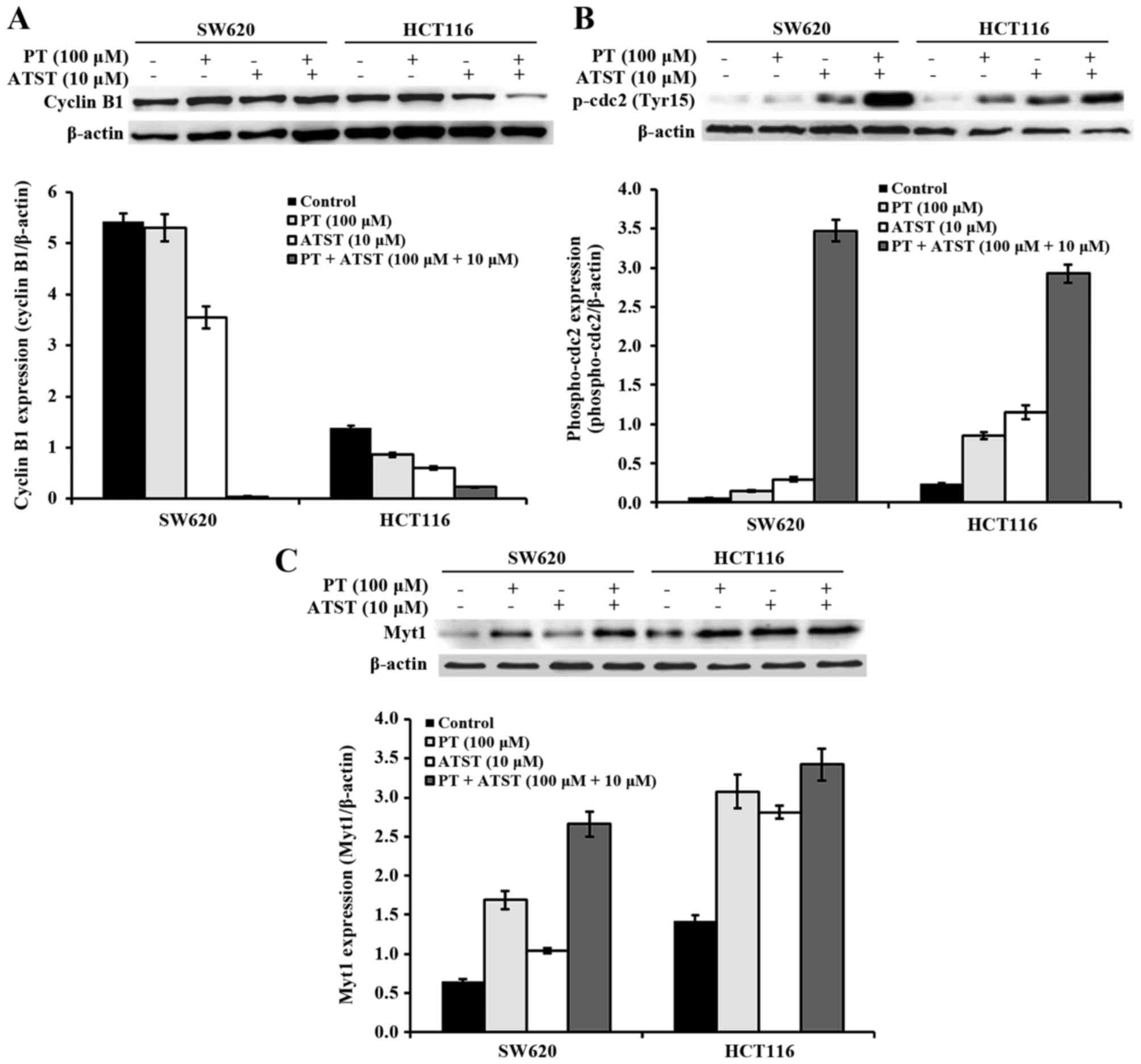
Combined PT and ATST treatment inhibits cdc2
activation. To further explore the mechanisms underlying the cell
cycle arrest, the activity of proteins that perform an important
role during progression at the S phase and the G2/M
checkpoint were examined (Fig. 5). In
both SW620 and HCT116 cells, the expression of cyclin B1 was not
significantly different between the control and PT-treated groups;
for ATST-treated cells, the expression of cyclin B1 was decreased
by ~50%. By contrast, combined treatments of PT and ATST markedly
suppressed the cyclin B expression in all cells (Fig. 5A). The phosphorylation of cdc2 at
Tyr15 was also analyzed, as dephosphorylation at this locus results
in the activation of cdc2. The results showed that the activity of
p-cdc2 were markedly upregulated following combined treatments
(Fig. 5B). Consistently, the activity
of Myt1, which is responsible for the phosphorylation of cdc2 at
Tyr15, showed a similar pattern (Fig.
5C). These results indicated that PT and ATST synergistically
inhibit the activation of cdc2 in both SW620 and HCT116 colon
cancer cells.
Discussion
Since increased 3-hydroxy-3-methylglutaryl coenzyme
A (HMG-CoA) levels have been observed in colon cell lines, previous
studies have evaluated the anti-cancer activity of statins. Statins
are competitive small-molecule inhibitors of HMG-CoA reductase, and
could prevent the transformation of HMG-CoA to mevalonate (24). Although in vitro data have
suggested that ATST could suppress HCT116 cell growth and induce
apoptosis, the effective doses of ATST in these experiments were
relatively higher (50 and 100 µM, respectively) (12,25). In
our experimental design, the maximum dose of ATST was only 25 µM.
As expected, the results showed that ATST exhibited little effect
on HCT116 cell growth and apoptosis at this relatively low dosage.
Previous evidence has suggested that the safe and tolerated
therapeutic dosage range of ATST is 10–80 mg/day (26). This dosage range is lower for ATST
when administered to exhibit a protective effect against colorectal
cancer. Our results suggested a combination strategy for the
prevention of colorectal cancer based on the synergy between ATST
and PT. Through this combination, the growth inhibition effect of
ATST would be substantially increased at a relatively low
dosage.
Similar to ATST, this enhancement effect is also
applicable to PT. The in vivo activities of phytochemicals
are usually restricted due to poor bioavailability. Although
previous studies have reported the anti-proliferative effects of PT
on HL-60, HT29 and HepG2 cells, the effective dosages of PT were
all >100 µM (18,20,22).
Concordantly, our results showed that, in both SW620 and HCT116
cells, PT could not efficiently inhibit cell growth unless the dose
was higher than 100 µM. Although previous studies have reported
that cytochalasin B could enhance the PT-induced apoptosis of HepG2
cells (21), and that PT can
potentiate the anticancer activity of paclitaxel (22), no prior research has focused on
synergy involving PT. The results of the present study demonstrated
that the antitumor efficacy of ATST could be enhanced at a
relatively low dosage through the synergistic action with PT, which
suggested the potential interaction of statins with other compounds
in the food matrix. This interaction affects the efficacy of
statins, and may explain the controversial results obtained in
prior studies regarding the associations between statin use and the
risk of colon cancer-associated mortality (27,28). As
the dietary composition is different for each individual, this can
result in varying statin efficacy. Conversely, different statins
have different antitumor effects. In six colorectal cancer cell
lines, including DLD1, HT29, SW620, HCT116, LoVo and colo320,
simvastatin and fluvastatin showed strong growth suppressive
effects. Atorvastatin demonstrated a relatively weak growth
suppressive effect, whereas no growth suppressive effect was
observed with pravastatin (29). This
may be another reason for the paradoxical results regarding the
antitumor effects of statins.
A close relationship is believed to exist between
the cell cycle and apoptosis in cancer cells (30). Numerous studies have reported
PT-induced apoptosis; however, few of these results involved the
cell cycle, especially in colon cancer cells. Some data supported
that the presence of HMG-CoA reductase inhibitors may influence the
cell cycle distribution of cancer cells. Known as the typical
statin family compound, ATST has been shown to induce colon cancer
cell cycle arrest in the G0/G1 phase when
combined with other compounds (11,12).
Therefore, we decided to determine whether treatments with PT and
ATST could regulate the cell cycle distribution of SW620 and HCT116
cells. The results partly confirmed our hypothesis, in that the
cell cycle was arrested, but also demonstrated that the cells were
arrested at the G2/M checkpoint and accompanied by an
increased cell population in the S phase, rather than arrested in
the G0/G1 phase. Specifically, cdc2 kinase
activation is the pivotal regulator mechanism responsible for the
G2/M checkpoint. Activation of cdc2 is controlled via
two steps: One is cyclin binding; the other is the
dephosphorylation of cdc2 at Tyr15, which is the core regulatory
step (31).
In the present study, we observed that PT and ATST
synergistically downregulated the expression of cyclin B1, which
indicated that formation of the cyclin B-cdc2 complex might be
inhibited. In addition, our data showed that the level of cdc2
phosphorylation at Tyr15 was markedly increased by combined
treatment with PT and ATST. These results demonstrated that the
cdc2 kinase was inactivated, possibly due to a failure to bind to
cyclin B and the increased levels of p-cdc2 at Tyr15. Myt1 protein
kinase is regarded as a negative modulator of cdc2, and carries out
the phosphorylation of cdc2 at Tyr15 (32). The p21 gene is an inhibitor of cyclin;
hyper-phosphorylation of p21 activates cdc2 kinase in the
G2/M transition (33,34).
Previous studies have shown that ATST can increase p21 levels in
A549 cells (35) and the pancreatic
cancer (36). However, Buranrat et
al reported opposing results, stating that ATST reduced p21
expression in KKU-100 cells and did not alter p21 expression in
KKU-M214 cells (37). In terms of the
synergistic effect, p21 levels were increased in HT29 and HCT116
cells following treatment with combined ATST and celecoxib
(11), and with combined ATST and
γ-tocotrienol (12). Therefore, the
p21 gene may be the potential regulatory target underlying the
G2/M phase arrest following the synergistic action of
ATST and PT; more in depth future investigations are warranted.
In summary, the present study demonstrated that PT
and ATST produce a powerful synergistic interaction in suppressing
colon cancer cell growth. This process was accomplished via the
synergistic induction of apoptosis and the arrest of the cell cycle
at the G2/M checkpoint, which resulted from
downregulated cdc2 activation following combined treatment.
Acknowledgements
The present study was conducted using grants
supported by the Chinese Academy of Agricultural Sciences (grant
no. 2014ZL041).
References
|
1
|
Baena R and Salinas P: Diet and colorectal
cancer. Maturitas. 80:258–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lippi G, Mattiuzzi C and Cervellin G: Meat
consumption and cancer risk: A critical review of published
meta-analyses. Crit Rev Oncol Hematol. 97:1–14. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Keshk WA, Zineldeen DH, Wasfy RE and
El-Khadrawy OH: Fatty acid synthase/oxidized low-density
lipoprotein as metabolic oncogenes linking obesity to colon cancer
via NF-kappa B in Egyptians. Med Oncol. 31:1922014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Davis-Yadley AH, Lipka S, Shen H, Devanney
V, Swarup S, Barnowsky A, Silpe J, Mosdale J, Pan Q, Fridlyand S,
et al: Ethnic disparities in the risk of colorectal adenomas
associated with lipid levels: A retrospective multiethnic study. J
Gastrointest Cancer. 46:29–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reverter M, Rentero C, Garcia-Melero A,
Hoque M, Vilà de Muga S, Alvarez-Guaita A, Conway JR, Wood P,
Cairns R, Lykopoulou L, et al: Cholesterol regulates Syntaxin 6
trafficking at trans-Golgi network endosomal boundaries. Cell Rep.
7:883–897. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sheng R, Kim H, Lee H, Xin Y, Chen Y, Tian
W, Cui Y, Choi JC, Doh J, Han JK, et al: Cholesterol selectively
activates canonical Wnt signalling over non-canonical Wnt
signalling. Nat Commun. 5:43932014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nielsen SF, Nordestgaard BG and Bojesen
SE: Statin use and reduced cancer-related mortality. N Engl J Med.
367:1792–1802. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Poynter JN, Gruber SB, Higgins PD, Almog
R, Bonner JD, Rennert HS, Low M, Greenson JK and Rennert G: Statins
and the risk of colorectal cancer. N Engl J Med. 352:2184–2192.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lytras T, Nikolopoulos G and Bonovas S:
Statins and the risk of colorectal cancer: An updated systematic
review and meta-analysis of 40 studies. World J Gastroenterol.
20:1858–1870. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiao H and Yang CS: Combination regimen
with statins and NSAIDs: A promising strategy for cancer
chemoprevention. Int J Cancer. 123:983–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiao H, Zhang Q, Lin Y, Reddy BS and Yang
CS: Combination of atorvastatin and celecoxib synergistically
induces cell cycle arrest and apoptosis in colon cancer cells. Int
J Cancer. 122:2115–2124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang Z, Xiao H, Jin H, Koo PT, Tsang DJ
and Yang CS: Synergistic actions of atorvastatin with
gamma-tocotrienol and celecoxib against human colon cancer HT29 and
HCT116 cells. Int J Cancer. 126:852–863. 2010.PubMed/NCBI
|
|
13
|
Lu G, Xiao H, You H, Lin Y, Jin H,
Snagaski B and Yang CS: Synergistic inhibition of lung
tumorigenesis by a combination of green tea polyphenols and
atorvastatin. Clin Cancer Res. 14:4981–4988. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bazzano LA, Serdula MK and Liu S: Dietary
intake of fruits and vegetables and risk of cardiovascular disease.
Curr Atheroscler Rep. 5:492–499. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu RH: Health benefits of fruit and
vegetables are from additive and synergistic combinations of
phytochemicals. Am J Clin Nutr. 78(3 Suppl): 517S–520S.
2003.PubMed/NCBI
|
|
16
|
Boyer J and Liu RH: Apple phytochemicals
and their health benefits. Nutr J. 3:52004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hyson DA: A comprehensive review of apples
and apple components and their relationship to human health. Adv
Nutr. 2:408–420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kobori M, Iwashita K, Shinmoto H and
Tsushida T: Phloretin-induced apoptosis in B16 melanoma 4A5 cells
and HL60 human leukemia cells. Biosci Biotechnol Biochem.
63:719–725. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iwashita K, Kobori M, Yamaki K and
Tsushida T: Flavonoids inhibit cell growth and induce apoptosis in
B16 melanoma 4A5 cells. Biosci Biotechnol Biochem. 64:1813–1820.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park SY, Kim EJ, Shin HK, Kwon DY, Kim MS,
Surh YJ and Park JH: Induction of apoptosis in HT-29 colon cancer
cells by phloretin. J Med Food. 10:581–586. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu CH, Ho YS, Tsai CY, Wang YJ, Tseng H,
Wei PL, Lee CH, Liu RS and Lin SY: In vitro and in vivo study of
phloretin-induced apoptosis in human liver cancer cells involving
inhibition of type II glucose transporter. Int J Cancer.
124:2210–2219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang KC, Tsai CY, Wang YJ, Wei PL, Lee CH,
Chen JH, Wu CH and Ho YS: Apple polyphenol phloretin potentiates
the anticancer actions of paclitaxel through induction of apoptosis
in human hep G2 cells. Mol Carcinog. 48:420–431. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gazzerro P, Proto MC, Gangemi G, Malfitano
AM, Ciaglia E, Pisanti S, Santoro A, Laezza C and Bifulco M:
Pharmacological actions of statins: A critical appraisal in the
management of cancer. Pharmacol Rev. 64:102–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang EH, Johnson LA, Eaton K, Hynes MJ,
Carpentino JE and Higgins PD: Atorvastatin induces apoptosis in
vitro and slows growth of tumor xenografts but not polyp formation
in MIN mice. Dig Dis Sci. 55:3086–3094. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Athyros VG, Tziomalos K, Karagiannis A and
Mikhailidis DP: Atorvastatin: Safety and tolerability. Expert Opin
Drug Saf. 9:667–674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gray RT, Loughrey MB, Bankhead P, Cardwell
CR, McQuaid S, O'Neill RF, Arthur K, Bingham V, McGready C, Gavin
AT, et al: Statin use, candidate mevalonate pathway biomarkers and
colon cancer survival in a population-based cohort study. Br J
Cancer. 116:1652–1659. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Voorneveld PW, Reimers MS, Bastiaannet E,
Jacobs RJ, van Eijk R, Zanders MMJ, Herings RMC, van Herk-Sukel
MPP, Kodach LL, van Wezel T, et al: Statin use after diagnosis of
colon cancer and patient survival. Gastroenterology.
153:470–479.e4. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishikawa S, Hayashi H, Kinoshita K, Abe M,
Kuroki H, Tokunaga R, Tomiyasu S, Tanaka H, Sugita H, Arita T, et
al: Statins inhibit tumor progression via an enhancer of zeste
homolog 2-mediated epigenetic alteration in colorectal cancer. Int
J Cancer. 135:2528–2536. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fisher D, Krasinska L, Coudreuse D and
Novák B: Phosphorylation network dynamics in the control of cell
cycle transitions. J Cell Sci. 125:4703–4711. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chow JP and Poon RY: The CDK1 inhibitory
kinase MYT1 in DNA damage checkpoint recovery. Oncogene.
32:4778–4788. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhan Q, Antinore MJ, Wang XW, Carrier F,
Smith ML, Harris CC and Fornace AJ Jr: Association with Cdc2 and
inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated
protein Gadd45. Oncogene. 18:2892–2900. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dash BC and El-Deiry WS: Phosphorylation
of p21 in G2/M promotes cyclin B-Cdc2 kinase activity. Mol Cell
Biol. 25:3364–3387. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin YC, Lin JH, Chou CW, Chang YF, Yeh SH
and Chen CC: Statins increase p21 through inhibition of histone
deacetylase activity and release of promoter-associated HDAC1/2.
Cancer Res. 68:2375–2383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mohammed A, Qian L, Janakiram NB,
Lightfoot S, Steele VE and Rao CV: Atorvastatin delays progression
of pancreatic lesions to carcinoma by regulating PI3/AKT signaling
in p48Cre/+ LSL-KrasG12D/+ mice. Int J Cancer. 131:1951–1962. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Buranrat B, Senggunprai L, Prawan A and
Kukongviriyapan V: Simvastatin and atorvastatin as inhibitors of
proliferation and inducers of apoptosis in human cholangiocarcinoma
cells. Life Sci. 153:41–49. 2016. View Article : Google Scholar : PubMed/NCBI
|