Introduction
Pancreatic cancer has one of the highest mortality
rates of all cancer types. It is estimated that >330,000 people
are diagnosed with pancreatic cancer annually worldwide (1). Despite the relatively low
epidemiological ranking of the disease, and although extensive
efforts are being made to improve the early diagnosis of the
disease, the prognosis of pancreatic cancer remains poor, with a
5-year survival rate of only 4%, making it the fourth-leading cause
of all cancer-associated mortality in the United States (2).
The most common type of pancreatic cancer is
adenocarcinoma (accounting for 95% of all cases), which originates
from the exocrine region of the pancreas and is classified as
pancreatic ductal adenocarcinoma (PDAC) (2). PDAC is insensitive to chemotherapy and
radiotherapy, meaning the identification of novel therapeutic
targets is imperative (3).
Fatty acid synthase (FASN) is a multifunctional
protein homodimer that converts acetyl coenzyme A (CoA) and
malonyl-CoA into palmitate, thus regulating lipogenesis (4). Overexpression of FASN has been found to
correlate with insulin resistance, type-2 diabetes and pancreatic
cancer (5). The overexpression of
FASN is an indicator of a poor patient prognosis, a high risk of
recurrence and poor survival in numerous cancer types, including
cancer of the breast (6), prostate
(7), lung (8) and pancreas (9).
FASN overexpression has been shown to cause
resistance to gemcitabine-based chemotherapy and radiotherapy in
pancreatic cancer patients (10);
however, the mechanism by which this happens remains unclear. The
Warburg effect is considered to be a possible mechanism for cancer
chemoresistance, with the tumor-specific pyruvate kinase M2 (PKM2)
protein essential for this effect. It has been reported that the
chemoresistance of pancreatic cancer to gemcitabine is
PKM2-dependent, and PKM2 is believed to be a therapeutic target of
gemcitabine-resistant pancreatic cancer (11). Indeed, the present study found that
FASN regulates PKM2 expression and glucose metabolism, leading to
gemcitabine chemoresistance in PDAC cells via the direct regulation
of PKM2. Collectively, the findings of the present study imply that
FASN upregulates PKM2 expression and induces gemcitabine resistance
in pancreatic cancer.
Materials and methods
Oncomine database analysis
mRNA microarray datasets of pancreatic cancer and
normal tissue samples were analyzed using the Oncomine microarray
gene expression database (www.oncomine.org). The parameters maintained including
the follow: P<0.0001; fold change >2; and the top 10% of
ranked genes.
Plasmids and reagents
Myc-tagged Myc-PKM2 was cloned into a pCMV vector.
To construct the Myc-PKM2 plasmid, the full length PKM2 gene was
amplified using 293T cells via PCR amplification and cloned into
the pCMV-Myc vector (Takara Bio, Inc., Otsu, Japan). Anti-PKM2
(cat. no. 4053; 1:1,000), anti-FASN (cat. no. 3189; 1:1,000) and
anti-cleaved caspase-3 (cat. no. 9662; 1:1,000) antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Anti-β-Tubulin (cat. no. sc-5274; 1:5,000) was purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Gemcitabine was
obtained from Eli Lilly (Surrey, UK) and dissolved in distilled
water. Pancreatic cancer cells were treated with 10 µM gemcitabine
and the control group was treated with equal amounts of distilled
water for 24 h. Cerulenin was purchased from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany) and 20 µM cerulenin was used to treat
pancreatic cancer cells for 24 h.
Cell culture and transfection
PDAC cell lines, PANC-1 and MIA PaCa-2, were
purchased from the American Type Culture Collection (Manassas, VA,
USA) and cultured in 5% CO2, at 37°C and in 95%
humidity. PANC-1 and MIA PaCa-2 cells were cultured in Dulbecco's
modified Eagle's medium supplemented with 10% fetal bovine serum
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 100 U/ml
penicillin and 100 µg/ml streptomycin (Thermo Fisher Scientific,
Inc.). Pancreatic cancer cells with or without FASN knockdown were
transfected with Myc-PKM2 plasmids (2 µg/1×106 cells)
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. A total of 24 h
post-transfection, cells were collected for further analyses.
Western blotting
Cells (1×106) were lysed with lysis
buffer (1% Nonidet P-40, 1X PBS, 0.1% sodium dodecyl sulfate and 1%
protease inhibitor cocktail), followed by protein quantification
using a bicinchoninic acid (BCA) assay. Samples were diluted in
loading buffer containing dithiothreitol and boiled for 5 min.
Equal amounts (50 µg) of protein for each sample was separated by
10% SDS-PAGE and transferred onto nitrocellulose membranes. The
membranes were immuno-blotted with the aforementioned specific
primary antibodies targeted at the protein of interest in 4°C
overnight. Subsequently, the membrane was wash three times with 1X
TBST and incubated with rabbit IgG (cat. no. MR-R100; 1:3,000) and
(mouse IgG; cat. no. MR-M100; 1:3,000) horseradish
peroxidase-conjugated secondary antibodies (both Shanghai
MRbiotech, Co., Ltd., Shanghai, China) for 1 h at room temperature,
and then visualized using SuperSignal West Pico Stable Peroxide
solution (Thermo Fisher Scientific, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
reagent (Thermo Fisher Scientific, Inc.). The cDNA was synthesized
using Superscript II reverse transcriptase (Thermo Fisher
Scientific, Inc.). qPCR was performed using IQ SYRB Green Supermix
and an iCycleriQTX detection system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The following thermocycling conditions were
maintained: Denaturing at 95°C for 20 sec; annealing at 58°C for 30
sec; and extension at 72°C for 30 sec (43 cycles). All signals were
normalized against GAPDH and the 2−∆∆Cq method was used
to quantify the fold change (12).
The primers used were as follows: FASN forward,
5′-GGTCTTGAGAGATGGCTTGC-3′ and reverse, 5′-AATTGGCAAAGCCGTAGTTG-3′;
PKM2 forward, 5′-TCGCATGCAGCACCTGATT-3′ and reverse,
5′-CCTCGAATAGCTGCAAGTGGTA-3′; and GAPDH forward,
5′-ACCCACTCCTCCACCTTTGAC-3′ and reverse,
5′-TGTTGCTGTAGCCAAATTCGTT-3′.
RNA interference
Lentivirus-based control and gene-specific short
hairpin RNAs (shRNAs) were purchased from Sigma-Aldrich (Merck
KGaA). Transfections were performed using Lipofectamine 2000
(Thermo Fisher Scientific, Inc.). A total of 2 µg of gene-specific
shRNA or shNT (control) were transfected into 293T cells
(5×105 cells). After 48 h transfection, the cultured
medium of 293T cells was collected and applied to pancreatic cancer
cells. Pancreatic cancer cells were cultured in 5% CO2,
at 37°C for 48 h, followed by puromycin (0.75 µg/ml; Sigma-Aldrich;
Merck KGaA) selection. Cells were collected 72 h post-transfection.
The knockdown efficiency was confirmed through western blotting or
PCR using the aforementioned method. shRNA sequences were as
follows: shFASN#1,
CCGGCCTACTGGATGCGTTCTTCAACTCGAGTTGAAGAACGCATCCAGTAGGTTTTTG;
shFASN#2,
CCGGGCTGCTAGATGTAGGTGTTAGCTCGAGCTAACACCTACATCTAGCAGCTTTTTG;
ShPKM2#1, CCGGCTTTCCTGTGTGTACTCTGTCCTCGAGGACAGAGTACAC
ACAGGAAAGTTTTTTG; shPKM2#2,
CCGGGTTCGGAGGTTTGATGAAATCCTCGAGGATTTCATCAAACCTCCGAACTTTTTTG.
Measurements of glucose consumption
and lactate production
Culturing medium was collected 24 h after plasmid
transfection or 48 h after lentivirus infection to allow for the
measurement of glucose and lactate concentrations. Glucose levels
were determined using a Glucose (GO) assay kit (Sigma-Aldrich;
Merck KGaA). Glucose consumption was defined as the difference
between the glucose concentration in fresh medium and collected
medium. Lactate levels were determined using a Lactate assay kit
(Eton Bioscience, Inc., San Diego, CA, USA). The optical densities
were measured at a wavelength of 570 nm in Molecular Devices
Spectramax 190 microplate reader (Marshall Scientific, Hampton, NH,
USA).
Caspase-3 activity measurement
The activity of caspase-3 was measured using a
Caspase-3 Colorimetric Protease assay (Thermo Fisher Scientific,
Inc.). Cells were counted and pellets of 3–5×106 cells
were produced per sample. Cells were lysed using 50 µl lysis
buffer, followed by protein quantification using a BCA assay.
Cytosolic extract was diluted to a concentration of 50–200 µg
protein per 50 µl in Cell Lysis Buffer (1–4 mg/ml). A total of 50
µl of 2X Reaction Buffer (containing 10 mM DTT) was added to each
sample, followed by 5 µl of the 4 mM DEVD-pNA substrate (200 µM
final concentration), which was then incubated at 37°C for 2 h in
the dark. Reactions were measured using a microplate reader at a
wavelength of 405 nm.
Cell proliferation assay
Cell growth was monitored by MTS Cell Proliferation
assay according to the manufacturer's instructions (Promega
Corporation, Madison, WI, USA). In brief, cells were plated in
96-well plates at a density of 1,000 cells per well. A total 20 µl
of CellTiter 96R AQueous One Solution Reagent (Promega Corporation)
was added to each cell. At 60 min after incubation (at 37°C in a
cell incubator), cell proliferation was measured using a microplate
reader at a wavelength of 490 nm.
Colony formation assay
Pancreatic cancer cells were seeded into a 6-well
plate at a density of 1×103 cells/well. Cells were
cultured in 5% CO2 at 37°C for 7 days. Cells were fixed
with methanol at room temperature for 20 min and stained with 0.1%
crystal violet at room temperature for 10 min. The number of clones
were counted to determine the efficiency of colony formation.
Statistical analysis
Microsoft Excel software (version 2013; Microsoft
Corporation, Redmond, WA, USA) was used for statistical analysis.
Each experiment was performed in triplicate or with more replicates
unless otherwise stated. Unpaired student's t-test was performed
for analysis of the statistical significance between groups. A
P-value of <0.05 was considered to indicate statistical
significance.
Results
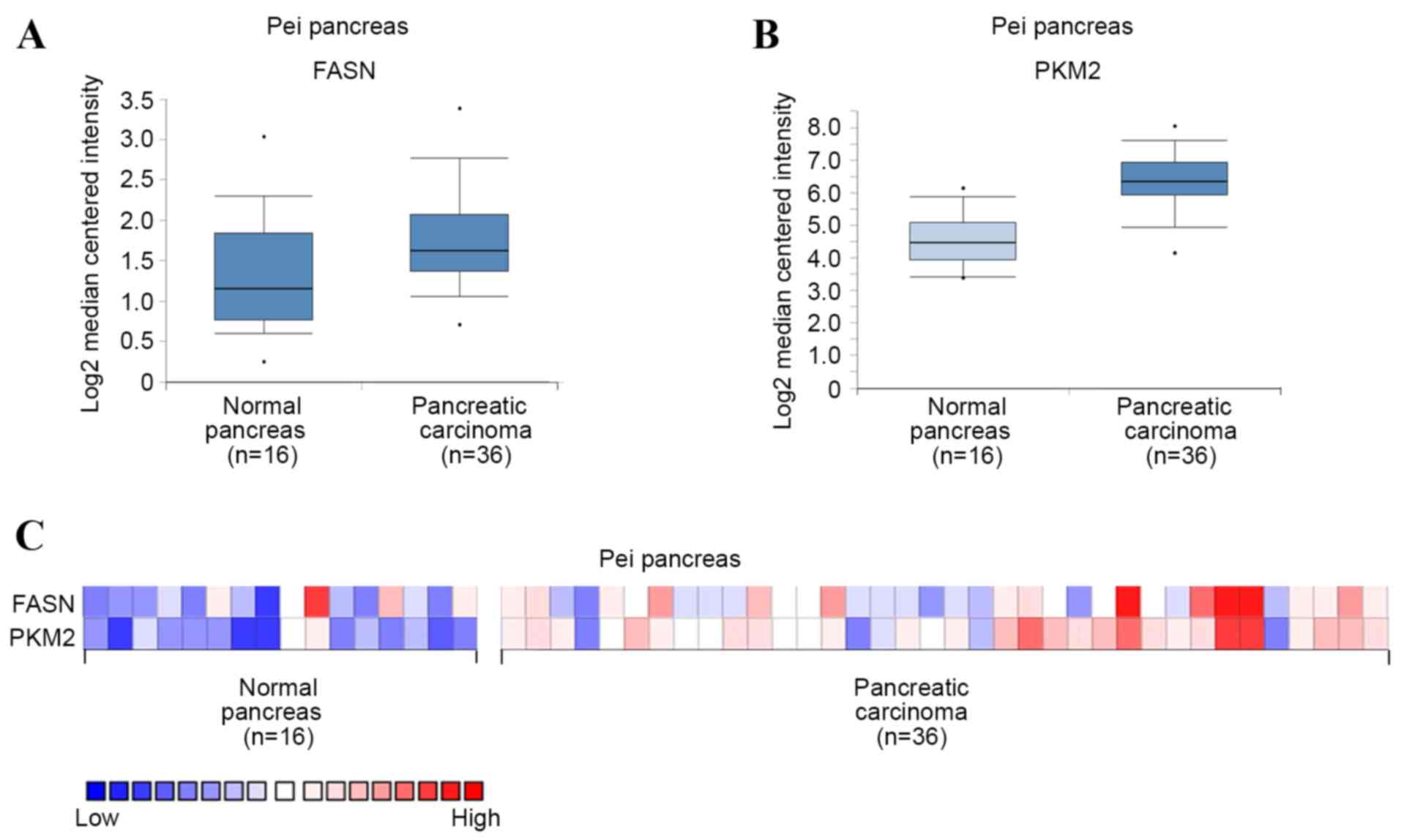
FASN and PKM2 are upregulated in
PDAC
To investigate the expression of FASN and PKM2 in
PDAC, mRNA levels were first analyzed in PDAC and normal pancreas
tissues using Oncomine microarray gene expression datasets
(www.oncomine.org) (13,14). It
was found that FASN and PKM2 were upregulated in PDAC tissues
compared with their normal counterparts, using the Pei pancreas
dataset in Oncomine (Fig. 1A and B).
PKM2 mRNA level was also demonstrated to potentially positively
correlate with that of FASN in each sample using the Pei pancreas
dataset (Fig. 1C). These results
suggest that FASN and PKM2 are upregulated in PDAC, and that PKM2
expression may be positively correlated with that of FASN.
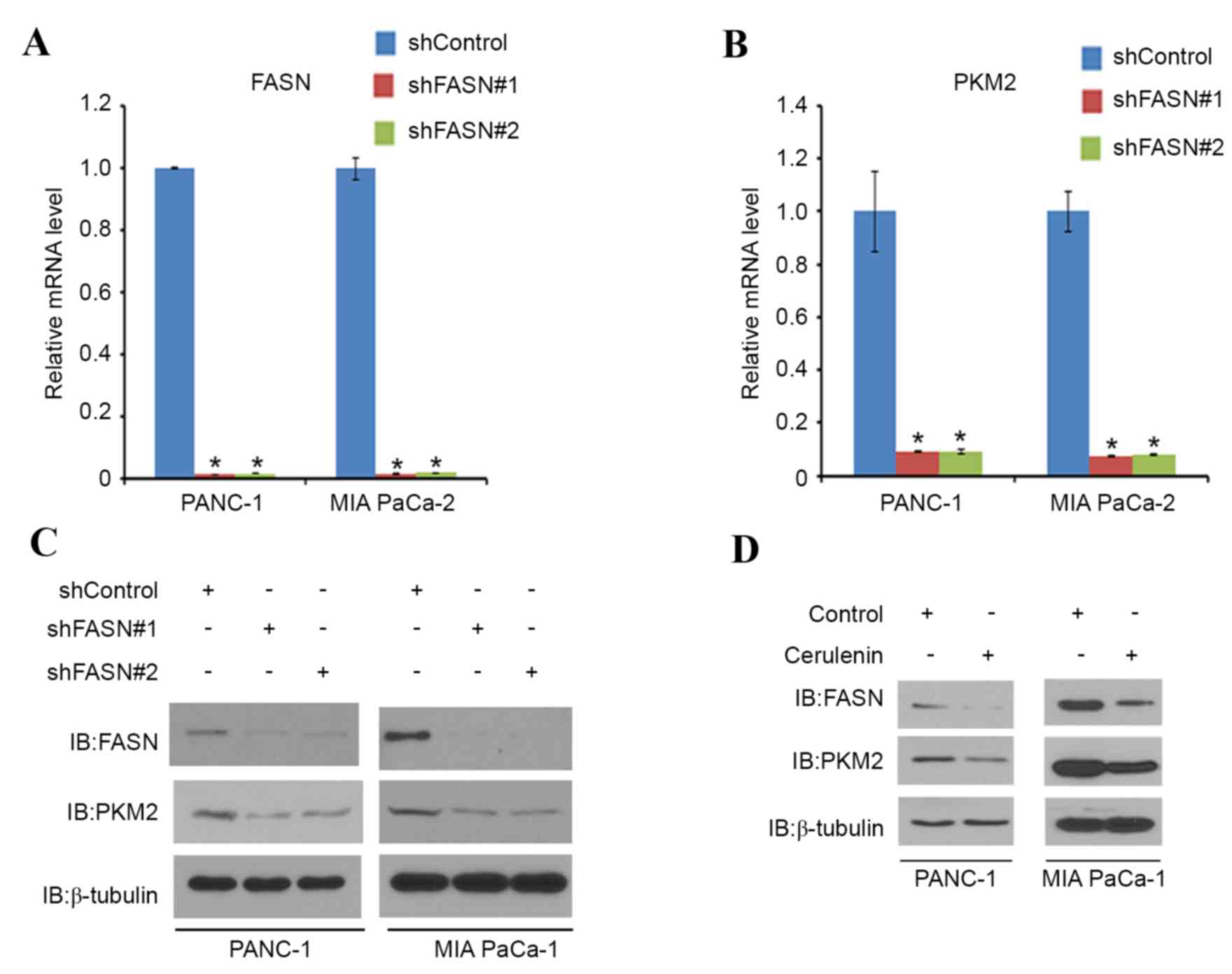
FASN regulates PKM2 expression at the
mRNA level
Next, the association between FASN and PKM2 was
examined in PDAC. PANC-1 and MIA PaCa-2 cells were treated with
non-specific (control) or FASN-specific shRNAs. When FASN was
effectively knocked down (Fig. 2A),
PKM2 mRNA (Fig. 2B) and protein
expression (Fig. 2C) was decreased in
these cells (Fig. 2A, C and D).
PANC-1 and MIA PaCa-2 cells were then treated with cerulenin, a
FASN-specific inhibitor. As expected, the protein level of FASN was
decreased in the two cell lines. Notably, the PKM2 protein level in
the two cell lines also decreased correspondingly (Fig. 2D). These results indicate that FASN
may regulate the expression of PKM2 in PDAC cell lines.
FASN regulates glucose metabolism
through PKM2 in PDAC cells
PKM2 is known to promote glucose metabolism in
cancer cells (15). To verify the
role of FASN in the PKM2-mediated regulation of glucose metabolism
in PDAC, PANC-1 and MIA PaCa-2 cells were treated with non-specific
or PKM2-specific shRNAs (Fig. 3A).
Glucose consumption and lactate production level were significantly
decreased in each cell line when PKM2 was knocked down (Fig. 3B and C). PANC-1 and MIA PaCa-2 cells
were then treated with non-specific or FASN-specific shRNAs
(Fig. 3D). Again, glucose consumption
and the lactate production level significantly decreased in the
cells that underwent FASN-knockdown (Fig.
3E and F). PKM2 overexpression also significantly increased the
glucose consumption and lactate production level in the two cell
lines; however, FASN-knockdown significantly diminished this
tendency (Fig. 3G and H). These
results imply that FASN may regulate glucose metabolism in PDAC
cells via PKM2.
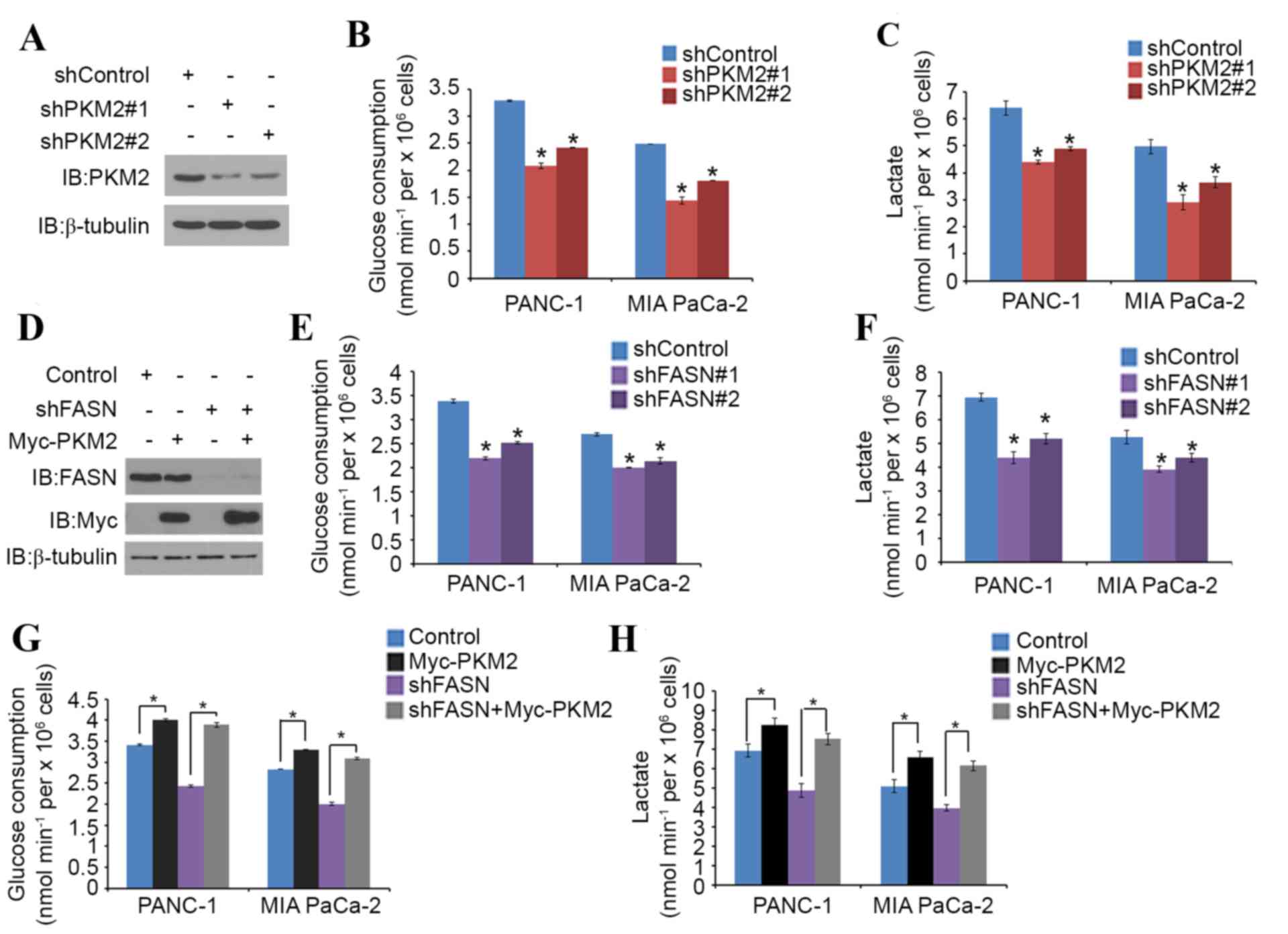 | Figure 3.FASN regulates glucose metabolism
through PKM2 in PDAC cells. (A-C) PANC-1 and MIA PaCa-2 cells were
transfected with PKM2 shRNA and negative control. After 48 h, cells
were harvested and (A) western blot analysis of the WCL was
performed, as well as measurement of (B) glucose consumption and
(C) L-Lactate production in the spent medium. Data are presented as
the mean ± SD from three replicates. (D) PANC-1 cells were
transfected with indicated plasmids and, after 24 h, cells were
harvested for western blot analysis. (E and F) Measurement of (E)
glucose consumption and (F) L-Lactate production in the spent
medium of PANC-1 and MIA PaCa-2 cells 48 h after transfection with
FASN shRNA and negative control. Data are presented as the mean ±
SD from three replicates. (G and H) Measurement of (G) glucose
consumption and (H) L-Lactate production in the spent medium of
PANC-1 and MIA PaCa-2 cells 48 h after transfection with the
indicated constructs. Data are presented as the mean ± SD from
three replicates. *P<0.05 vs. shControl or shFASN+Myc-PKM2
groups. FASN, fatty acid synthase; PKM2, pyruvate kinase M2; shRNA,
short hairpin RNA; SD, standard deviation; IB, immunoblot; Myc,
V-myc avian myelocytomatosis viral oncogene homolog. |
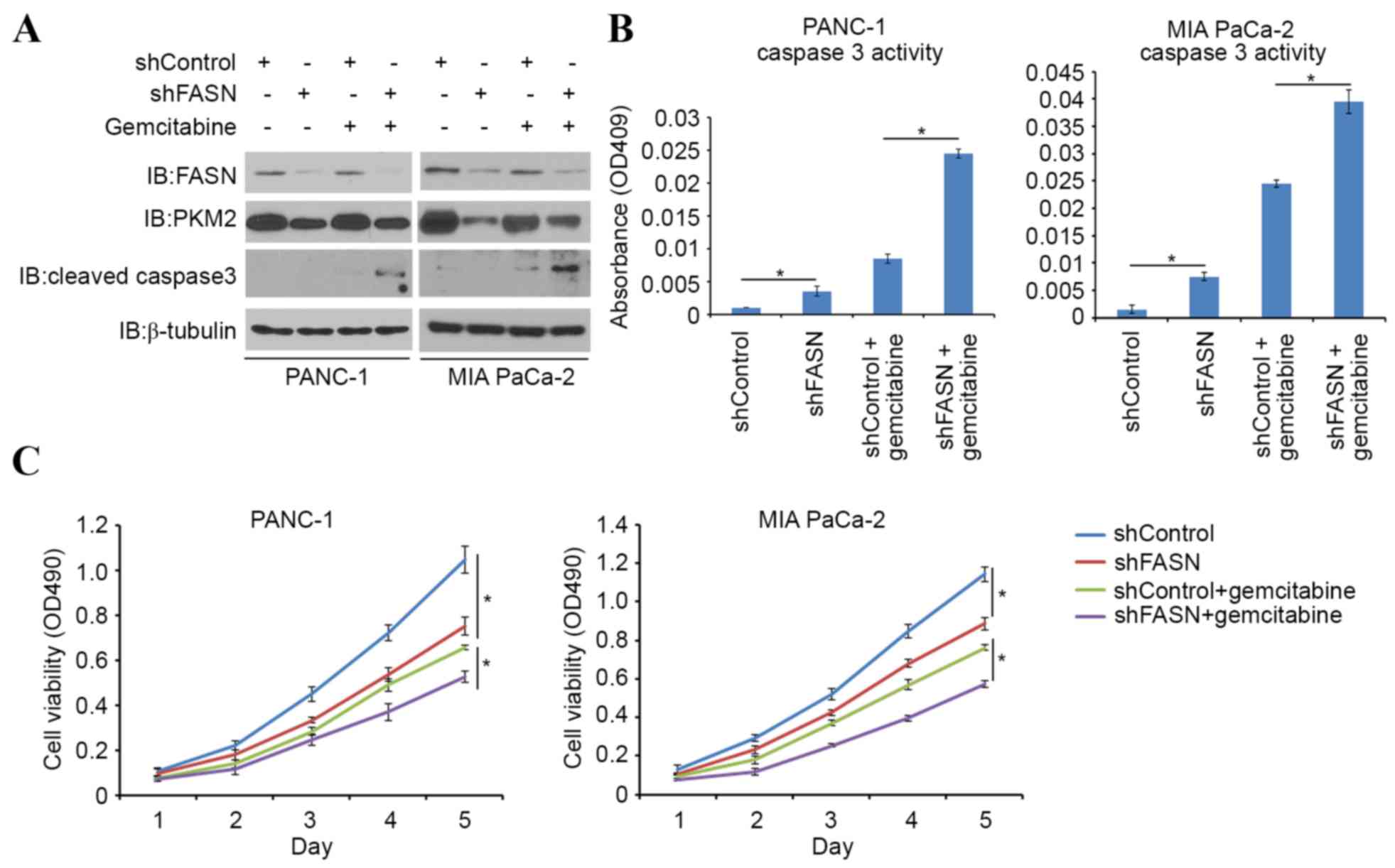
FASN induces gemcitabine
chemoresistance in PDAC cells via PKM2
Gemcitabine is one of the first-line therapeutic
agents for the treatment of pancreatic cancer; however, it has
limited efficacy in advanced pancreatic cancer owing to
chemoresistance. One possible mechanism of cancer chemoresistance
is through the Warburg effect, in which tumor-specific PKM2 may
play a pivotal role. Indeed, it has been reported that PKM2 is
implicated in gemcitabine chemoresistance (11). In order to investigate the role of
FASN in sensitizing PDAC cells to gemcitabine-induced apoptotic
death, PDAC cells were treated with gemcitabine alone or in
combination with FASN-targeted shRNAs. FASN-knockdown led to an
increase in cleaved caspase-3 expression (a pro-apoptotic protein),
as well as in caspase-3 activity induced by gemcitabine (Fig. 4A and B). Cell viability was also
decreased in FASN-knockdown cells, compared with controls when
treated with gemcitabine (Fig. 4C).
Consistent with previous findings (10), the present study also indicates that
FASN serves a significant role in gemcitabine resistance in PDAC
cells.
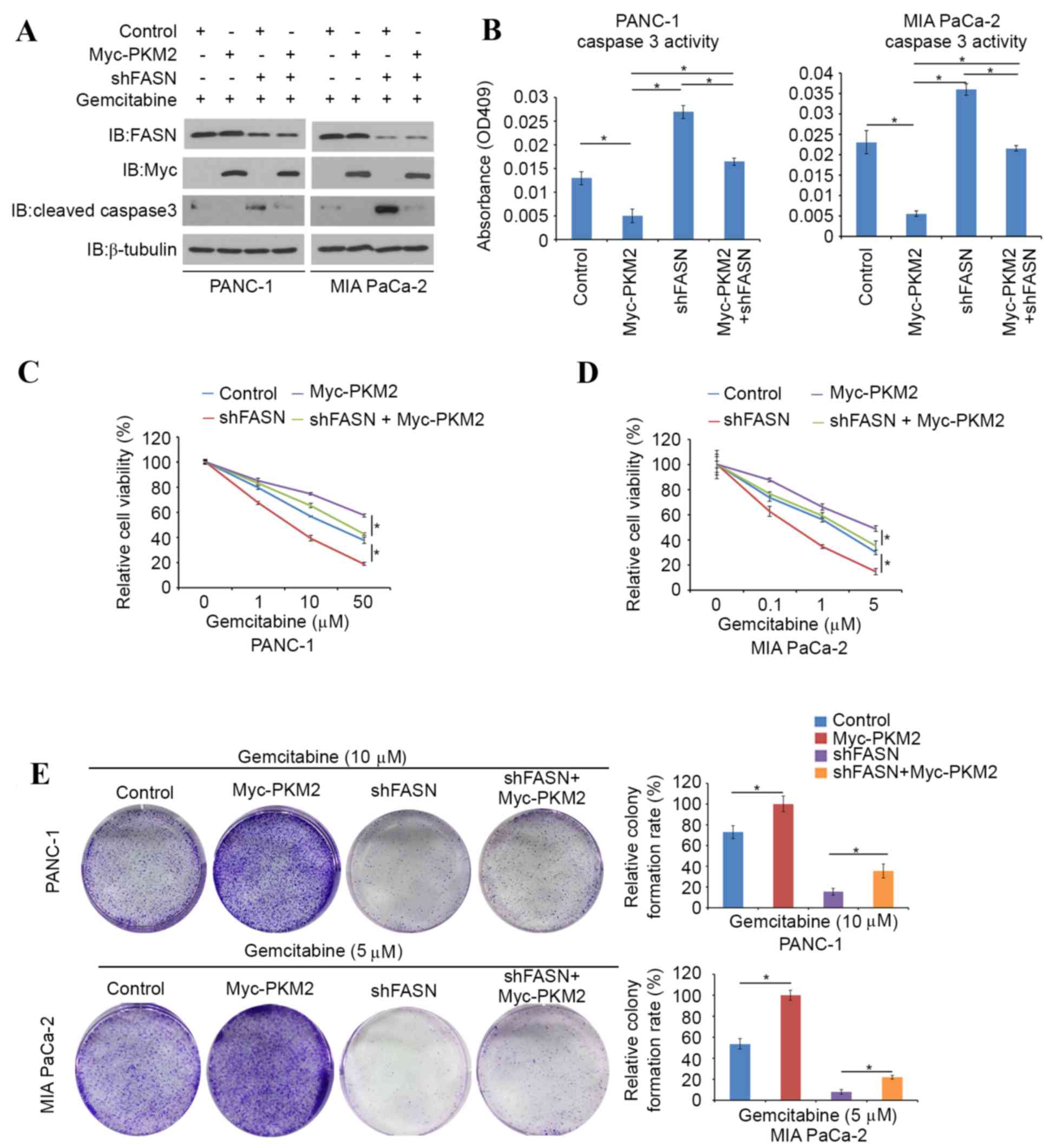
A PKM2-expressing plasmid was transfected into
FASN-knockdown PDAC cells and the evaluation of caspase 3 activity
was repeated. It was found that cleaved caspase-3 protein levels
and caspase-3 activity were markedly decreased in
PKM2-overexpressing PDAC cells (Fig. 5A
and B). Cell viability assays were then performed in
PKM2-overexpressing, FASN-knockdown or PKM2-overexpression and
FASN-knockdown double-treated PANC-1 and MIA PaCa-2 cells in the
presence of different concentration of gemcitabine. Higher
concentrations of gemcitabine were required to suppress cell growth
in PKM2-overexpressing PANC-1 and MIA PaCa-2 cells compared with
controls, whereas FASN-knockdown diminished the effects of PKM2
overexpression (Fig. 5C and D). In
agreement with this observation, a colony formation assay was
performed in the presence of 10 µM gemcitabine. In this assay, the
colony forming ability of FASN-knockdown PDAC cells was inhibited
by gemcitabine, and the effect of gemcitabine resistance induced by
PKM2-overexpression was reduced by FASN-knockdown (Fig. 5E). Collectively, these data indicate
that inhibition of FASN reduces gemcitabine chemoresistance in PDAC
cells, at least in part through the regulation of PKM2.
Discussion
Pancreatic cancer is a highly aggressive disease,
with an incidence rate approximately equal to the mortality rate
(16). Surgical resection followed by
radiotherapy and/or chemotherapy is currently the first-line
treatment; however, >80% of patients with pancreatic cancer have
unresectable lesions or present with metastatic disease at the time
of diagnosis (17). The underlying
mechanism for this aggression is unknown; however, alternations in
cancer metabolism are believed to contribute to the tumor
progress.
On the basis of the Warburg effect, tumor cells have
the capacity to produce energy through glycolysis despite lacking a
sufficient oxygen supply. Reprogrammed glucose metabolism is one of
the hallmarks of cancer (18).
Pyruvate kinases are rate-limiting enzymes for aerobic glycolysis
in tumor cells, with increased PKM2 expression not only increasing
glycolysis but also acting as a nucleus protein kinase to regulate
gene expression and promote tumor progression (19). PKM2 functions as an oncogene by
promoting cell survival and invasion in PDAC, meaning that it may
be a therapeutic target for PDAC (20).
In the current study, FASN and PKM2 were upregulated
in pancreatic cancer compared with normal pancreatic tissue, and
PKM2 expression was positively correlated with FASN expression in
PDAC. The present study has confirmed that FASN upregulates the
mRNA and protein levels of PKM2, and increases the
glucose-consumption rate in pancreatic cancer cells. These
observations indicate that FASN regulates PKM2 expression and
promotes the Warburg effect in pancreatic cancer cells.
FASN, a key lipogenic enzyme, is overexpressed in a
number of human cancer types and functions as a metabolic oncogene
in pancreatic cancer. FASN has been shown to be consistently
upregulated in pancreatic cancer (16). Previous studies have shown that FASN
overexpression may contribute to gemcitabine and radiotherapy
resistance (10), protect cancer
cells against hypoxic conditions and harsh microenvironments
(21), and increase the metabolic
rate and fatty acid oxidation in cancer cells (22). However, the detailed molecular
mechanism by which FASN induces treatment resistance is
unclear.
Pancreatic cancer is a heterogeneous disease and
numerous factors contribute to resistance to gemcitabine treatment.
One such factor is increased PKM2 expression (11). The experiments described in the
present study demonstrated that FASN-knockdown reduced PKM2
expression and alleviated gemcitabine resistance, whereas
PKM2-overexpression eliminated the effect induced by
FASN-knockdown. These results indicate that in PDAC cells, FASN
induces gemcitabine resistance through regulation of PKM2.
The present study demonstrated that FASN upregulated
PKM2 expression and glucose metabolism, contributing to gemcitabine
resistance in PDAC cells. Therefore, FASN may represent a novel
potential therapeutic target for patients with PDAC.
Acknowledgements
This study was supported by the Scientific Research
Training Program for Young Talents of Union Hospital, Tongji
Medical College, Huazhong University of Science and Technology.
References
|
1
|
The Lancet Oncology, . Pancreatic cancer
in the spotlight. Lancet Oncol. 15:2412014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blum R and Kloog Y: Metabolism addiction
in pancreatic cancer. Cell Death Dis. 5:e10652014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bian Y, Yu Y, Wang S and Li L:
Up-regulation of fatty acid synthase induced by EGFR/ERK activation
promotes tumor growth in pancreatic cancer. Biochem Biophys Res
Commun. 463:612–617. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Little JL and Kridel SJ: Fatty acid
synthase activity in tumor cells. Subcell Biochem. 49:169–194.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Menendez JA, Vazquez-Martin A, Ortega FJ
and Fernandez-Real JM: Fatty acid synthase: Association with
insulin resistance, type 2 diabetes, and cancer. Clin Chem.
55:425–438. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuhajda FP, Jenner K, Wood FD, Hennigar
RA, Jacobs LB, Dick JD and Pasternack GR: Fatty acid synthesis: A
potential selective target for antineoplastic therapy. Proc Natl
Acad Sci USA. 91:6379–6383. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shurbaji MS, Kalbfleisch JH and Thurmond
TS: Immunohistochemical detection of a fatty acid synthase (OA-519)
as a predictor of progression of prostate cancer. Hum Pathol.
27:917–921. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Piyathilake CJ, Frost AR, Manne U, Bell
WC, Weiss H, Heimburger DC and Grizzle WE: The expression of fatty
acid synthase (FASE) is an early event in the development and
progression of squamous cell carcinoma of the lung. Hum Pathol.
31:1068–1073. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alo PL, Amini M, Piro F, Pizzuti L,
Sebastiani V, Botti C, Murari R, Zotti G and Di Tondo U:
Immunohistochemical expression and prognostic significance of fatty
acid synthase in pancreatic carcinoma. Anticancer Res.
27:2523–2527. 2007.PubMed/NCBI
|
|
10
|
Yang Y, Liu H, Li Z, Zhao Z, Yip-Schneider
M, Fan Q, Schmidt CM, Chiorean EG, Xie J, Cheng L, et al: Role of
fatty acid synthase in gemcitabine and radiation resistance of
pancreatic cancers. Int J Biochem Mol Biol. 2:89–98.
2011.PubMed/NCBI
|
|
11
|
Kim DJ, Park YS, Kang MG, You YM, Jung Y,
Koo H, Kim JA, Kim MJ, Hong SM, Lee KB, et al: Pyruvate kinase
isoenzyme M2 is a therapeutic target of gemcitabine-resistant
pancreatic cancer cells. Exp Cell Res. 336:119–129. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pei H, Li L, Fridley BL, Jenkins GD,
Kalari KR, Lingle W, Petersen G, Lou Z and Wang L: FKBP51 affects
cancer cell response to chemotherapy by negatively regulating Akt.
Cancer Cell. 16:259–266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lai IL, Chou CC, Lai PT, Fang CS, Shirley
LA, Yan R, Mo X, Bloomston M, Kulp SK, Bekaii-Saab T and Chen CS:
Targeting the Warburg effect with a novel glucose transporter
inhibitor to overcome gemcitabine resistance in pancreatic cancer
cells. Carcinogenesis. 35:2203–2213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Witkiewicz AK, Nguyen KH, Dasgupta A,
Kennedy EP, Yeo CJ, Lisanti MP and Brody JR: Co-expression of fatty
acid synthase and caveolin-1 in pancreatic ductal adenocarcinoma:
Implications for tumor progression and clinical outcome. Cell
Cycle. 7:3021–3025. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zimmon DS and Ferstenberg R: Pancreatic
carcinoma. N Engl J Med. 326:17821992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wong N, Ojo D, Yan J and Tang D: PKM2
contributes to cancer metabolism. Cancer Lett. 356:184–191. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li C, Zhao Z, Zhou Z and Liu R: PKM2
Promotes cell survival and invasion under metabolic stress by
enhancing warburg effect in pancreatic ductal adenocarcinoma. Dig
Dis Sci. 61:767–773. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Furuta E, Pai SK, Zhan R, Bandyopadhyay S,
Watabe M, Mo YY, Hirota S, Hosobe S, Tsukada T, Miura K, et al:
Fatty acid synthase gene is up-regulated by hypoxia via activation
of Akt and sterol regulatory element binding protein-1. Cancer Res.
68:1003–1011. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Legaspi A, Jeevanandam M, Starnes HF Jr
and Brennan MF: Whole body lipid and energy metabolism in the
cancer patient. Metabolism. 36:958–963. 1987. View Article : Google Scholar : PubMed/NCBI
|