Introduction
Hepatocellular carcinoma (HCC) is the third leading
cause of cancer-associated mortalities worldwide (1). The transforming growth factor-β1
(TGF-β1)/Smad signaling pathway serves an important role in HCC
development. In the canonical TGF-β1/Smad signaling pathway, TGF-β
receptor type I (TβR-I) activated by TGF-β1 phosphorylates
receptor-regulated (R)-Smads, including Smad2 and Smad3 at the
C-terminal, producing phosphorylated (p)Smad2C and pSmad3C
(2). Subsequently, pSmad2C and
pSmad3C oligomerize with Smad4 to form the Smad2/3/4 complex, which
then translocates into the nucleus and regulates target gene
expression (2).
The canonical Smad signaling pathway is integrated
into a complex network of cross-talks with other signaling
pathways, particularly the mitogen-activated protein kinase (MAPK)
signaling pathways, including extracellular signal-regulated kinase
(ERK), c-Jun N-terminal kinase (JNK) and p38 kinase pathways. In
the cytoplasm, MAPKs phosphorylate R-Smads at the linker region,
producing pSmad2L and pSmad3L. This regulatory phosphorylation at
the linker region inhibits the translocation into the nucleus and
the transcriptional activity of R-Smads that have been
phosphorylated at the C-terminal (3).
JNK/pSmad3L upregulates the expression of plasminogen activator
inhibitor type 1 (PAI1), which is a downstream target gene of the
TGF-β1/Smad signaling pathway (4,5). This has
been demonstrated to facilitate hepatocytic cell invasion and
reduce the tumor suppressive activity of pSmad3C in liver cancer
(6,7).
It has been suggested that the phosphorylated isoforms of R-Smad
are essential for the interaction between MAPKs and the TGF-β1/Smad
signaling pathway (8).
MAPK signaling regulates the subcellular
distribution of pSmads, in addition to regulating the
phosphorylation of Smads (9). This
regulation can be context-dependent. The data of previous studies
revealed that ERK activation inhibits Smad2/3 nuclear translocation
and that the ERK inhibitor, U0126, can restore the accumulation of
Smad2/3 in the nucleus (10,11). Furthermore, ERK or p38 inhibitors are
able to inhibit the nuclear translocation of pSmad2/3, which is
induced by cyclosporine A (12).
Inhibition of JNK1 activation prevents TGF-β1-induced Smad3
activation and nuclear translocation (13). However, this regulation is also
cell-type specific. The p38 inhibitor SB203580 inhibits
TGF-β-dependent translocation of Smad2/3 to the nucleus in
myofibroblasts or human dental pulp cells, but not in Burkitt
lymphoma cells (14–16). Our previous study reported that in
keloid fibroblasts, the ERK inhibitor PD98059 inhibited the nuclear
accumulation of pSmad2/3 (17).
However, the effect of MAPK inhibitors on the translocation of
R-Smads in HepG2 cells remains unclear.
Our previous data demonstrated that MAPK specific
inhibitors, including ERK inhibitor (PD98059), JNK inhibitor
(SP600125) or p38 inhibitor (SB203580), could inhibit the
transcription of PAI1 in HepG2 cells (18). The phosphorylation of R-Smads was
decreased following treatment with SP600125 and SB203580, but was
scarcely affected upon PD98059 treatment, indicating that PD98059
affects the expression of PAI1 via interfering with the nuclear
accumulation of pSmad2/3 (18). Thus,
the present study aimed to investigate the effect of MAPK signaling
on the nucleocytoplasmic distribution of pSmad2/3 in HepG2
cells.
There are four distinct mechanisms that may explain
the nuclear accumulation of Smads in response to TGFβ: Increase in
nuclear import, decrease in nuclear export, release from
cytoplasmic anchoring or establishment of nuclear anchoring
(19). In the current study, the
nuclear import activity of Smads was investigated. In the
conventional nuclear import progress, cargo proteins bind different
importin (Imp)β proteins to form complexes, with or without the aid
of Impα protein, which then translocate into the nucleus. Imp7 or 8
are two members of the Impβ family, responsible for transporting
pSmad2/3 and Smad4 (20,21). The role of Imp7 or 8 in the regulation
of the MAPK signaling pathway associated with the translocation of
activated R-Smads was also investigated in the present study. In
order to confirm this hypothesis, the intracellular distribution of
pR-Smads and Smad4, and the subcellular localization of Imp7/8 were
determined in HepG2 cells treated with three different MAPK
inhibitors and TGF-β1.
Materials and methods
Cell culture
The human HCC HepG2 cell line was purchased from the
Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). HepG2 cells were grown as sub-confluent
monolayer cultures in Dulbecco's modified Eagle's medium (DMEM;
cat. no. SH30022.01; Hyclone; GE Healthcare, Logan, UT, USA)
supplemented with 10% fetal bovine serum (cat. no. 11011-8611;
Zhejiang Tianhang Biological Technology Co., Ltd., Hangzhou,
China). Cells were incubated at 37°C with 5% CO2. The
experiment was performed at the log phase of growth after the cells
had been plated for 24 h. HepG2 cells were starved overnight in a
humidified 5% CO2 incubator at 37°C in serum-free DMEM,
in the absence or presence of 10 µM ERK inhibitor (PD98059), JNK
inhibitor (SP600125) or p38 inhibitor (SB203580) (all EMD
Millipore, Billerica, MA, USA) for 5 h in a humidified 5%
CO2 incubator at 37°C; subsequently cells were treated
with 9 pM TGF-β1 (R&D Systems, Inc., Minneapolis, MN, USA) for
1 h in a humidified 5% CO2 incubator at 37°C. The cells
in the control groups were added to an equal volume of serum-free
medium.
Immunofluorescence analysis
To detect the intracellular localization of Smads,
HepG2 cells were grown on slides in 24-well plates and then treated
under the aforementioned conditions. After fixing with 4%
paraformaldehyde for 30 min at room temperature, the cells were
permeabilized, and blocked with 0.1% saponin and 0.5% bovine serum
albumin in PBS for 30 min at 4°C. Subsequently, samples were
incubated with the specified primary antibody (dilution, 1:50)
overnight at 4°C. The primary antibodies used were as follows:
Domain-specific antibodies directed against pR-Smads [αpSmad2C
(Ser465/467) (cat. no. 3108) and αpSmad3C (Ser423/425) (cat. no.
9520) (both from Cell Signaling Technology, Inc., Danvers, MA,
USA); αpSmad2L (Ser249/254) and αpSmad3L (Ser207/212) (both
provided by Dr K. Matsuzaki, Kansai Medical University, Osaka,
Japan)]; mouse monoclonal anti-Smad4 antibody (cat. no. sc-7966;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA); rabbit anti-Imp 7
(cat. no. ab99273) and rabbit anti-Imp 8 antibodies (cat. no.
ab72109) (both from Abcam, Cambridge, UK). Subsequently, cells were
incubated with fluorescein isothiocyanate-conjugated goat
immunoglobulin G (IgG) anti-rabbit (cat. no. ZF-0311) or anti-mouse
(cat. no. ZF-0312) (1:100 dilution; both from OriGene Technologies,
Inc., Beijing, China) antibody for 2 h at room temperature. Next,
slides were mounted with 80% phosphoglycerol, and images were
captured under a fluorescence microscope (Olympus, Tokyo, Japan).
In a single experiment, ≥100 stained cells/sample were
analyzed.
Immunoblot analysis
To determine Imp7 and Imp8 expression,
immunoblotting was performed using the aforementioned rabbit
anti-Imp 7 and rabbit anti-Imp 8 primary antibodies diluted in
Tris-buffered saline solution/0.1% Tween 20 (TBST) (1:10,000), and
mouse anti-GAPDH (cat. no. 97166; Cell Signaling Technology, Inc.).
Total protein was extracted from the HepG2 cells using Western blot
and IP Cell Lysis reagent (cat. no. P0013; Beyotime Institute of
Biotechnology, Haimen, China). The samples were subjected to 12%
SDS-PAGE (10 µg/lane) and then transferred onto polyvinylidene
difluoride (PVDF) membranes (EMD Millipore). Non-specific antibody
binding was blocked using 5% skimmed milk powder dissolved in TBST.
PVDF membranes were incubated with the primary antibodies overnight
at 4°C, followed by incubation with horseradish
peroxidase-conjugated affinipure goat IgG anti-rabbit (cat. no.
ZB-2301) or anti-mouse (cat. no. ZB-2305) (both from OriGene
Technologies, Inc.) diluted in TBST (1:10,000) for 1 h at room
temperature. After being washed three times with TBST, the
immunoreactive proteins were visualized using an enhanced
chemiluminescence reagent (GE Healthcare, Chicago, IL, USA) and
autoradiography. Densitometric analysis was performed using
Quantity One software (version 4.62; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Statistical analysis
All cell experiments were performed three times. All
data are presented as the mean ± standard deviation. Statistical
analyses were performed using SPSS software (version 17.0; SPSS,
Inc., Chicago, IL, USA). Experimental and control values were
compared using the unpaired Student's t-test or one-way analysis of
variance followed with the post-hoc Fisher's least significant
difference test for multiple comparisons where appropriate.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of three MAPK-specific
inhibitors on TGF-β1-stimulated nuclear translocation of pSmad2⁄3
and Smad4 in HepG2 cells
Immunofluorescence analysis was used to detect the
intracellular localization of pSmad2⁄3 at the C-terminal and linker
region using corresponding anti-pSmad2/3 antibodies. Per sample,
≥100 stained HepG2 cells were analyzed.
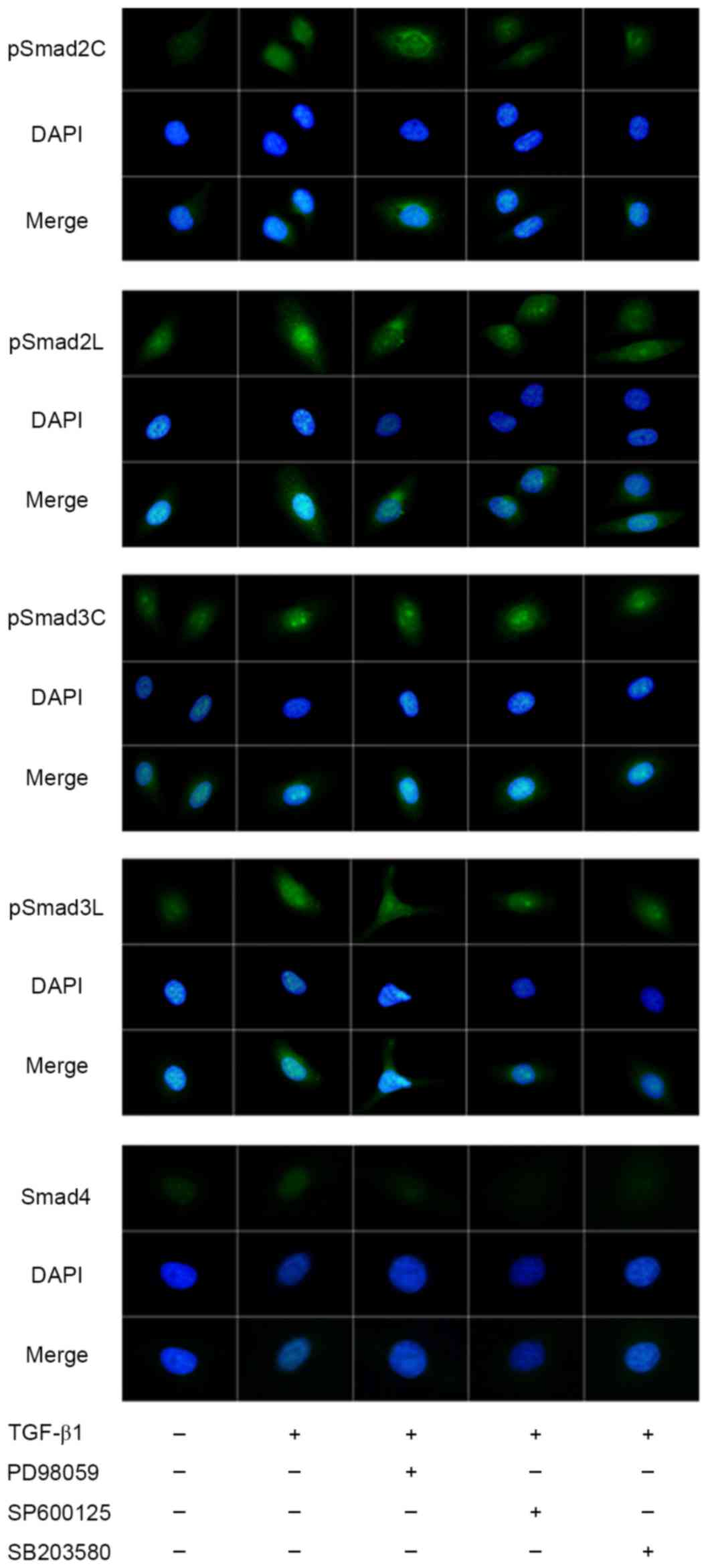
Intracellular location of pSmad2/3 and Smad4
expression in HepG2 cells is presented in Fig. 1. pSmad2C, pSmad2L, pSmad3C and pSmad3L
expression increased and was translocated into the nuclei following
TGF-β1 treatment. The presence of ERK inhibitor PD98059 only
inhibited the phosphorylation of Smad2C and had no notable
influence on the phosphorylation of the other three pR-Smads.
However, it affected the nuclear accumulation of pSmad2C, pSmad2L,
pSmad3C and pSmad3L. The presence of JNK inhibitor SP600125
inhibited the phosphorylation of Smad2C, Smad2L and Smad3L, and
affected their nuclear accumulation, but had no notable influence
on the phosphorylation of Smad3C, despite affecting its nuclear
accumulation. The p38 inhibitor SB203580 inhibited the
phosphorylation of Smad2C and Smad2L, but had no notable influence
on the phosphorylation of Smad3C, despite affecting its nuclear
accumulation. The phosphorylation of Smad3L was inhibited, but the
distribution in the cytoplasm and nuclei was scarcely affected in
the presence of p38 inhibitor. Nuclear accumulation of Smad4
increased following stimulation with TGF-β1. All three MAPK
inhibitors inhibited the translocation of Smad4 into the
nucleus.
Effects of three MAPK-specific
inhibitors on TGF-β1-stimulated nuclear translocation and
expression of Imp7/8 in HepG2 cells
Intracellular localization of Imp7 and Imp8 was
examined through immunofluorescence microscopy. Per sample, ≥100
stained HepG2 cells were analyzed.
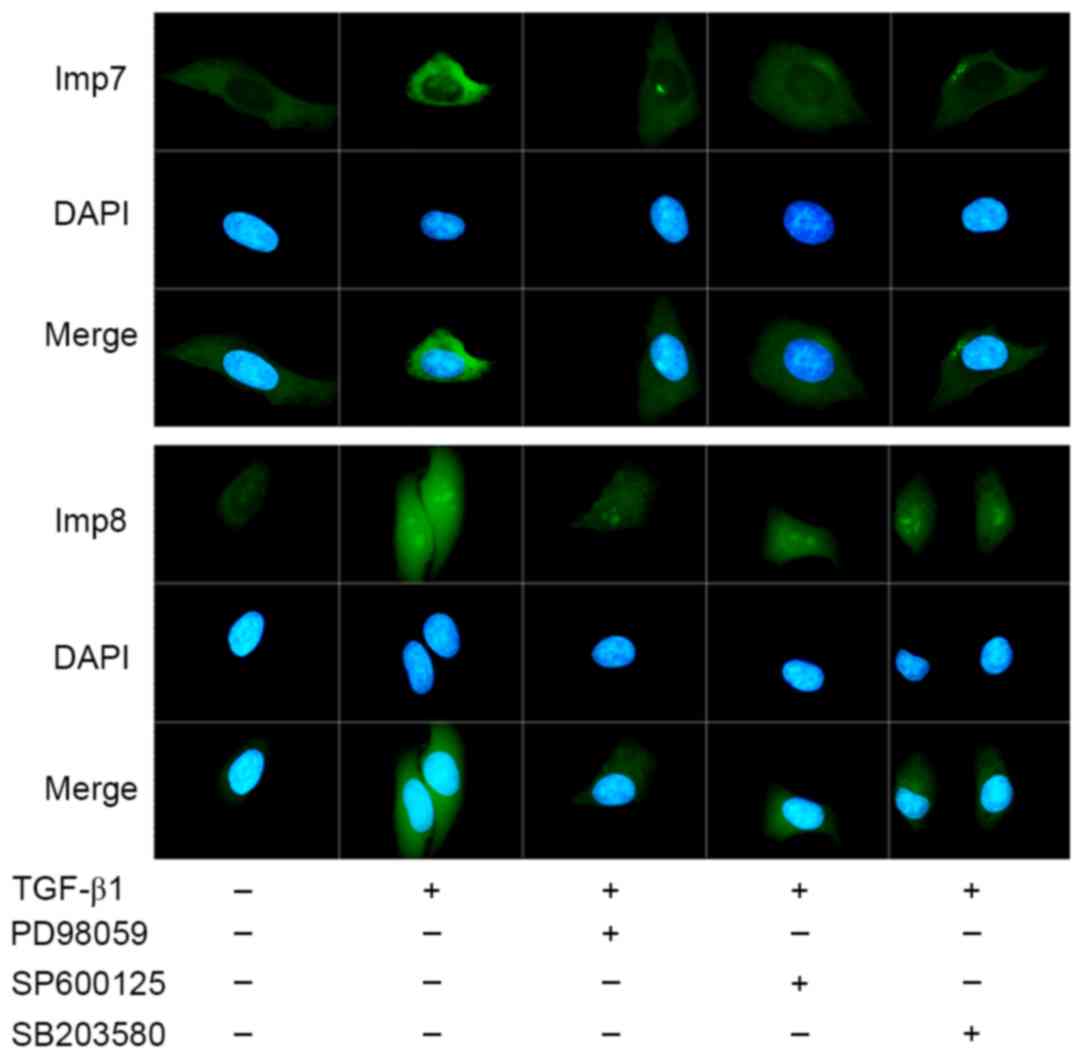
As presented in Fig.
2, Imp7 and Imp8 expression increased and was translocated into
the nuclei under TGF-β1 treatment in HepG2 cells. The presence of
ERK, JNK and p38 inhibitors inhibited the expression of Imp7 and
Imp8. Nuclear accumulation of Imp7 was affected in the presence of
three inhibitors, particularly p38 inhibitor. The presence of ERK
or JNK inhibitors decreased the nuclear accumulation of Imp8, and
the p38 inhibitor had no notable influence on the nuclear
accumulation of Imp8.
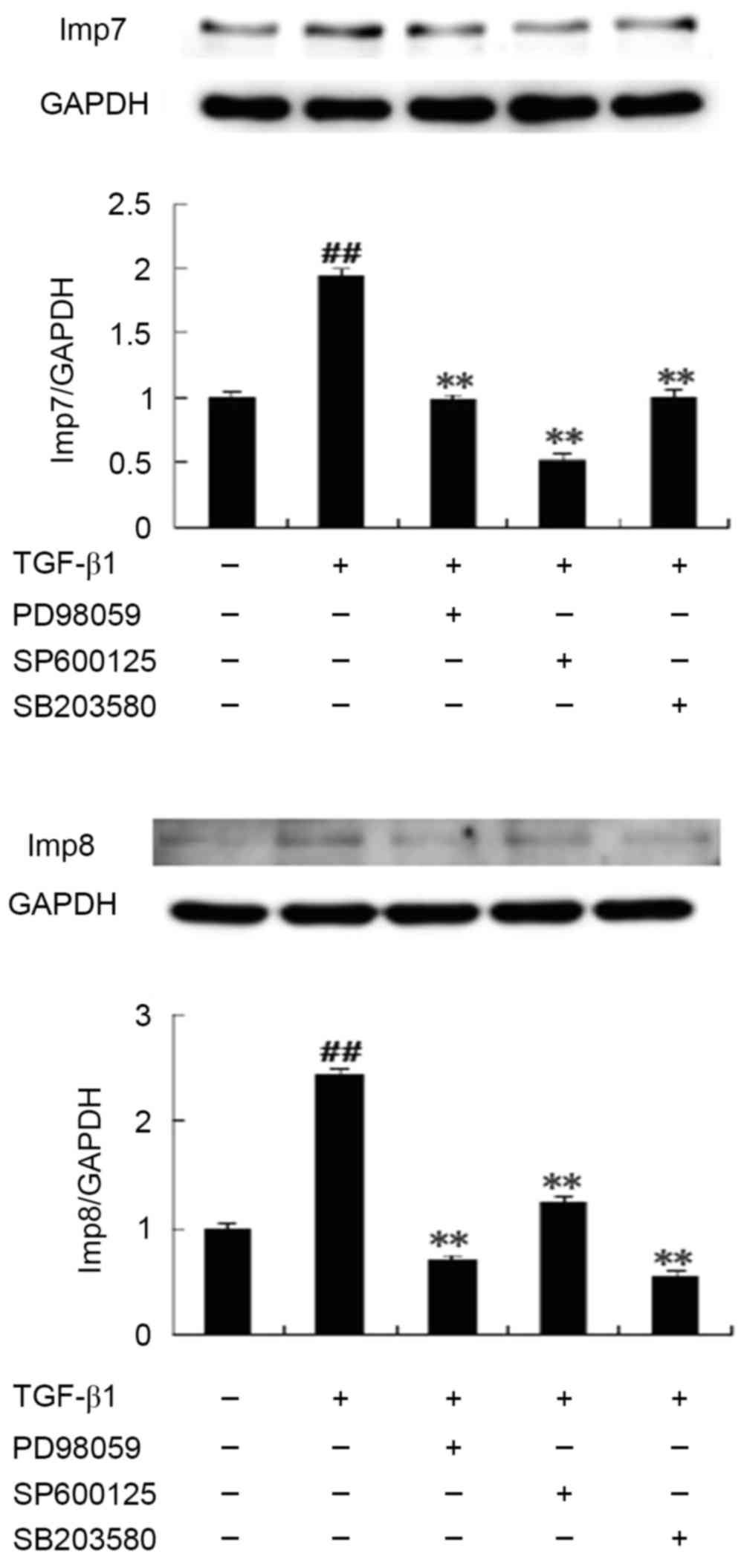
Immunoblotting results revealed that the expression
of Imp7/8 was significantly upregulated following TGF-β1 treatment
compared with untreated control HepG2 cells. In the presence of
SB203580, PD98059 and SP600125, the expression of Imp7/8 was
significantly inhibited compared with the TGF-β1-treated group
(Fig. 3).
Discussion
Following stimulation with TGF-β1, R-Smads can be
phosphorylated at the C-terminal by TβR-I or at the linker region
by MAPK, to produce different phosphorylated isoforms, including
pSmad2C, pSmad2L, pSmad3C and pSmad3L, with distinct
transcriptional responses to regulate different physiological and
pathological processes (22).
MAPK/pSmad3L conveys mitogenic signals, while
TβR-I/pSmad3C conveys cytostatic signals. pSmad3L and pSmad3C
signals oppose each other, regulating the balance between cell
growth and inhibition. In tumor cells, pSmad3L signaling impairs
tumor-suppressive pSmad3C signaling, transmitting oncogenic TGF-β1
signaling (6,23–25).
Similarly, in HepG2 cells, TGF-β1 promotes cell proliferation and
increases the phosphorylation of oncogenic Smad3L, demonstrating an
oncogenic effect (18). In a previous
study, the increased cell proliferation rate was markedly inhibited
by JNK inhibitor or p38 inhibitor, which also inhibited pSmad3L
expression. However, ERK inhibitor did not affect the proliferation
of HepG2 cells and pSmad3L expression (18). In the current study, the change in
fluorescence intensity of pSmad3L was consistent with the
aforementioned results in previous study mentioned above (18). JNK inhibitor or p38 inhibitor were
demonstrated to inhibit pSmad3L distribution in the nucleus.
Furthermore, ERK inhibitor reduced the nuclear accumulation of
pSmad3L, while reducing the nuclear accumulation of pSmad3C. The
effect of ERK inhibitor on the nuclear accumulation of pSmad3L or
pSmad3C may reach a balance, without the functional effect.
In the advanced stage of tumor progression, the role
of TGF-β1 switches to tumor promotion, inducing tumor cell
invasion. PAI1, a downstream target of the TGF-β/Smad signaling
pathway (4,5), facilitates cell migration and invasion
by enhancing cell adhesion (7). PAI1
induction requires pSmad2L (Ser-245/250/255)/C and pSmad3L
(Ser-213) activity (15,26–28). We
previously demonstrated that TGF-β1 induced the expression of
pSmad2C, pSmad2L and pSmad3L, and increased PAI1 expression and the
invasiveness of HepG2 cells (18).
All three MAPK inhibitors suppressed the invasiveness of the cells
and PAI1 expression. Among them, JNK or p38 inhibitor inhibited the
expression of pSmad2C, pSmad2L and pSmad3L, but ERK inhibitor only
inhibited the expression of pSmad2L, and did not affect the
expression of pSmad2C or pSmad3L (18). In the present study, the
immunofluorescence results demonstrated that the nuclear
distribution of pSmad2C and pSmad3L decreased following treatment
with PD98059, although the expression of pSmad2C and pSmad3L
remained unchanged, suggesting that ERK inhibitor may inhibit the
invasiveness of HepG2 cells and PAI1 expression via regulation of
pR-Smad transport.
R-Smads-Smad4 complexes are essential for TGFβ1
signaling, and Smad4 is an essential partner in these complexes
(2). Following the formation of
heterocomplexes with Smad4, pSmad3L and pSmad3C enter the nucleus
to transduce signaling (29).
pSmad2C/L undergoes translocation to the nucleus where it binds to
the pSmad3L/Smad4 complex (15,30). The
cell invasion-induced effect of pSmad2C/L and pSmad3L requires
Smad4 and complex formation. In addition, the nuclear accumulation
of Smad4 is dependent on R-Smad accumulation (31–33).
Furthermore, the intracellular distribution of Smad4 reflects the
distribution of pR-Smads or R-Smads-Smad4 complexes. In the present
study, following treatment with JNK or p38 inhibitor, the formation
of Smad2/3/4 complexes was inhibited as pR-Smad expression was
inhibited, subsequently the nuclear accumulation of Smad4
decreased. Although ERK inhibitor did not inhibit the Smad2/3/4
complexes, it reduced the nuclear distribution of Smad4, further
confirming the effect of ERK inhibitor on the translocation of
Smad2/3/4 complexes.
Smads are continuously shuttling between the
cytoplasm and the nucleus even in unstimulated cells. TGF-β
promotes Smad2, 3 and 4 accumulating in the nucleus, reaching a
maximum concentration after ~45 min (34), as was observed in the current study.
C-terminal phosphorylation is a prerequisite for Smads to
accumulate in the nucleus. The data of in vitro experiments
demonstrated that Smad3 is imported into the nucleus more
efficiently following phosphorylation (35). Schmierer et al (36) proposed a mathematical model to
understand the mechanism of nucleocytoplasmic shuttling of Smads,
which requires that the import of Smad complexes into the nucleus
should be ~5 times faster compared with the import of monomeric
Smads. It has been suggested that the phosphorylation of R-Smads
and the formation of Smad2/3/4 complexes are important to the
nuclear import of Smads, which may explain why JNK or p38
inhibitors inhibit the translocation of Smads into the nucleus.
However, ERK inhibitor also inhibited the nuclear accumulation of
Smads, with little influence on the phosphorylation of R-Smads and
the formation of Smad2/3/4 complexes.
It has been reported that the subcellular
distribution of representative cargo proteins is similar to that of
Impβ (37). In the present study, it
was observed that the nuclear accumulation of Imp7 or 8 was
impaired by MAPK inhibitors, similar to that of Smads, suggesting
that MAPK inhibitors regulate Smads import by affecting Imp7 or
8.
Previous studies have reported that the knockdown of
Imp7 and Imp8 inhibits TGF-β-induced Smad2/3 nuclear translocation,
while overexpression of Imp8 increases the concentration of Smad3
or 4 in the nucleus (20,21). The expression levels of Imp7 or Imp8
directly affect the nuclear translocation of Smads. The data of the
present study demonstrated that all three inhibitor types were able
to significantly decrease the expression of Imp7 or Imp8. Thus,
this suggests that inhibiting Imp7 or Imp8 is an important
mechanism in regulating Smad translocation by MAPK inhibitors.
In conclusion, the results of the present study
demonstrated that MAPK inhibitors, particularly ERK inhibitor,
regulate the TGF-β1/Smad signaling pathway by reducing the nuclear
accumulation of Smads. Inhibiting Imp7 or Imp8 is an important
mechanism in regulating Smad translocation by MAPK inhibitors.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81573652 and
81374012) and the Natural Science Foundation of Anhui Province
(grant no. 1508085QH168).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Heldin CH and Moustakas A: Role of Smads
in TGFβ signaling. Cell Tissue Res. 347:21–36. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kretzschmar M, Doody J, Timokhina I and
Massagué J: A mechanism of repression of TGFbeta/Smad signaling by
oncogenic Ras. Genes Dev. 13:804–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tahashi Y, Matsuzaki K, Date M, Yoshida K,
Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y and Inoue
K: Differential regulation of TGF-beta signal in hepatic stellate
cells between acute and chronic rat liver injury. Hepatology.
35:49–61. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matsuzaki K, Murata M, Yoshida K, Sekimoto
G, Uemura Y, Sakaida N, Kaibori M, Kamiyama Y, Nishizawa M,
Fujisawa J, et al: Chronic inflammation associated with hepatitis C
virus infection perturbs hepatic transforming growth factor beta
signaling, promoting cirrhosis and hepatocellular carcinoma.
Hepatology. 46:48–57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Freytag J, Wilkins-Port CE, Higgins CE,
Higgins SP, Samarakoon R and Higgins PJ: PAI-1 mediates the
TGF-beta1+EGF-induced ‘scatter’ response in transformed human
keratinocytes. J Invest Dermatol. 130:2179–2190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wrighton KH, Willis D, Long J, Liu F, Lin
X and Feng XH: Small C-terminal domain phosphatases dephosphorylate
the regulatory linker regions of Smad2 and Smad3 to enhance
transforming growth factor-beta signaling. J Biol Chem.
281:38365–38375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Javelaud D and Mauviel A: Crosstalk
mechanisms between the mitogen-activated protein kinase pathways
and Smad signaling downstream of TGF-beta: Implications for
carcinogenesis. Oncogene. 24:5742–5750. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Janda E, Lehmann K, Killisch I, Jechlinger
M, Herzig M, Downward J, Beug H and Grünert S: Ras and TGF[beta]
cooperatively regulate epithelial cell plasticity and metastasis:
Dissection of Ras signaling pathways. J Cell Biol. 156:299–313.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kfir S, Ehrlich M, Goldshmid A, Liu X,
Kloog Y and Henis YI: Pathway- and expression level-dependent
effects of oncogenic N-Ras: p27(Kip1) mislocalization by the
Ral-GEF pathway and Erk-mediated interference with Smad signaling.
Mol Cell Biol. 25:8239–8250. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iwayama H, Sakamoto T, Nawa A and Ueda N:
Crosstalk between smad and mitogen-activated protein kinases for
the regulation of apoptosis in cyclosporine a-induced renal tubular
injury. Nephron Extra. 1:178–189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Q, Zhang Y, Mao H, Chen W, Luo N, Zhou
Q, Chen W and Yu X: A crosstalk between the Smad and JNK signaling
in the TGF-β-induced epithelial-mesenchymal transition in rat
peritoneal mesothelial cells. PLoS One. 7:e320092012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang FM, Hu T and Zhou X: p38
mitogen-activated protein kinase and alkaline phosphatase in human
dental pulp cells. Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 102:114–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Furukawa F, Matsuzaki K, Mori S, Tahashi
Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki
Y, et al: p38 MAPK mediates fibrogenic signal through Smad3
phosphorylation in rat myofibroblasts. Hepatology. 38:879–889.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abécassis L, Rogier E, Vazquez A, Atfi A
and Bourgeade MF: Evidence for a role of MSK1 in transforming
growth factor-beta-mediated responses through p38alpha and Smad
signaling pathways. J Biol Chem. 279:30474–30479. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He S, Liu X, Yang Y, Huang W, Xu S, Yang
S, Zhang X and Roberts MS: Mechanisms of transforming growth factor
beta(1)/Smad signalling mediated by mitogen-activated protein
kinase pathways in keloid fibroblasts. Br J Dermatol. 162:538–546.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boye A, Kan H, Wu C, Jiang Y, Yang X, He S
and Yang Y: MAPK inhibitors differently modulate TGF-β/Smad
signaling in HepG2 cells. Tumour Biol. 36:3643–3651. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hill CS: Nucleocytoplasmic shuttling of
Smad proteins. Cell Res. 19:36–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu L, Yao X, Chen X, Lu P, Zhang B and Ip
YT: Msk is required for nuclear import of TGF-{beta}/BMP-activated
Smads. J Cell Biol. 178:981–994. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao X, Chen X, Cottonham C and Xu L:
Preferential utilization of Imp7/8 in nuclear import of Smads. J
Biol Chem. 283:22867–22874. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsuzaki K: Smad phospho-isoforms direct
context-dependent TGF-β signaling. Cytokine Growth Factor Rev.
24:385–399. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Murata M, Matsuzaki K, Yoshida K, Sekimoto
G, Tahashi Y, Mori S, Uemura Y, Sakaida N, Fujisawa J, Seki T, et
al: Hepatitis B virus X protein shifts human hepatic transforming
growth factor (TGF)-beta signaling from tumor suppression to
oncogenesis in early chronic hepatitis B. Hepatology. 49:1203–1217.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nagata H, Hatano E, Tada M, Murata M,
Kitamura K, Asechi H, Narita M, Yanagida A, Tamaki N, Yagi S, et
al: Inhibition of c-Jun NH2-terminal kinase switches Smad3
signaling from oncogenesis to tumor-suppression in rat
hepatocellular carcinoma. Hepatology. 49:1944–1953. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kawamata S, Matsuzaki K, Murata M, Seki T,
Matsuoka K, Iwao Y, Hibi T and Okazaki K: Oncogenic Smad3 signaling
induced by chronic inflammation is an early event in ulcerative
colitis-associated carcinogenesis. Inflamm Bowel Dis. 17:683–695.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Velden JL, Alcorn JF, Guala AS, Badura EC
and Janssen-Heininger YM: c-Jun N-terminal kinase 1 promotes
transforming growth factor-β1-induced epithelial-to-mesenchymal
transition via control of linker phosphorylation and
transcriptional activity of Smad3. Am J Respir Cell Mol Biol.
44:571–581. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sekimoto G, Matsuzaki K, Yoshida K, Mori
S, Murata M, Seki T, Matsui H, Fujisawa J and Okazaki K: Reversible
Smad-dependent signaling between tumor suppression and oncogenesis.
Cancer Res. 67:5090–5096. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hough C, Radu M and Doré JJ: Tgf-beta
induced Erk phosphorylation of smad linker region regulates smad
signaling. PLoS One. 7:e425132012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mori S, Matsuzaki K, Yoshida K, Furukawa
F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa
M, et al: TGF-beta and HGF transmit the signals through
JNK-dependent Smad2/3 phosphorylation at the linker regions.
Oncogene. 23:7416–7429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsuzaki K, Kitano C, Murata M, Sekimoto
G, Yoshida K, Uemura Y, Seki T, Taketani S, Fujisawa J and Okazaki
K: Smad2 and Smad3 phosphorylated at both linker and COOH-terminal
regions transmit malignant TGF-beta signal in later stages of human
colorectal cancer. Cancer Res. 69:5321–5330. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
De Bosscher K, Hill CS and Nicolás FJ:
Molecular and functional consequences of Smad4 C-terminal missense
mutations in colorectal tumour cells. Biochem J. 379:209–216. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen HB, Rud JG, Lin K and Xu L: Nuclear
targeting of transforming growth factor-beta-activated Smad
complexes. J Biol Chem. 280:21329–21336. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reguly T and Wrana JL: In or out? The
dynamics of Smad nucleocytoplasmic shuttling. Trends Cell Biol.
13:216–220. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Inman GJ, Nicolás FJ and Hill CS:
Nucleocytoplasmic shuttling of Smads 2, 3 and 4 permits sensing of
TGF-beta receptor activity. Mol Cell. 10:283–294. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kurisaki A, Kose S, Yoneda Y, Heldin CH
and Moustakas A: Transforming growth factor-beta induces nuclear
import of Smad3 in an importin-beta1 and Ran-dependent manner. Mol
Biol Cell. 12:1079–1091. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schmierer B, Tournier AL, Bates PA and
Hill CS: Mathematical modeling identifies Smad nucleocytoplasmic
shuttling as a dynamic signal-interpreting system. Proc Natl Acad
Sci USA. 105:pp. 6608–6613. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang J, Ito H, Wate R, Ohnishi S, Nakano
S and Kusaka H: Altered distributions of nucleocytoplasmic
transport-related proteins in the spinal cord of a mouse model of
amyotrophic lateral sclerosis. Acta Neuropathol. 112:673–680. 2006.
View Article : Google Scholar : PubMed/NCBI
|