Introduction
Heart failure affected 40 million people worldwide
in 2015 (1). In developing countries,
the morbidity and mortality of cardiovascular disease continue to
increase annually, which threatens the lives and quality of life of
patients (2). Until now, ventricular
remodeling has been considered as an important pathological process
in the occurrence and development of heart failure (3). The main pathological manifestations of
ventricular remodeling are myocardial microcirculation, apoptosis
and necrosis of myocardial cells, progressive myocardial collagen
network remodeling and myocardial interstitial fibrosis (4). Myocardial fibrosis represents excessive
deposition of extracellular matrix in normal myocardial tissue
caused by various pathological factors. The characteristics of
myocardial fibrosis are numerous, including enhanced collagen
concentration and volume fraction, the imbalance in the proportion
of various types of collagen, and disordered arrangement of
collagen (5). The pathological basis
of myocardial fibrosis is that the percentage of myocardial
interstitium in myocardial tissue is increased. Cardiac muscle
fiber connective tissue is mainly comprised of fibroblasts,
myocardial fibroblasts, valvular mesenchymal cells and
extracellular matrix. Additionally, extracellular matrix of the
myocardium, such as collagen, is mainly synthesized by fibroblasts
(6,7).
However, in the past, research has focused on the study of
myocardial cells, ignoring the importance of myocardial fibroblasts
(MFBs). MFBs have a strong ability to split and to secrete matrix
proteins, collagen-I (COL-I) and COL-III under various pathological
conditions, including hypoxic-ischemic injury, local inflammatory
cytokine stimulation and neuroendocrine factor secretion (8,9).
Therefore, MFBs serve an important role in the myocardial fibrosis
of MFBs.
Bone morphogenetic proteins (BMPs) serve as a
multi-potent family of proteins regulating the growth and
differentiation of cells. Studies have demonstrated that BMPs may
reverse fibrosis progression (10,11). BMP2
overexpression significantly suppressed the fibrosis induced by
transforming growth factor β1 (TGF-β1) in renal interstitial
fibroblasts (12). Furthermore, it
has been proven that upregulation of BMP2 may enhance myocardial
fibrotic signaling through activation of the Smurf1/Smad6 complex
(13). Additionally, a large volume
of evidence has suggested that the Smad family is associated with
the pathological mechanisms underlying fibrosis (14–16).
Therefore, it was hypothesized that BMP2 and Smad may participate
in the development and progression of myocardial fibrosis.
SPARC-related modular calcium binding 1 (SMOC1)
represents a vital member of the SPARC matricellular protein family
that regulates cell-matrix interactions through binding to
cell-surface receptors, including growth factors and the components
of extracellular matrix (17,18). SMOC1 is widely expressed in numerous
tissue types, and it has been revealed that SMOC1 is mainly located
at the basement membrane (18,19). A
previous study has demonstrated that Xenopus SMOC protein,
also known as the ortholog of human SMOC1, acted as a BMP
antagonist (20). These results
suggested that human SMOC1 may also regulate BMP signaling.
However, knowledge is insufficient regarding the biological
function of SMOC1 in BMP pathway regulation in myocardial
fibrosis.
The present study analyzed the association between
SMOC1 silencing and the fibrosis of mouse MFBs. Furthermore, the
exact roles and molecular mechanisms of SMOC1 silencing and the
BMP2/Smad pathway in the fibrosis of angiotensin II (Ang
II)-treated MFBs were also investigated.
Materials and methods
Cell culture, genes and plasmids
Mouse MFBs (MIC-iCell-c002, iCell Bioscience Inc.,
Shanghai) were maintained in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) in a 5% CO2 atmosphere at 37°C. The
cells were observed with an inverted microscope (×40
magnification). si-SMOC1 (GGGAAGTCAAGGTCAGTACG) or si-negative
control (NC) (GGGAAGTCAAGGTCAGTACG), was cloned into the
psiRNA-h7SK vector (Biovector Science Lab, Inc.). The plasmid (1
µg) was transfected into the cells with Lipofectamine 3000
(Invitrogen; Thermo Fisher Scientific, Inc.). The cells were
incubated at 37°C for 6 h; and then the cells were transferred into
fresh DMEM and maintained for 18 h at 37°C. Then the cells were
used to perform the following experiments.
Grouping
Six treatment groups were created in the present
research, including a control group (MFBs without treatment), a
negative control (NC) group (MFBs transfected with an empty
vector), an si-SMOC1 group (MFBs transfected with si-SMOC1), an Ang
II group [MFBs treated with 1 µM Ang II (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 18 h at 37°C], an Ang II+NC group (MFBs
transfected with an empty vector and then treated with 1 µM Ang II)
and an Ang II+si-SMOC1 group (MFBs transfected with si-SMOC1 and
then treated with 1 µM Ang II).
Cell viability analysis
A Cell Counting kit-8 assay (CCK-8; Beyotime
Institute of Biotechnology, Haimen, China) was performed to measure
cell viability. Approximately 6×104 MFBs/ml in the
logarithmic phase were seeded into 96-well plates and maintained in
an incubator at 37°C in 5% CO2 for 12 h. Subsequently,
cells were divided into the aforementioned treatment groups. Cells
were maintained for 12, 24 and 48 h, respectively. Subsequently, 10
µl CCK-8 reagent was added to each well and cells were maintained
for 3 h at 37°C. A microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) was used to measure the absorbance at 450 nm.
Cell viability was evaluated by the percentage of surviving cells
compared with the control.
Flow cytometry
MFBs were trypsinized by 0.25% trypsin (Beyotime
Institute of Biotechnology) and collected in Eppendorf tubes. Cells
were then washed with PBS. Subsequently, cells were re-suspended
with serum-free Dulbecco's modified Eagle medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 2′-7′-dichlorofluorescin
diacetate (D6883, Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at
a density of 1×106 cells/ml and incubated for 0.5 h at
37°C. Following centrifugation at 224 × g for 1 min at room
temperature, the supernatant was discarded and cells were
collected. Cells were re-suspended with PBS and a FACSCalibur flow
cytometer with CellQuest software version 3.3 (BD Biosciences,
Franklin Lakes, NJ, USA) was used to assess the reactive oxygen
species (ROS) content.
Detection of malondialdehyde (MDA),
lactate dehydrogenase (LDH) and superoxide dismutase (SOD)
The following kits were used: LDH assay kit (Abcam,
Cambridge, UK), MDA assay kit (Beyotime Institute of Biotechnology,
Haimen, China) and SOD assay kit (Sigma-Aldrich; Merck KGaA). All
assays were performed according to the manufacturer's protocol.
ELISA
The expression of TGF-β1, COL-I and COL-III were
determined by ELISA kits: Mouse TGF-β1 ELISA Kit (E-EL-M0051c;
Elabscience, Wuhan, Hubei, China), COL1 ELISA kit (E-EL-M0325c;
Elabscience) and COL-III ELISA kit (E-EL-M0316; Elabscience).
According to the manufacturer's protocol, MFBs
(6×103/well) were added into 96-well plate. The plates
were sealed with adhesive tape and maintained at 37°C for 90 min.
Subsequently, 100 µl biotinylated antibody fluids were added. The
plates were sealed with adhesive tape and maintained at 37°C for 60
min. Next, 100 µl HRP conjugate enzyme binding solutions were
added. The plates were sealed with adhesive tape and maintained at
37°C for 30 min. Substrate regent (100 µl) was added and plates
were maintained for 10–15 min in the dark at 37°C. Subsequently,
stop solution was added and mixed in for 10 min immediately. The
OD450 value was detected using a microplate reader (Bio-Rad
Laboratories, Inc.).
Western blot analysis
The proteins were isolated with NP40 lysis buffer
(Beyotime Institute of Biotechnology). The protein concentration
was measured by bicinchoninic assay protein assay kit (Pierce;
Thermo Fisher Scientific Inc.). 20 µg protein was separated by 12%
SDS-PAGE and then transferred onto polyvinylidene difluoride
membranes (EMD Millipore, Billerica, MA, USA). The membranes were
blocked with 5% skimmed milk at room temperature for 2 h. The
primary antibodies were incubated with the membranes at 4°C
overnight. Western blotting was performed using the following
specific antibodies: Rabbit anti-mouse anti-SMOC1 (dilution, 1:500;
catalog no., ab200219; Abcam, Cambridge, MA, USA), rabbit
anti-mouse anti-fibronectin (FN) (dilution, 1:1,000; catalog no.,
ab131390; Abcam), rabbit anti-mouse anti-TGF-β1 (dilution, 1:1,000;
catalog no., ab92486; Abcam), rabbit anti-mouse anti-COL-I
(dilution, 1:500; catalog no., ab64883; Abcam), rabbit anti-mouse
anti-COL-III (dilution, 1:5,000; catalog no., ab7778; Abcam),
rabbit anti-mouse anti-BMP2 (dilution, 1:500; catalog no., ab14933;
Abcam), rabbit anti-mouse anti-Smad2 (dilution, 1:1,000; catalog
no., ab33875; Abcam), rabbit anti-mouse anti-p-Smad2 (dilution,
1:1,000; catalog no., ab53100; Abcam), and rabbit anti-mouse
anti-actin (dilution, 1:5,000; catalog no., ab179467; Abcam). The
membranes were subsequently incubated with a horseradish
peroxidase-conjugated goat anti-rabbit secondary antibody
(dilution, 1:2,000; catalog no., ab205718; Abcam) at room
temperature for 1 h. Enhanced chemiluminescent (ECL) reagents (EMD
Millipore) and an ECL system (GE Healthcare, Chicago, IL, USA) were
used to assess the results. The density of the blots was analyzed
by Quantity One software version 4.6.9 (Bio-Rad Laboratories).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured MFBs using
TRIzol reagent (Thermo Fisher Scientific, Inc.). RNA was reverse
transcribed to cDNA using a Reverse Transcription kit
(Sigma-Aldrich; Merck KGaA), according to the manufacturer's
protocols. RT-qPCR was performed using SYBR-Green PCR master mix
(Vazyme, Piscataway, NJ, USA) on ABI 7500 Thermocycler (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: 5 min pretreatment at 95°C; 94°C for 15
sec, 60°C for 45 sec (35 cycles); final extension at 76°C for 10
min and maintenance at 4°C. Actin was used as the control of the
input RNA level. The method of quantification was according to
2−∆∆Cq method (21). The
primers used, designed by Invitrogen; Thermo Fisher Scientific,
Inc., were as follows: SMOC1 forward, 5′-CCAAGCCCAAGAAATGTGCC-3′
and reverse, 5′-AGTCCTGTCTCCTCGGAGTT-3′ (227 bp); FN forward,
5′-TGACAACTGCCGTAGACCTG-3′ and reverse, 5′-CACTGGGGTGTGGATTGACC-3′
(232 bp); TGF-β1 forward, 5′-GTCCAAACTAAGGCTCGCCA-3′ and reverse,
5′-ATAGATGGCGTTGTTGCGGT-3′ (202 bp); COL-I forward,
5′-ATTTGTGCGTCGGTTGGGTA-3′ and reverse, 5′-GTTGTGTTCTGAAGCCACGG-3′
(298 bp); COL-III, forward, 5′-GCTACAGGCCTTTTGTTGGC-3′ and reverse,
5′-CCACAGAATGGGTGGGAGAC-3′ (222 bp); and actin, forward,
5′-TGCCCGGTGCTTTAGACTAC-3′ and reverse, 5′-AAATAATGAACCCAGCCAGCC-3′
(171 bp).
Statistical analysis
The results of the present study are presented as
the mean ± standard error of the mean of at least three independent
experiments. All of the experimental data were analyzed by one-way
analysis of variance following Tukey's test. P<0.05 was
considered to indicate a statistically significant difference.
GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA) was
used for data analysis.
Results
Identification of mouse MFBs
Over the course of the present study, mouse MFBs
were cultured with DMEM containing 10% FBS. As demonstrated in
Fig. 1, the morphology of the MFBs
was spindle-shaped and polygonal. Furthermore, large nuclei and
clear cytoplasms were observed in the MFBs. Therefore, MFBs were
harvested and used for the subsequent experiments.
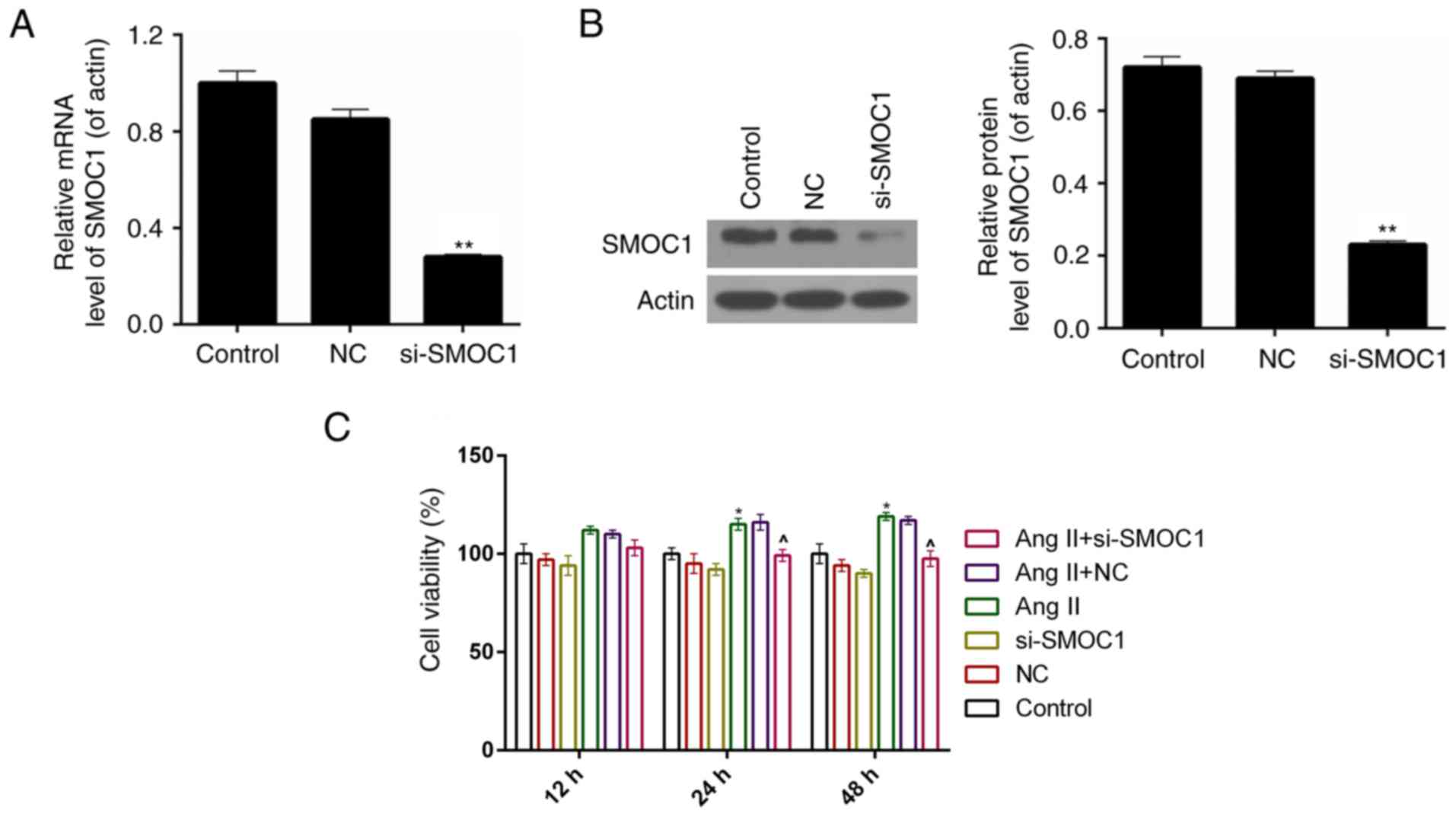
SMOC1 silencing suppresses the cell
viability of Ang II-treated MFBs
A siRNA vector targeting SMOC1, si-SMOC1, was
constructed in the present study. The knockdown efficiency was ~75%
in MFBs following stable transfection with si-SMOC1 (P<0.01;
Fig. 2A). According to the results of
western blot analysis, it was revealed that, following transfection
with si-SMOC1, the protein expression level of SMOC1 was
significantly reduced (P<0.01; Fig.
2B). Therefore, a CCK-8 assay was performed to measure the cell
viability of MFBs separated into the six treatment groups described
earlier. The results demonstrated that, the cell viability was
inhibited in si-SMOC1 group compared to Ang II group (Fig. 2C).
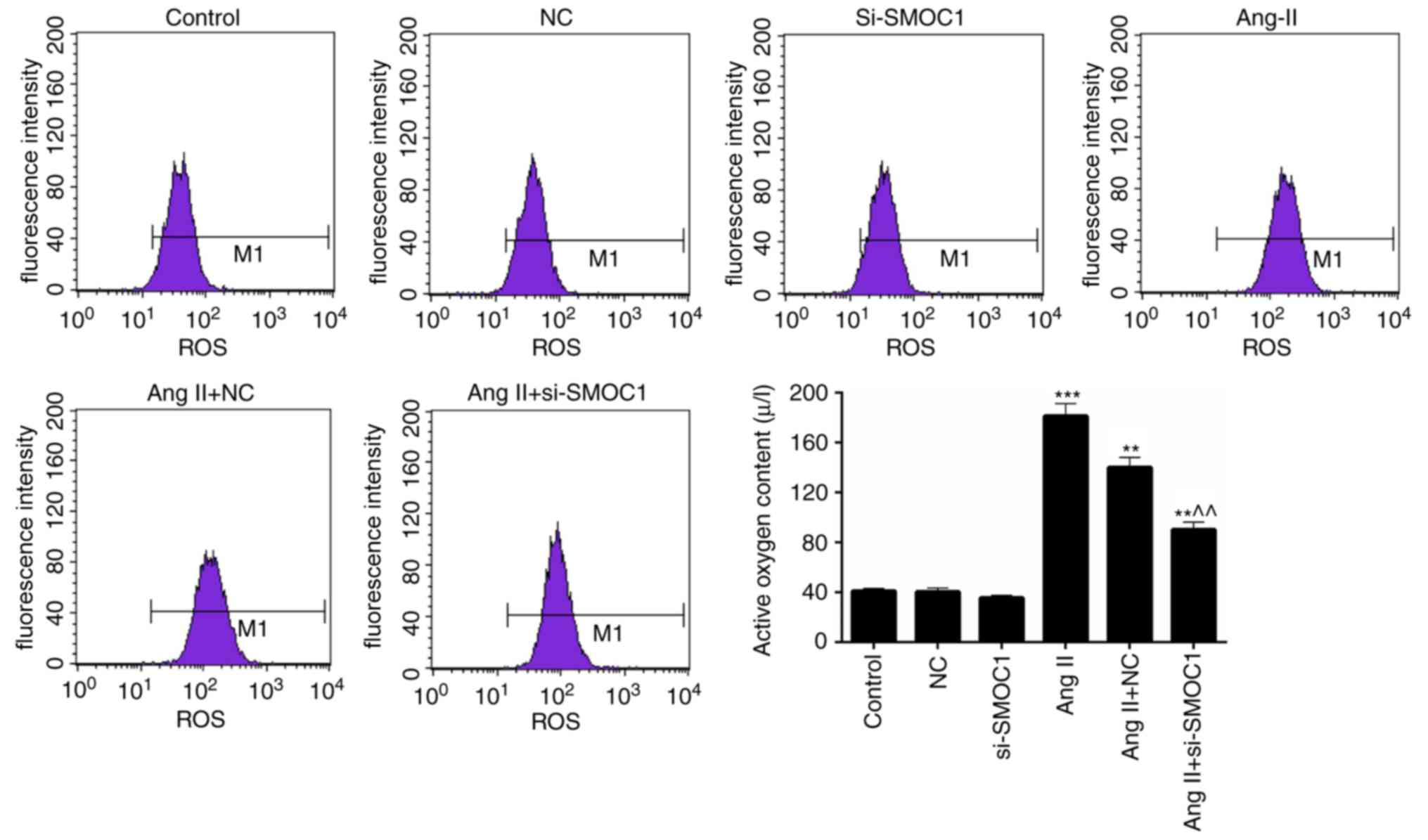
SMOC1 silencing reduces the ROS
content in Ang II-treated MFBs
Additionally, the ROS content in MFBs of the
previously described treatment groups was also assessed. A
significant increase in ROS content was observed in the Ang II
group compared with the NC group (P<0.01; Fig. 3). However, it was also revealed that
the ROS content in Ang II-induced MFBs was significantly reduced by
transfection with si-SMOC1 (P<0.01; Fig. 3). Therefore, it has been determined
that SMOC1 silencing is able to reduce the production of ROS in
MFBs treated with Ang II.
SMOC1 silencing mitigates oxidative
stress in MFBs treated with Ang II
The MDA and LDH content in MFBs treated with Ang II
were significantly higher than in the NC group (P<0.01; Fig. 4A-B). However, significant decrease in
MDA and LDH content in Ang II-treated MFBs transfected with
si-SMOC1 were observed (P<0.05; Fig.
4A-B). Nevertheless, Ang II was revealed to inhibit the
activity of SOD in MFBs. In the SMOC1-silenced group, the SOD
activity in Ang II-treated MFBs was significantly enhanced
(P<0.05; Fig. 4C). In summary, it
was confirmed that SMOC1 silencing reduced the MDA and LDH content,
whereas enhancing SOD activity in Ang II-induced MFBs. Therefore,
SMOC1 silencing mitigated oxidative stress in MFBs induced by Ang
II.
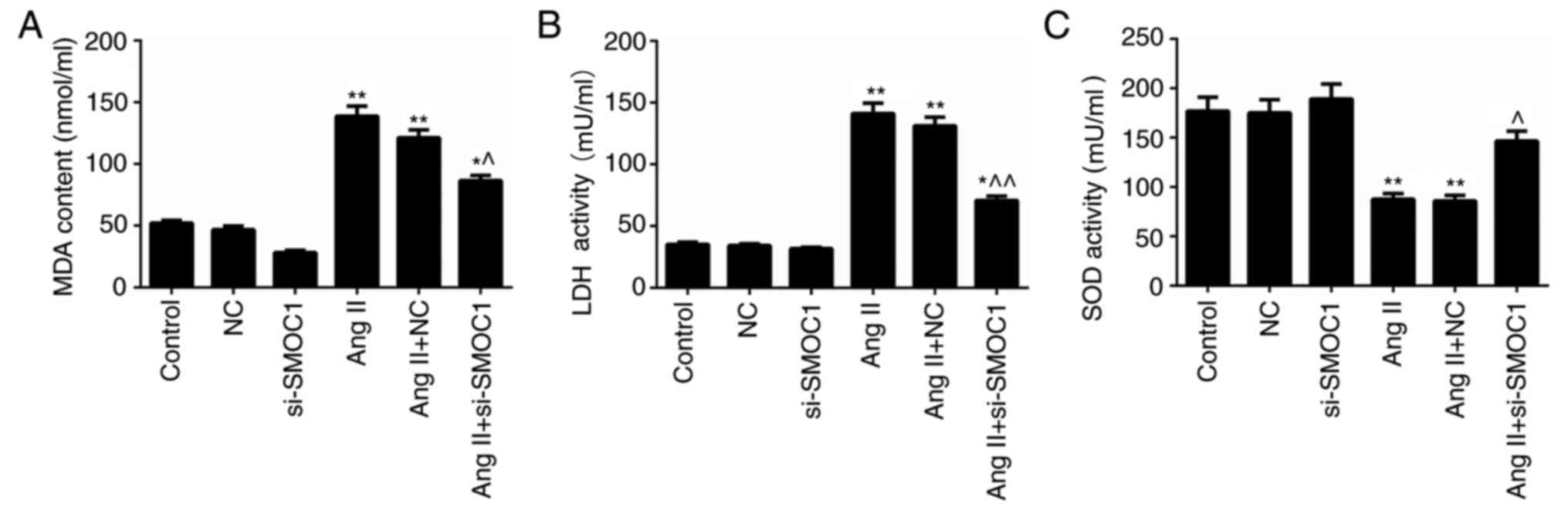 | Figure 4.SMOC1 silencing mitigates the
oxidative stress in MFBs induced by Ang II. ELISA was performed to
evaluate the (A) MDA content, (B) LDH content and (C) SOD activity
in MFBs, MFBs transfected with an empty vector, and Ang II-induced
MFBs transfected with an empty vector or si-SMOC1. *P<0.05 and
**P<0.01 vs. NC; ^P<0.05 and
^^P<0.01 vs. Ang II+NC. SMOC1, SPARC-related modular
calcium binding 1; MFBs, myocardial fibroblasts; Ang II,
angiotensin II; MDA, malondialdehyde; LDH, lactate dehydrogenase;
SOD, superoxide dismutase; si, small interfering RNA; NC, negative
control. |
SMOC1 silencing downregulates the
expression levels of fibrosis-associated proteins
Furthermore, the present study investigated the
molecular mechanisms underlying fibrosis in MFBs and the expression
levels of fibrosis-associated proteins, including FN, TGF-β1, COL-I
and COL-III in MFBs. On the basis of ELISA data, it was revealed
that SMOC1 silencing significantly decreased the expression of
TGF-β1, COL-I and COL-III in Ang II-treated MFBs (P<0.05;
Fig. 5A). Furthermore, the RT-qPCR
results indicated that the expression levels of FN, TGF-β1, COL-I
and COL-III in Ang II-treated MFBs were significantly downregulated
in response to si-SMOC1 (P<0.05; Fig.
5B). Western blot analysis results also revealed similar trends
in the expression of fibrosis-associated proteins in MFBs from each
group (Fig. 5C). Based on these
results, it was confirmed that SMOC1 silencing suppressed fibrosis
of Ang II-treated MFBs through downregulating the expression levels
of FN, TGF-β1, COL-I and COL-III.
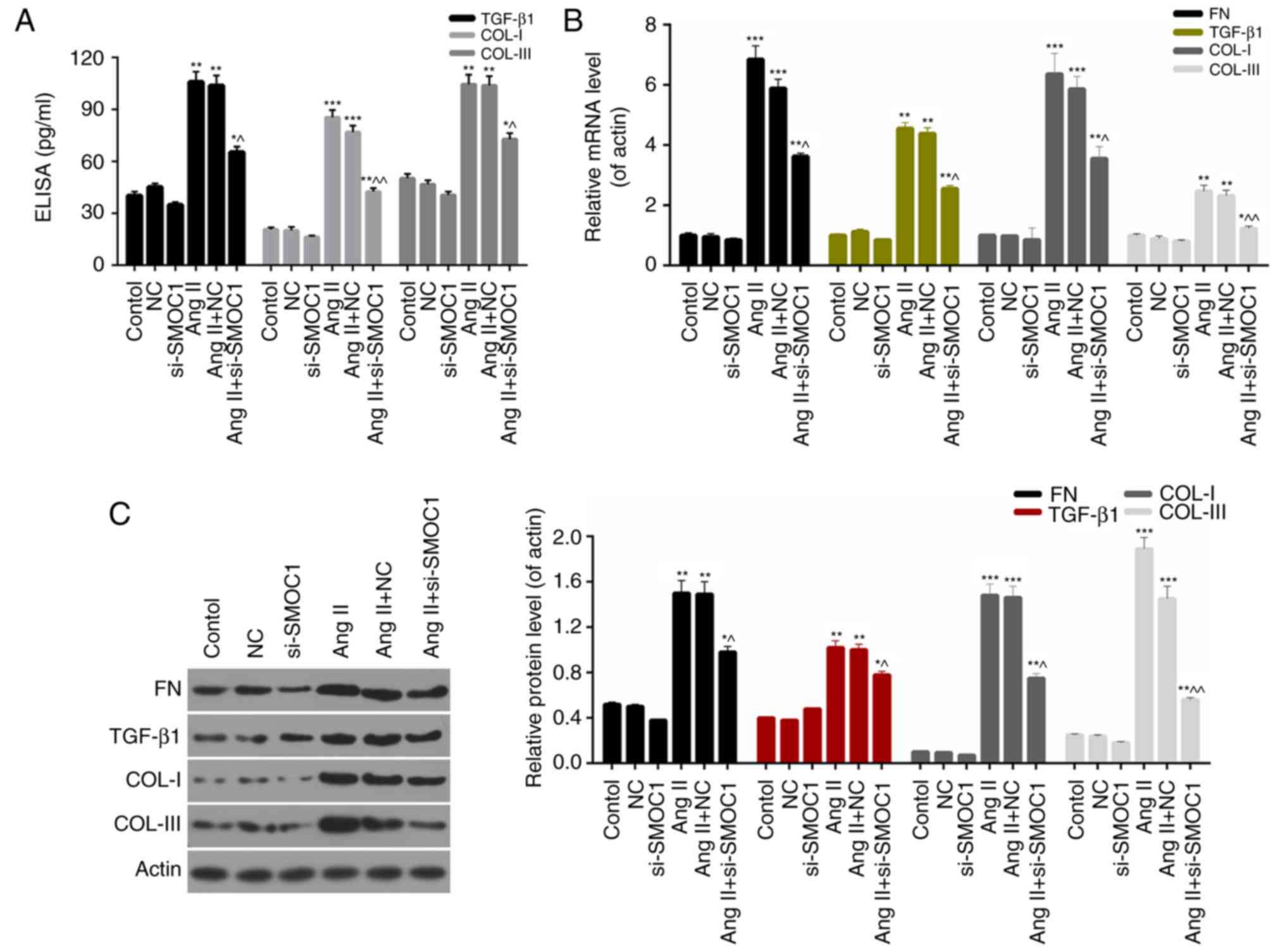 | Figure 5.SMOC1 silencing downregulates the
expression levels of fibrosis-associated proteins. (A) ELISA was
performed to measure the TGF-β1, COL-I and COL-III expression in
MFBs, MFBs transfected with an empty vector, and Ang II-induced
MFBs transfected with an empty vector or si-SMOC1. (B) Reverse
transcription-quantitative polymerase chain reaction and (C)
western blot analysis were performed on the expression levels of
FN, TGF-β1, COL-I and COL-III in MFBs, MFBs transfected with an
empty vector, and Ang II-induced MFBs transfected with an empty
vector or si-SMOC1. *P<0.05, **P<0.01, and ***P<0.001 vs.
NC; ^P<0.05 and ^^P<0.01 vs. Ang II+NC.
SMOC1, SPARC-related modular calcium binding 1; FN, fibronectin;
TGF-β1, transforming growth factor β1; COL, collagen; Ang II,
angiotensin II; MFBs, myocardial fibroblasts; si, small interfering
RNA; NC, negative control. |
SMOC1 silencing affects the BMP2/Smad
signaling pathway
Finally, the expression levels of BMP2,
phosphorylated Smad2 and Smad2 in MFBs from each group were
evaluated. Western blot analysis results revealed that the
expression levels of BMP2 and phosphorylated Smad2 in Ang
II-treated MFBs were significantly downregulated in response to
si-SMOC1 (P<0.05; Fig. 6).
Therefore, it was determined that SMOC1 silencing was able to
suppress the phosphorylation of Smad2 in Ang II-treated MFBs.
Additionally, there was no significant difference in the Smad2
expression in MFBs from each group (Fig.
6). Therefore, it was concluded that SMOC1 silencing affected
the BMP2/Smad pathway in Ang II-treated MFBs.
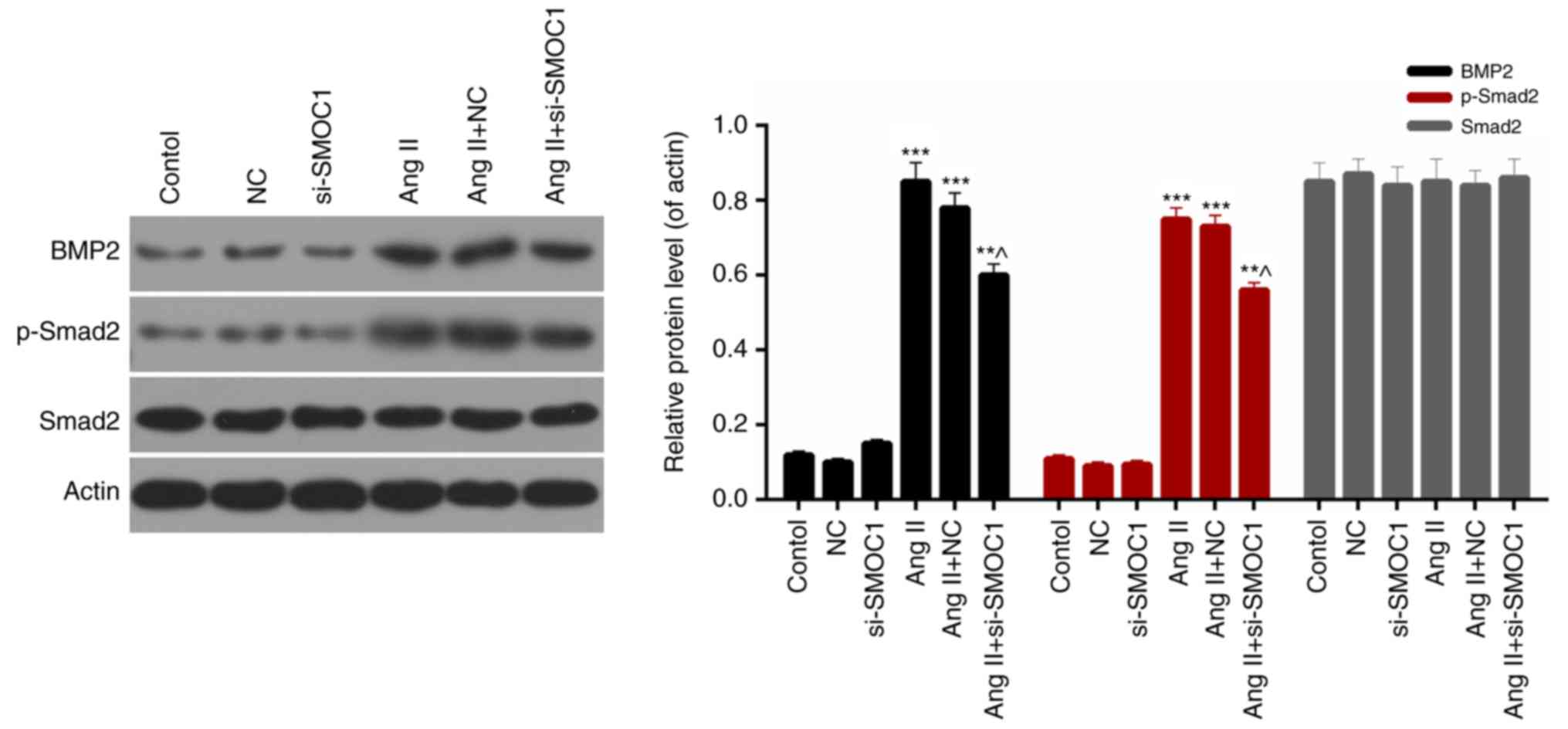 | Figure 6.SMOC1 silencing affects the BMP2/Smad
pathway. Western blot analysis was performed to measure the
expression levels of BMP2, Smad2 and p-Smad2 in MFBs, MFBs
transfected with an empty vector, and Ang II-induced MFBs
transfected with an empty vector or si-SMOC1. **P<0.01 and
***P<0.001 vs. NC; ^P<0.05 vs. Ang II+NC. SMOC1,
SPARC-related modular calcium binding 1; BMP2, bone morphogenetic
protein 2; p-, phosphorylated; MFBs, myocardial fibroblasts; si,
small interfering RNA; Ang II, angiotensin II; NC, negative
control. |
Discussion
Ang II serves as the most crucial component of the
renin-angiotensin system, which is usually associated with
hypertension and renal failure (22).
It has been demonstrated that Ang II is able to increase the
expression level of TGF-β1, promote the DNA synthesis of
fibroblasts and facilitate the proliferation of fibroblasts
(23,24). Numerous studies have demonstrated that
Ang II not only induced atherosclerosis by promoting the migration
of human umbilical vein endothelial cells to the intima (25), but also led to hepatic fibrosis via
promoting hepatic stellate cell migration and myocardial fibrosis
via accelerating the migration of MFBs (26,27).
Therefore, Ang II was selected as the inducer to establish the
model of myocardial fibrosis on mouse MFBs.
SMOC1, a member of the matricellular protein family,
is mainly expressed in the basement membrane of different tissues
(18,19,28,29).
Knowledge is limited regarding the roles of SMOC1 in physiology or
pathophysiology, while studies have proven that the expression of
SMOC1 was enhanced in several types of cancer (30,31). To
the best of our knowledge, the function of SMOC1 in the prevention
and therapy of myocardial fibrosis has not yet been studied. In the
present study, the plasmid cloned with SMOC1 siRNA was prepared and
named si-SMOC1. The knockdown efficiency of SMOC1 was ~75% in MFBs
following stable transfection with si-SMOC1, according to the
RT-qPCR and western blot analyses. Initially, the cell viability of
untreated MFBs, MFBs transfected with an empty vector, and Ang
II-induced MFBs transfected with an empty vector or si-SMOC1 was
assessed. The results revealed that Ang II markedly enhanced the
cell viability of MFBs, while SMOC1 silencing suppressed the cell
viability of Ang II-induced MFBs.
Oxidative stress refers to the overproduction of
highly reactive molecules, including ROS, when the organism is
subjected to various harmful stimuli, which exceeds the scavenging
activity of the organism and further results in tissue damage
(32). As a second messenger of
intracellular signal transduction, ROS is often involved in cell
proliferation, apoptosis and accumulation. In vivo,
physiological quantities of ROS can destroy pathogenic
microorganisms and possess defensive physiological functions.
However, sustained high concentrations of ROS may cause oxidation
reactions with the surrounding macromolecular substances, and
further impair the structure and function of cells (33,34). In
the cardiovascular system, Ang II has been identified to cause the
activation of NADPH oxidase to produce ROS, and to be involved in
the migration of vascular smooth muscle cells (35). Therefore, the content of ROS and the
levels of oxidative stress markers in the MFBs from all the
treatment groups were evaluated. Based on the results, it was
confirmed that SMOC1 silencing significantly reduced the ROS
content in Ang II-induced MFBs. In addition, it was revealed that
SMOC1 silencing also lessened the content of MDA and LDH, and
strengthened the activity of SOD in Ang II-induced MFBs. According
to these results, it was concluded that SMOC1 silencing reduced
oxidative stress in Ang II-induced MFBs.
In order to investigate the accurate roles and
mechanisms of SMOC1 in myocardial fibrosis, several
fibrosis-associated proteins were further selected as the objects
of the present study. Based on previous studies, the expression
levels of FN, TGF-β1, COL-I and COL-III in MFBs treated with Ang II
and transfected with si-SMOC1 were assessed (34–37). In
accordance with the ELISA data, it was revealed that SMOC1
silencing markedly decreased the TGF-β1, COL-I and COL-III
expression levels in Ang II-induced MFBs. Additionally, RT-qPCR and
western blot analysis results also suggested that SMOC1 silencing
markedly downregulated the expression levels of FN, TGF-β1, COL-I
and COL-III in Ang II-induced MFBs. Therefore, it was confirmed
that SMOC1 silencing was able to downregulate the expression levels
of fibrosis-associated proteins in Ang II-induced MFBs. Therefore,
it was determined that SMOC1 silencing suppressed the fibrosis of
MFBs induced by Ang II. Studies have reported that BMPs and Smad
family proteins were associated with the development and
progression of myocardial fibrosis (13,36–38).
However, the regulatory mechanism of the BMP2/Smad pathway in MFBs
remains unclear. Therefore, the present study measured the
expression levels of BMP2, phosphorylated Smad2 and Smad2 in MFBs
treated with Ang II and si-SMOC1. The western blot results
indicated that Ang II increased the expression level of BMP2 and
the phosphorylation of Smad2 in MFBs. Additionally, it was revealed
that SMOC1 silencing decreased the expression of BMP2 and the
phosphorylation of Smad2 in MFBs in Ang II-induced MFBs. However,
no significant difference in Smad2 expression was observed. Taken
together, these results indicated that SMOC1 silencing affected the
BMP2/Smad pathway in Ang II-induced MFBs. Based on the
aforementioned results, it was hypothesized that SMOC1 silencing
suppressed the fibrosis of MFBs induced by Ang II by affecting the
BMP2/Smad pathway. Nevertheless, the present study only
investigated the effects of SMOC1 silencing on Ang II-induced
myocardial fibrosis. At present, knowledge regarding the roles and
mechanisms of SMOC1 overexpression on Ang II-induced myocardial
fibrosis remains insufficient. Future studies should consider the
effects of overexpressing SMOC1.
Taken together, the results of the present study
demonstrated that SMOC1 silencing suppressed the Ang II-induced
myocardial fibrosis of MFBs through affecting the BMP2/Smad
pathway. Additionally, the observations of the present study
provided novel insight for comprehending the pathogenesis of
myocardial fibrosis and enabling an alternative approach for the
therapy of myocardial fibrosis.
In summary, the present study indicated that SMOC1
silencing suppressed the Ang II-induced myocardial fibrosis of MFBs
through affecting the BMP2/Smad pathway. The results of the present
study have crucial influence on the mechanisms of SMOC1 and Ang
II-induced MFBs. The potential effects of SMOC1 on the myocardial
fibrosis of Ang II-induced MFBs suggested that SMOC1 may be an
effective target for myocardial fibrosis therapies.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated and/or analyzed during this study
are included in this published article
Authors' contributions
YW wrote the main manuscript and analyzed the data.
XW performed the experiments. YW designed the study. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
GBD 2016 Disease and Injury Incidence and
Prevalence Collaborators. Global, regional, and national incidence,
prevalence, and years lived with disability for 328 diseases and
injuries for 195 countries, 1990–2016: A systematic analysis for
the Global Burden of Disease Study 2016. Lancet. 390:1211–1259.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dunlay SM and Roger VL: Understanding the
epidemic of heart failure: Past, present, and future. Curr Heart
Fail Rep. 11:404–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lebrin F, Goumans MJ, Jonker L, Carvalho
RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM and ten
Dijke P: Endoglin promotes endothelial cell proliferation and
TGF-beta/ALK1 signal transduction. Embo J. 23:4018–4028. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McCain ML, Agarwal A, Nesmith HW, Nesmith
AP and Parker KK: Micromolded gelatin hydrogels for extended
culture of engineered cardiac tissues. Biomaterials. 35:5462–5471.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Daniels A, van Bilsen M, Goldschmeding R,
van der Vusse GJ and van Nieuwenhoven FA: Connective tissue growth
factor and cardiac fibrosis. Acta Physiol. 195:321–338. 2009.
View Article : Google Scholar
|
|
6
|
Fan D, Takawale A, Lee J and Kassiri Z:
Cardiac fibroblasts, fibrosis and extracellular matrix remodeling
in heart disease. Fibrogenesis Tissue Repair. 5:152012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Segura AM, Frazier OH and Buja LM:
Fibrosis and heart failure. Heart Fail Rev. 19:173–185. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fan D, Takawale A, Basu R, Patel V, Lee J,
Kandalam V, Wang X, Oudit GY and Kassiri Z: Differential role of
TIMP2 and TIMP3 in cardiac hypertrophy, fibrosis, and diastolic
dysfunction. Cardiovasc Res. 103:268–280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma Y, Halade GV and Lindsey ML:
Extracellular matrix and fibroblast communication following
myocardial infarction. J Cardiovasc Transl Res. 5:848–857. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang YL, Liu YS, Chuang LY, Guh JY, Lee
TC, Liao TN, Hung MY and Chiang TA: Bone morphogenetic protein-2
antagonizes renal interstitial fibrosis by promoting catabolism of
type I transforming growth factor-beta receptors. Endocrinology.
150:727–740. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zeisberg M, Bottiglio C, Kumar N, Maeshima
Y, Strutz F, Muller GA and Kalluri R: Bone morphogenic protein-7
inhibits progression of chronic renal fibrosis associated with two
genetic mouse models. Am J Physiol Renal Physiol. 285:F1060–F1067.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang YL, Ju HZ, Liu SF, Lee TC, Shih YW,
Chuang LY, Guh JY, Yang YY, Liao TN, Hung TJ and Hung MY: BMP-2
suppresses renal interstitial fibrosis by regulating
epithelial-mesenchymal transition. J Cell Biochem. 112:2558–2565.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang S, Sun A, Li L, Zhao G, Jia J, Wang
K, Ge J and Zou Y: Up-regulation of BMP-2 antagonizes
TGF-β1/ROCK-enhanced cardiac fibrotic signalling through activation
of Smurf1/Smad6 complex. J Cell Mol Med. 16:2301–2310. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Flanders KC: Smad3 as a mediator of the
fibrotic response. Int J Exp Pathol. 85:47–64. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim KK, Wei Y, Szekeres C, Kugler MC,
Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg
JA and Chapman HA: Epithelial cell alpha3beta1 integrin links
beta-catenin and Smad signaling to promote myofibroblast formation
and pulmonary fibrosis. J Clin Invest. 119:213–224. 2009.PubMed/NCBI
|
|
16
|
Lan HY: Diverse roles of TGF-beta/Smads in
renal fibrosis and inflammation. Int J Biol Sci. 7:1056–1067. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bornstein P and Sage EH: Matricellular
proteins: Extracellular modulators of cell function. Curr Opin Cell
Biol. 14:608–616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vannahme C, Smyth N, Miosge N, Gosling S,
Frie C, Paulsson M, Maurer P and Hartmann U: Characterization of
SMOC-1, a novel modular calcium-binding protein in basement
membranes. J Biol Chem. 277:37977–37986. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gersdorff N, Müller M, Schall A and Miosge
N: Secreted modular calcium-binding protein-1 localization during
mouse embryogenesis. Histochem Cell Biol. 126:705–712. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thomas JT, Canelos P, Luyten FP and Moos M
Jr: Xenopus SMOC-1 Inhibits bone morphogenetic protein
signaling downstream of receptor binding and is essential for
postgastrulation development in Xenopus. J Biol Chem.
284:18994–19005. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Phillips MI and Kagiyama S: Angiotensin II
as a pro-inflammatory mediator. Curr Opin Investig Drugs.
3:569–577. 2002.PubMed/NCBI
|
|
23
|
Marshall RP, McAnulty RJ and Laurent GJ:
Angiotensin II is mitogenic for human lung fibroblasts via
activation of the type 1 receptor. Am J Respir Crit Care Med.
161:1999–2004. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Uhal BD, Kim JK, Li X and Molina-Molina M:
Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary
fibrosis: Autocrine mechanisms in myofibroblasts and macrophages.
Curr Pharm Des. 13:1247–1256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu M, Chen D, Li D, Ding H, Zhang T, Xu T
and Zhang Y: Luteolin inhibits angiotensin II-induced human
umbilical vein endothelial cell proliferation and migration through
downregulation of Src and Akt phosphorylation. Circ J. 77:772–779.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rosin NL, Sopel M, Falkenham A, Myers TL
and Legare JF: Myocardial migration by fibroblast progenitor cells
is blood pressure dependent in a model of angII myocardial
fibrosis. Hypertens Res. 35:449–456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang L, Zhu QJ, Zhou W, Ye J, Qian W, Zhu
R, Hu TH and Hou XH: Effect of beta-elemene on the proliferation,
migration and RhoA expression of hepatic stellate cells induced by
angiotensin II. Zhonghua Gan Zang Bing Za Zhi. 16:748–751. 2008.(In
Chinese). PubMed/NCBI
|
|
28
|
Bradshaw AD: Diverse biological functions
of the SPARC family of proteins. Int J Biochem Cell Biol.
44:480–488. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi YA, Lim J, Kim KM, Acharya B, Cho JY,
Bae YC, Shin HI, Kim SY and Park EK: Secretome analysis of human
BMSCs and identification of SMOC1 as an important ECM protein in
osteoblast differentiation. J Proteome Res. 9:2946–2956. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boon K, Edwards JB, Eberhart CG and
Riggins GJ: Identification of astrocytoma associated genes
including cell surface markers. BMC Cancer. 4:392004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brellier F, Ruggiero S, Zwolanek D,
Martina E, Hess D, Brown-Luedi M, Hartmann U, Koch M, Merlo A, Lino
M and Chiquet-Ehrismann R: SMOC1 is a tenascin-C interacting
protein over-expressed in brain tumors. Matrix Biol. 30:225–233.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–462. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kliment CR, Englert JM, Gochuico BR, Yu G,
Kaminski N, Rosas I and Oury TD: Oxidative stress alters syndecan-1
distribution in lungs with pulmonary fibrosis. J Biol Chem.
284:3537–3545. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Papaharalambus CA and Griendling KK: Basic
mechanisms of oxidative stress and reactive oxygen species in
cardiovascular injury. Trends Cardiovasc Med. 17:48–54. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Montezano AC, Callera GE, Yogi A, He Y,
Tostes RC, He G, Schiffrin EL and Touyz RM: Aldosterone and
angiotensin II synergistically stimulate migration in vascular
smooth muscle cells through c-Src-regulated redox-sensitive RhoA
pathways. Arterioscler Thromb Vasc Biol. 28:1511–1518. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun B, Huo R, Sheng Y, Li Y, Xie X, Chen
C, Liu HB, Li N, Li CB, Guo WT, et al: Bone morphogenetic protein-4
mediates cardiac hypertrophy, apoptosis, and fibrosis in
experimentally pathological cardiac hypertrophy. Hypertension.
61:352–360. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Voloshenyuk TG, Landesman ES, Khoutorova
E, Hart AD and Gardner JD: Induction of cardiac fibroblast lysyl
oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling.
Cytokine. 55:90–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang B, Hao J, Jones SC, Yee MS, Roth JC
and Dixon IM: Decreased Smad 7 expression contributes to cardiac
fibrosis in the infarcted rat heart. Am J Physiol Heart Circ
Physiol. 282:H1685–H1696. 2002. View Article : Google Scholar : PubMed/NCBI
|