Introduction
Bone homeostasis is maintained by a balance between
bone resorption by osteoclasts and bone formation by osteoblasts
(1). Osteoblasts not only play a
central role in bone formation by synthesizing multiple bone matrix
proteins, but also regulate osteoclast maturation by soluble
factors like receptor activator of nuclear factor-κB ligand (RANKL)
(2) and cognate interaction,
resulting in bone resorption (3). The
chemokine CXCL12 and its receptor CXCR4 play a key role in
regulating hematopoietic stem cells and cell migratory function
during morphogenesis (4). Osteoblasts
express both the CXCL12 ligand and the CXCR4 receptor, provide
regulatory feedback, and play a multifunctional role in regulating
bone formation and resorption (5).
Recent evidence has shown that the ambient air
pollution may be associated with increased bone turnover rate in
children (6). High levels of
osteocalcin, an osteoblast-specific gene expressed by fully
differentiated osteoblasts (7), have
been associated with long-term exposure to ambient air pollution
(6). Among the pollutants,
2,3,7,8-tetrachlorodi-benzo-p-dioxin (TCDD) are by-products in the
manufacturing of chlorophenols and chlorophenoxy herbicides and in
other common processes such as combustions of fossil fuel. Evidence
indicates that TCDD may promote liver and skin tumor formation
through the inhibition of tumor senescence (8).
The aryl hydrocarbon receptor (AhR) is a
ligand-dependent transcription factor whose activity is modulated
by xenobiotics and physiological ligands (9). Ligand binding induces a conformational
change in AhR, thereby exposing a nuclear translocation site
leading to pathologic effects in both animals and humans (10). As a ligand to AHR, TCDD elicits
toxicities through AhR (11) and on
TCDD exposure, the activated AhR translocates to the nucleus, is
dimerized by the Ah receptor nuclear translocator, and binds to
dioxin response elements (DRE) in the genome (12). Studies on conditional AHR deficient
mice demonstrate that AhR signal transduction is involved in the
development of normal bone phenotype and in the manifestation of
TCDD-induced bone alterations (13).
Evidence indicates that dioxins may interfere with differentiation
of osteoblasts and osteoclasts in TCDD concentrations as low as 100
fM (14). Currently, dioxin and
dioxin-like chemicals are not thought to be directly genotoxic.
Their carcinogenic effect is likely the result of their tumor
promoting activity produced by activation of the AhR (15).
Osteosarcoma is the most common primary malignant
bone tumor affecting children and young adolescents (16). Composed of malignant osteoblasts
producing immature bone or osteoid tissue, osteosarcomas can be
subdivided histologically according to different cell forms
(17). Several risk factors have been
described, including high bone turnover issue, previous bone
diseases, or heritable syndromes that predispose to osteosarcoma
(17,18). Environmental factors, such as
radiation (19), is reported to
increase the risk of osteosarcoma, but other environmental
alterations are not well characterized.
Therefore, in the present study, we investigated the
effects of TCDD treatment on the human osteosarcoma cell line,
MG-63 cells and explored the AhR, RANKL and CXCR4/CXCL12 axis
expressions on MG-63 cells in response to TCDD. We propose that
TCDD, a ubiquitous environmental contaminant, may be associated
with the aggressiveness of osteosarcoma. The results suggest that
TCDD stimulation may mediate RANKL, COX-2, PGE2 and CXCR4 axis
expressions and modulate the aggressiveness of osteosarcoma. The
results further reveal novel signaling paradigms and regulatory
networks that expand the understanding of the complex mechanisms
involved in maintaining the homeostasis of MG-63 under TCDD
stimulations.
Materials and methods
MG-63 osteosarcoma cell line
culture
The osteosarcoma cell line, MG-63 cell (20), was purchased from the American Type
Culture Collection (ATCC) Manassas, VA, USA, and cultivated in
Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS,
according to the supplier's recommendations. The cells were
cultured in a humidified incubator at 37°C, using a standard
mixture of 95% air and 5% CO2. For passage, the cells
were detached with trypsin/EDTA and subsequently re-plated and
maintained in culture for 2–4 days before use. At 70% confluence,
the cells were treated with 0.1, 1 or 10 nM TCDD (Sigma-Aldrich,
St. Louis, MO, USA) diluted in toluene, or with an equivalent
volume of toluene alone (0.2% vol/vol).
Cell growth and viability assays
Cells (100 µl, 2×104 cells/ml) were
plated in 96-well plates and allowed to attach for 24 h. The cells
were then treated with 0, 0.1, 1, or 10 nM of TCDD for 24 and 72 h.
The proportion of viable cells was determined by MTT assay
following the manufacturer's instructions. Briefly, the cells were
incubated with MTT (0.5 g/l) for 4 h. The formazan precipitate was
dissolved in 200 µl dimethyl sulfoxide (DMSO) and the absorbance at
550 nm was measured with a Benchmark microplate reader. The tumor
inhibitory rate was calculated as (1 - OD experimental group/OD
control group) × 100%. The experiments were performed in
triplicate.
Analysis of apoptosis and cell cycle
arrest
Apoptosis induced by TCDD was quantified using the
Annexin V-FITC Apoptosis Detection kit I (BD Biosciences, Franklin
Lakes, NJ, USA). The MG-63 cells were treated with 0, 0.1, 1 or 10
nM of TCDD for 24 and 72 h, and then washed twice with cold
hosphate-buffered saline (PBS) and re-suspended in 1× binding
buffer at a concentration of 1×106 cells/ml. The
solution (100 µl, 1×105 cells) was transferred to a 5-ml
culture tube and 5 µl Annexin V-FITC and 5 µl propidium iodide (PI)
were added. The cells were gently vortexed and incubated for 15 min
at room temperature (25°C) in the dark. The 1× binding buffer (400
µl) was added to each tube. Samples were analyzed by flow cytometry
within 1 h. Total cell apoptosis was defined as the sum of cells in
early and late apoptosis. The experiments were performed in
triplicate.
Antibodies and flow cytometry study
for CXCR4 expressions
Briefly, 2×105 cells were incubated for
10 min at 4°C and stained with specific CXCR4 mouse anti-human
monoclonal primary antibodies conjugated with FITC (cat. no.
sc-12764 FITC; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
based on the manufacturer's recommendations. To measure the CXCR4
expression on MG-63 cells, data were acquired on a FACSCalibur flow
cytometer (BD Biosciences) using CellQuest software (BD
Biosciences). At least 10,000 cells were analyzed per sample.
Enzyme-linked immune-sorbent assay
(ELISA)
Cells 2×105 were treated with 0, 0.1, 1,
or 10 nM of TCDD for 24 and 72 h, and then cultured in 24-well
plates in medium (500 µl) for 24 h. The Prostaglandin E2 ELISA Kit
(cat. no. KA4522; Abnova, Taipei, Taiwan) was used to measure PGE2
metabolites in supernatants according to the manufacturer's
protocol. The CXCL12 levels were quantified by commercially
available ELISA kits (cat. no. DSA00; R&D Systems, Inc.,
Minneapolis, MN, USA), according to the manufacturer's
specifications. All samples were run in batches to minimize
inter-assay variability, assayed in duplicate, and quantitated
using a standard curve method.
Western blot analysis
The MG-63 cells (5×105) were plated and
treated with 0, 0.1, 1, or 10 nM of TCDD for 24 h, and then lysed
with a buffer composed of 50 mmol/l Tris-HCl (pH 8.0), 150 mmol/l
NaCl, 0.1% Triton X-100, 0.01 g/l aprotinin, and 0.05 g/l
phenylmethylsulfonyl fluoride. Protein was quantified by the
Bradford method and equal amounts (30 µg) of protein were loaded
and electrophoresed on a 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis mini-gel. Proteins were transferred to a
polyvinylidene fluoride (PVDF) membrane and pre-blocked with casein
PBS and 0.05% Tween-20 for 1 h at room temperature. The membranes
were incubated with mouse monoclonal antibody against AHR (1:500;
cat. no. SC-133088; Santa Cruz Biotechnology, Inc.), rabbit
polyclonal antibody against COX-2 (1:1,000; cat. no. ab52237),
mouse monoclonal antibody against RANKL (1:250; cat. no. ab45039)
(both from Abcam, Cambridge, UK), and mouse monoclonal antibody
against β-actin (1:10,000; cat. no. MAB1501; EMD Millipore,
Billerica, MA, USA) incubate at 4 Celsius degree overnight.
Horseradish peroxidase-conjugated goat against mouse IgG antibody
(1:1,000; cat. no. AP-124P; EMD Millipore) or goat against rabbit
IgG antibody (1:750; cat. no. AP-132P; EMD Millipore) were used
with incubation time of 1 h at room temperature. Protein bands were
visualized with enhanced chemiluminescence reagent (ECL Kit,
Amersham, UK), and were then exposed to Kodar XAR film.
RT-QPCR and QPCR
Total RNA was isolated from cells using RNeasy kits
(Qiagen, Inc., Valencia, CA, USA) according to the manufacturer's
instructions. cDNA was reverse-transcribed from isolated RNA by
incubating 200 ng DNase-treated RNA with the first strand synthesis
kit from Advanced Biotechnologies Ltd. (Surrey, UK) following the
manufacturer's instructions. The QPCR was performed in a Rotor-Gene
6000 System (Corbett Life Science; Qiagen, Inc.). The PCR mixtures
contained 0.5 µM each of specific forward and reverse primers:
5′-GGCCACATCCGGGACATCACAGA-3 (sense) and
5′-GGGGGATGGTGAAGGGGACGAA-3′ (antisense); for cytochrome P450 1A1
(Cyp1a1); 5′-GTCAACGGATTTGGCGTATT-3′ (sense) and
5′-AAAGTTGTCATGGATGACCTTGGC-3′ (antisense) for GAPDH.
The samples were denatured at 95°C for 10 min,
followed by 30 cycles for Cyp1a1 (95°C for 15 sec, 58°C for 5 sec,
and 72°C for 10 sec), 30 cycles for GAPDH (95°C for 15 sec, 58°C
for 5 sec, and 72°C for 10 sec). Melting curves were obtained at
the end of amplification by cooling the samples to 65°C for 15 sec
followed by further cooling to 40°C for 30 sec. Data was analyzed
by the standard curve method of absolute quantification method
(21) using Corbett analysis
software. In RT-QPCR, data was normalized with GAPDH.
Statistical analysis
The data presented were obtained from three separate
experiments using cell cultures. Each experiment was performed in
triplicate. One-way ANOVA (analysis of variance) for mixed design
was used to compare hemodynamic values of more than two different
experimental groups. If variance among groups was noted, a
Bonferroni test was used to determine any significant difference
between specific points within groups. The data were analyzed by
Student's t-test for paired or unpaired data. For data with even or
uneven variation, a Mann-Whitney U test or Wilcoxon signed rank
test was used for unpaired or paired data, respectively. The
GraphPad Prism (version 5.0; GraphPad Software, San Diego, CA, USA)
was used for all statistical analyses. Data are presented as the
mean ± standard error or the mean. Statistical significance was set
at P<0.05.
Results
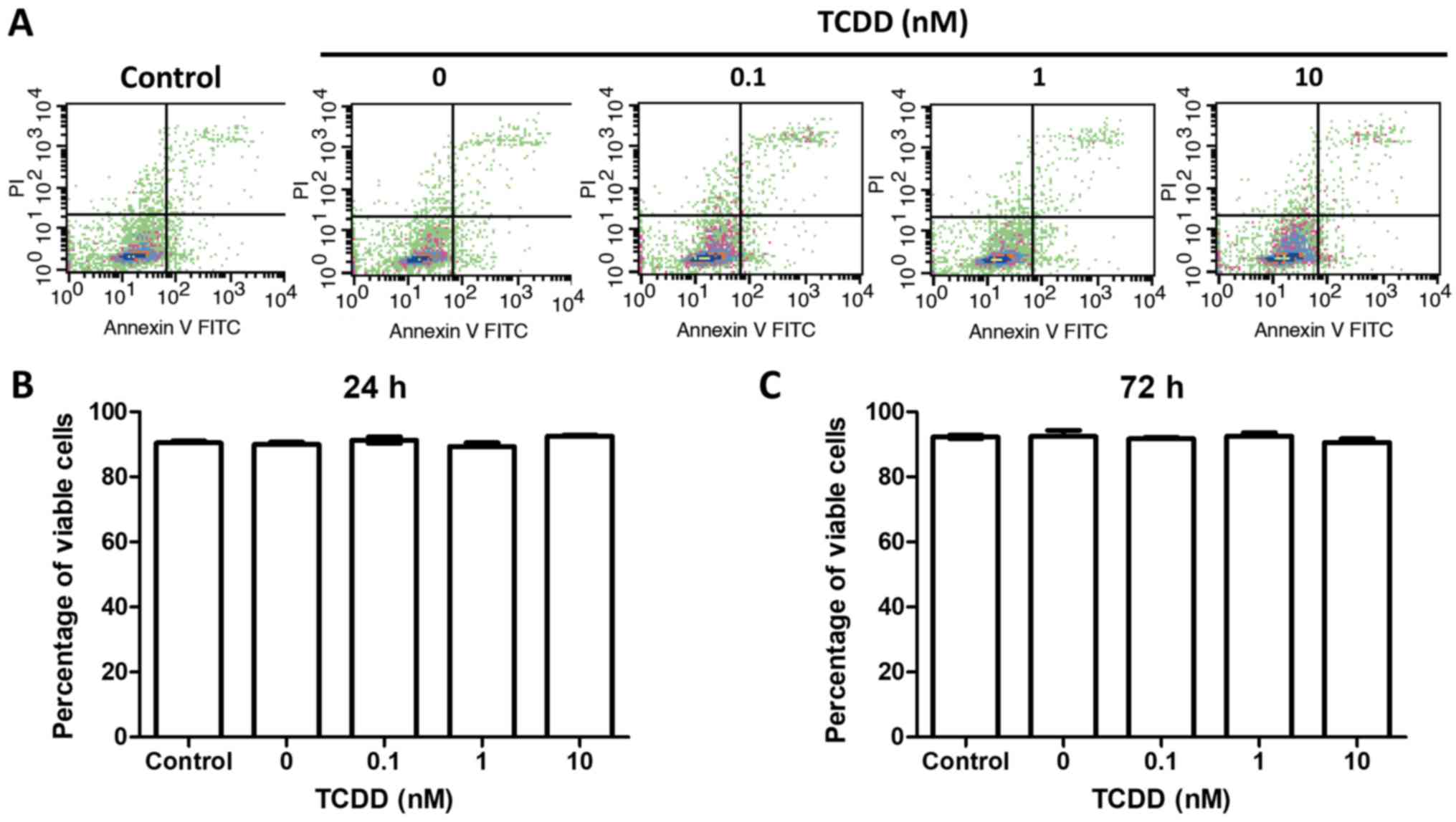
TCDD treatment did not affect MG-63
survival
To test the hypothesis that TCDD treatment was not
associated with survival, the MG-63 cells were treatment with
toluene or 0.1, 1 or 10 nM TCDD. The viable cells were counted at
24 and 72 h later by Annexin V and PI measurements via
flow-cytometry. There were no statistically significant changes
before 72 h of culture (Fig. 1).
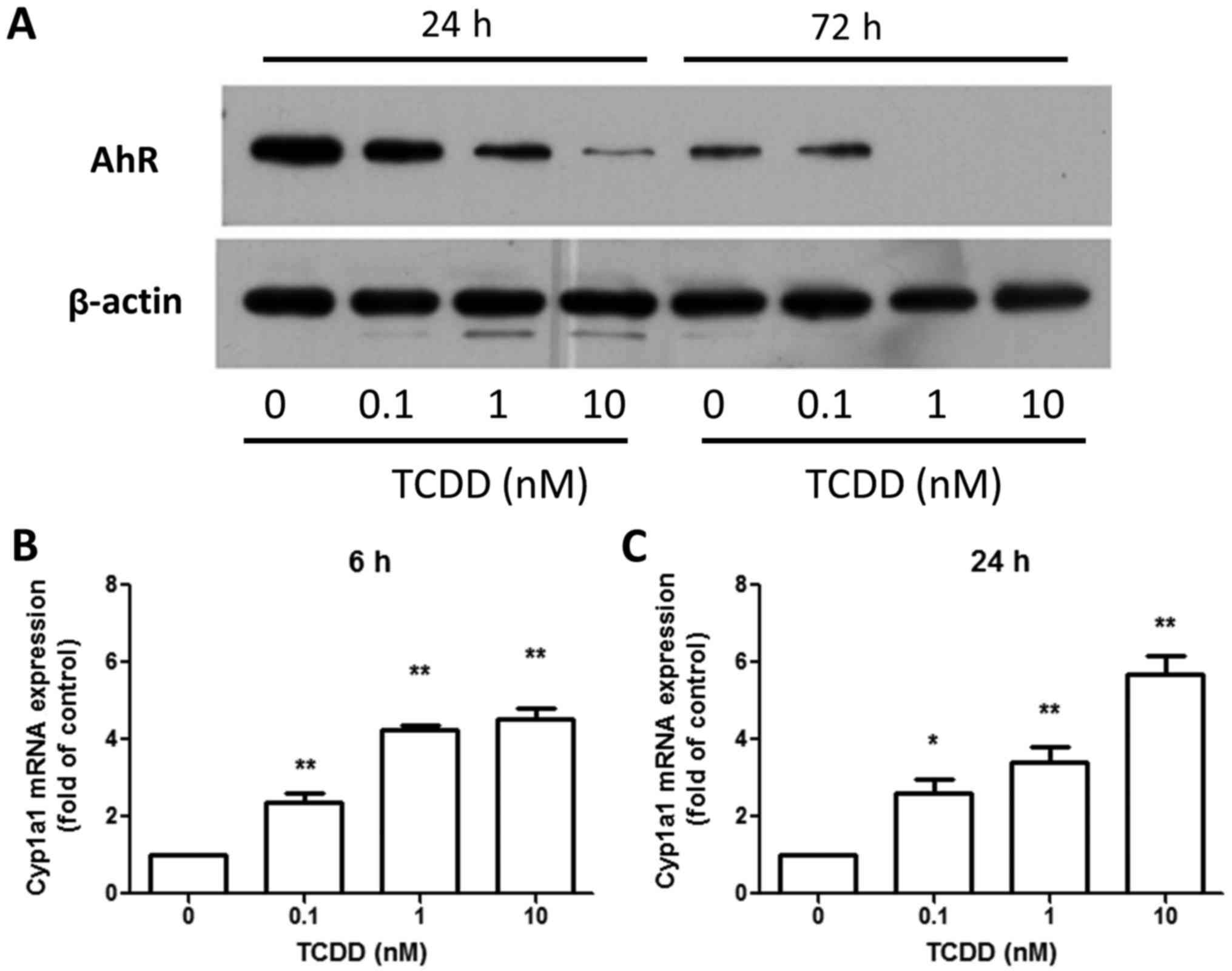
MG-63 cells expressed a functional AhR
under TCDD treatment
The amount of AhR protein was investigated in MG-63
cells treated with toluene or 0.1, 1 or 10 nM TCDD for 24 and 72 h.
The protein was detected in MG-63 cells and the TCDD treatment
decreased the amounts of AhR protein (Fig. 2A) in a dose-dependent manner. This, in
turn, was reported to be controlled by the ubiquitin-proteasome
pathway (22). However, TCDD
treatment increased the expression of the AhR-regulated gene,
Cyp1a1, after 6 and 24 h (Fig. 2B and
C), indicating that AhR pathway was responsive to TCDD and
participated in DRE-mediated signaling in MG-63 cells.
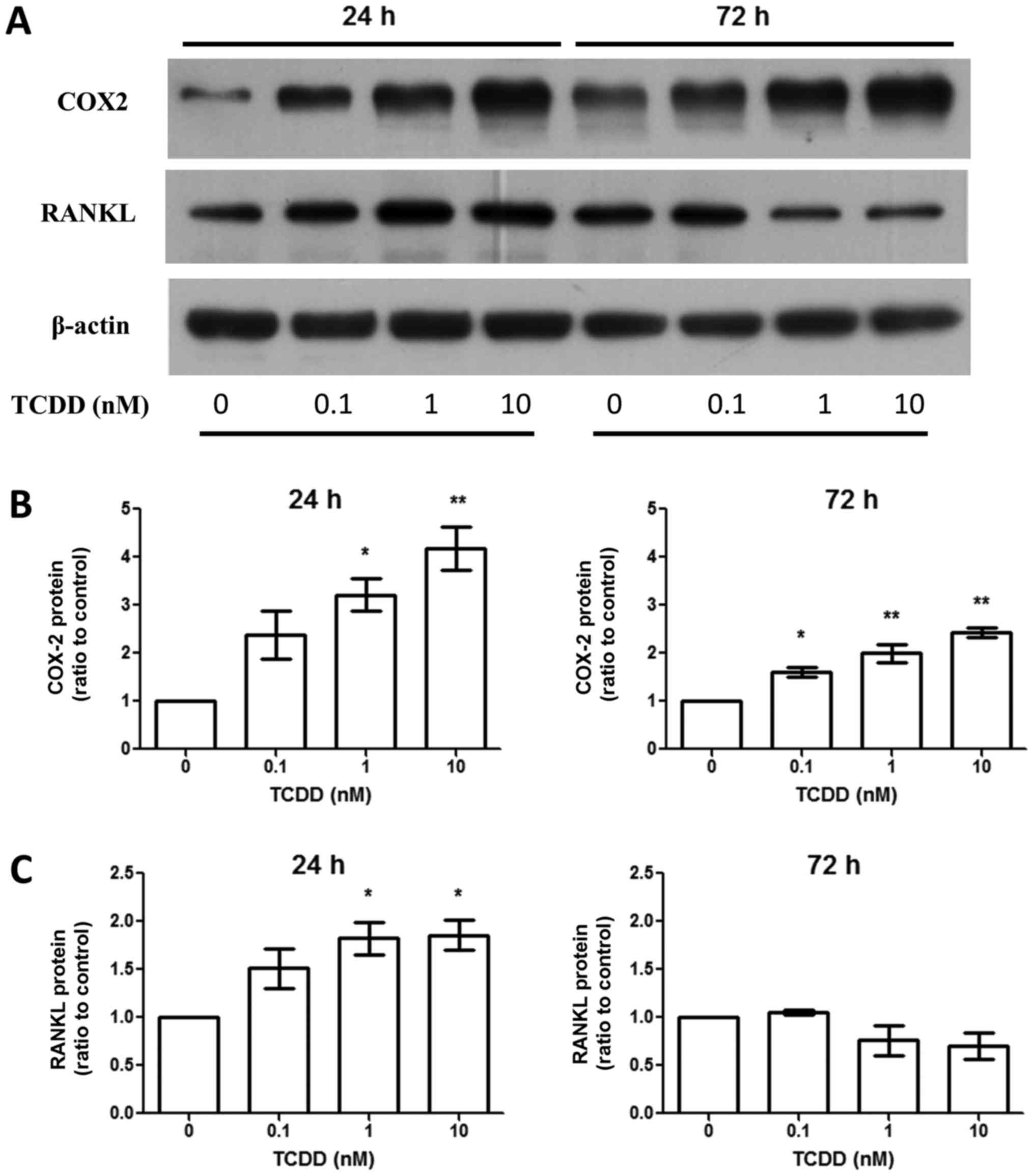
TCDD treatment increased
cyclooxygenase-2 (COX-2) and RANKL production
Aside from AhR activation, activated MG-63 by TCDD
upregulated RANKL expression around 24 h at the protein level
(Fig. 3). This was linked to the
regulation of bone resorption through the effects on osteoclasts.
However, the effects were not seen after TCDD treatment for 72 h
(Fig. 3C). The COX-2 expressions were
elevated at the protein level (Fig.
3B) after TCDD treatment for 24 and 72 h.
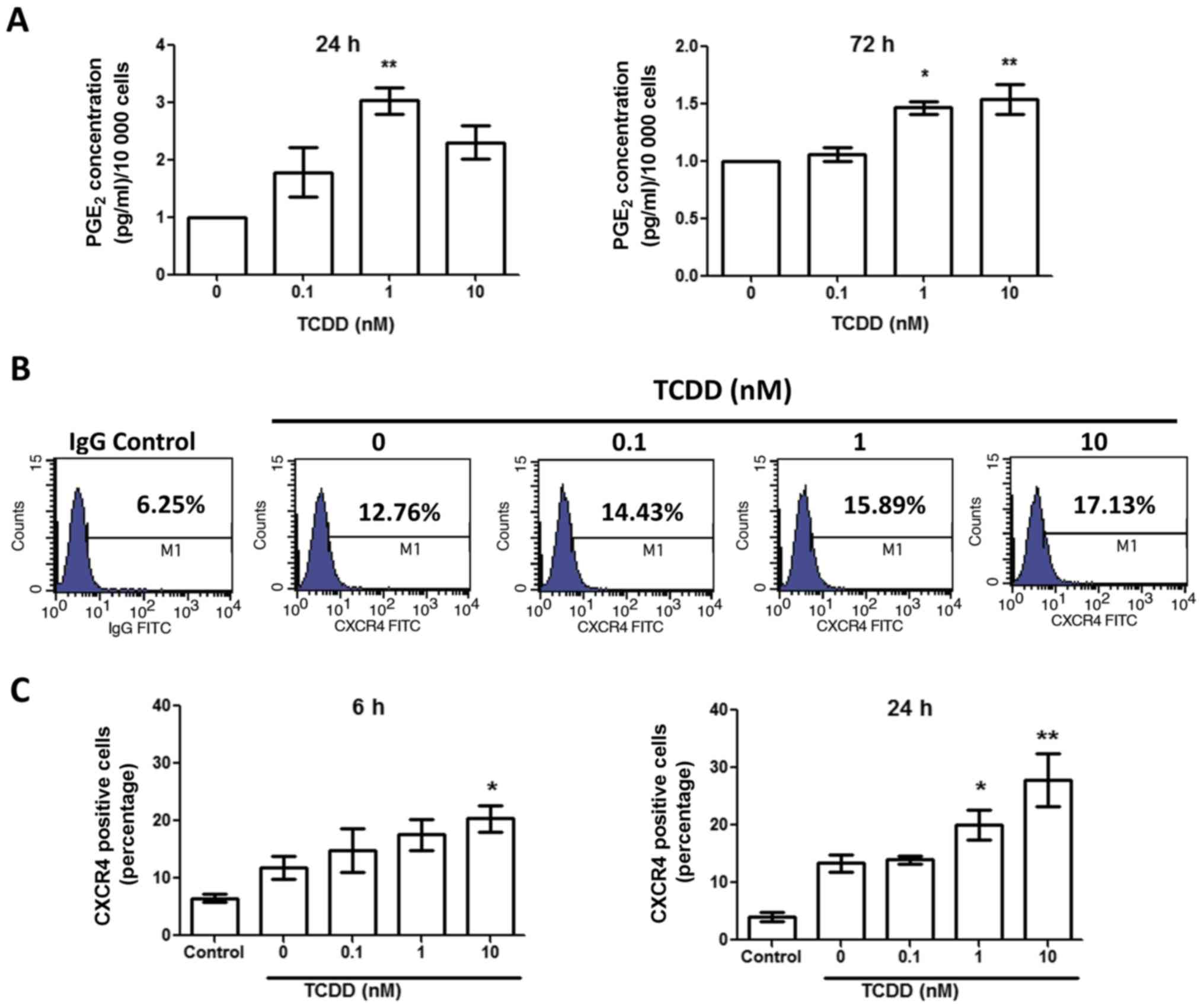
TCDD treatment increased prostaglandin
E2 (PGE2)
To test the impact of TCDD treatment on MG-63 cells
activation mediated by the COX-2/PGE2 cascade, the PGE2 level was
measured by ELISA after treated with toluene, 0.1, 1 and 10 nM TCDD
for 24 and 72 h (Fig. 4A). There was
a marked increase in PGE2 levels after 24 and 72 h in the presence
of TCDD. Together with the elevated COX-2 level, this finding
suggests that TCDD treatment may promote the activation of the
COX-2/PGE2 pathways.
TCDD treatment increased CXCR4
expressions
To determine if TCDD treatment enhanced CXCR4
expression, MG-63 cells were cultured for 6 and 24 h with toluene,
0.1, 1 and 10 nM TCDD. The CXCR4 expressions were then measured by
flow cytometry (Fig. 4B). The results
demonstrated that CXCR4 expressions were upregulated in a
relatively dose-dependent manner at 6 and 24 h (Fig. 4C).
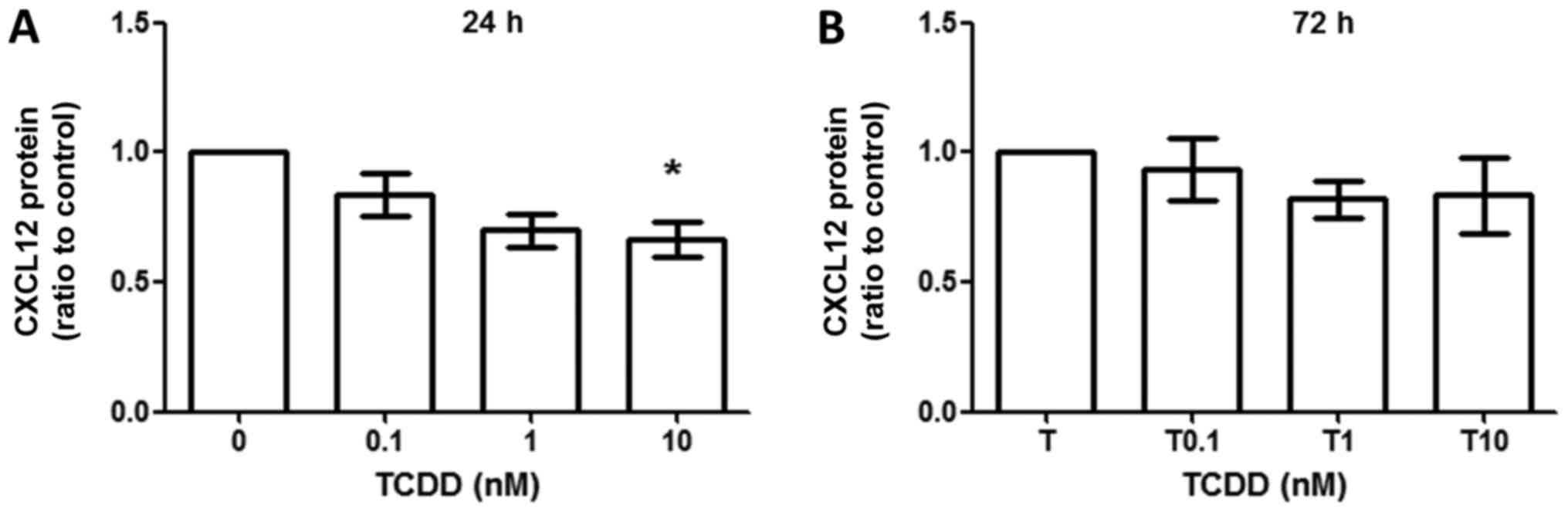
TCDD treatment did not increase CXCL12
production
CXCL12 production was measured by ELISA method,
which revealed that CXCL12 production did not change between
toluene- and TCDD-treated cells at 24 h (Fig. 5A) and 72 h (Fig. 5B). However, there were trends towards
decreased production of CXCL12 when treated under 10 nM TCDD at 24
h (ratio compared to vehicle control, 0.701±0.109, P<0.05).
Effects of AhR Antagonists on TCDD
induced AhR, RANKL, COX-2, PGE2, CXCR4 and CXCL12 changes in MG-63
cells
To study whether the TCDD induced protein changes
are AhR pathway related, AhR antagonist CH-223191 (10 µm,
Selleckchem, Houston, USA) blocks the TCDD-mediated suppression of
AhR (Fig. 6B), RANKL (Fig. 6D), and COX-2 (Fig. 6C) expressions after treated by TCDD
for 72 h. Meanwhile, CH-223191 also abrogated TCDD induced
increasements of PGE2 (Fig. 7A)
levels and CCR4 (Fig. 7B) expressions
on MG-63 cells. However, there were no significant changes on TCDD
induced CXCL12 (Fig. 7C) expressions
when treated with CH-223191.
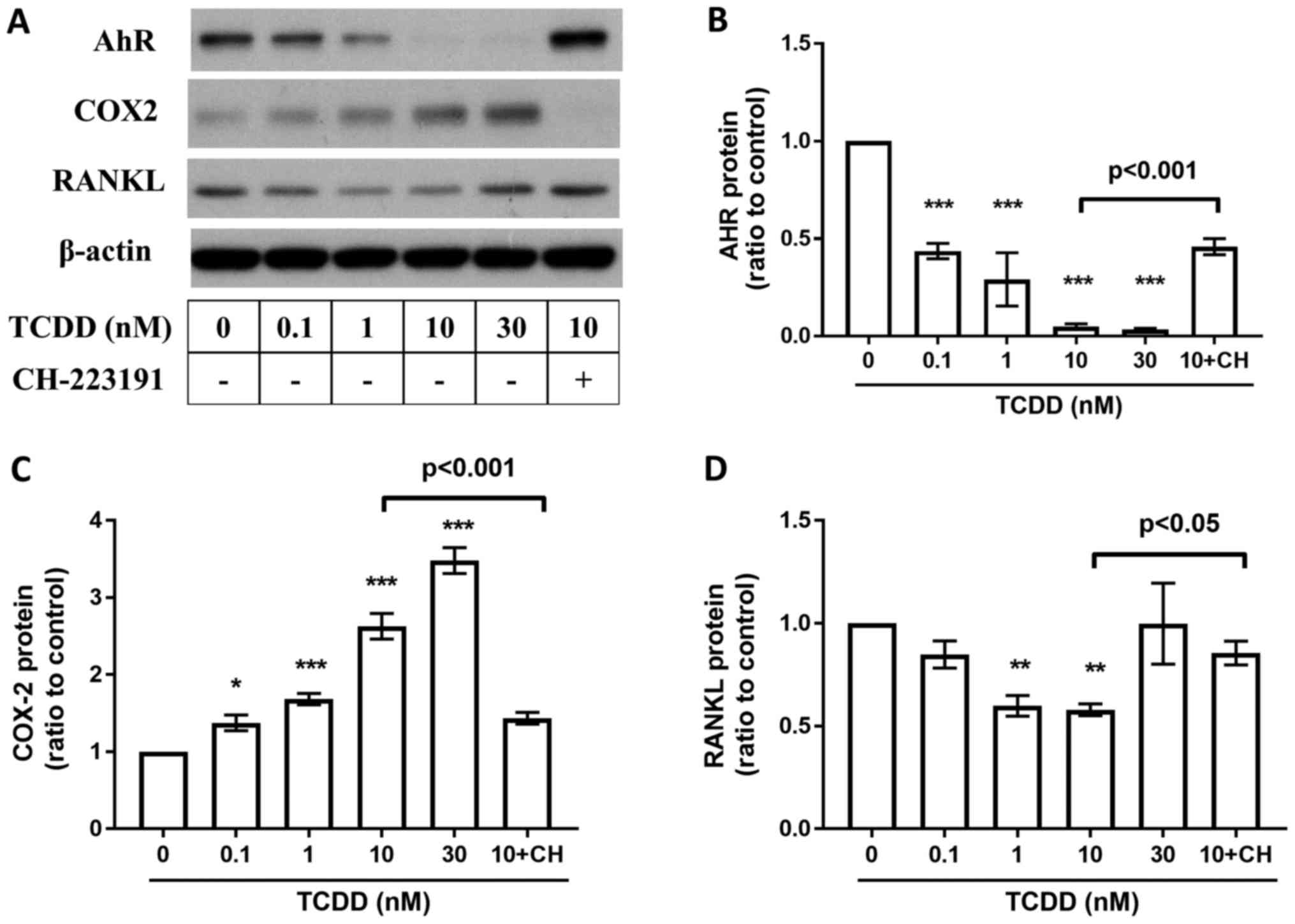 | Figure 6.Effects of AhR Antagonists on AhR,
COX-2, RANKL, and PGE2, CXCR4, CXCL12 expressions. MG-63 cells were
cultured for 72 h with toluene (vehicle control), 10 nM TCDD and 10
nM TCDD plus CH-223191 for 72 h. (A) AhR, COX-2 and RANKL protein
levels were measured by western blot analysis. (B) AhR protein
levels at 72 h. (C) COX-2 protein levels at 72 h. (D) RANKL protein
levels at 72 h. Data are presented as the mean ± standard error of
the mean (n=3). *P<0.05, **P<0.01 and ***P<0.005 vs. the
toluene-treated control (n=3). CH, 10 µM CH-223191; TCDD,
2,3,7,8-tetrachlorodibenzo-p-dioxin; COX-2, cyclooxygenase-2;
RANKL, receptor activator of nuclear factor-κB ligand. |
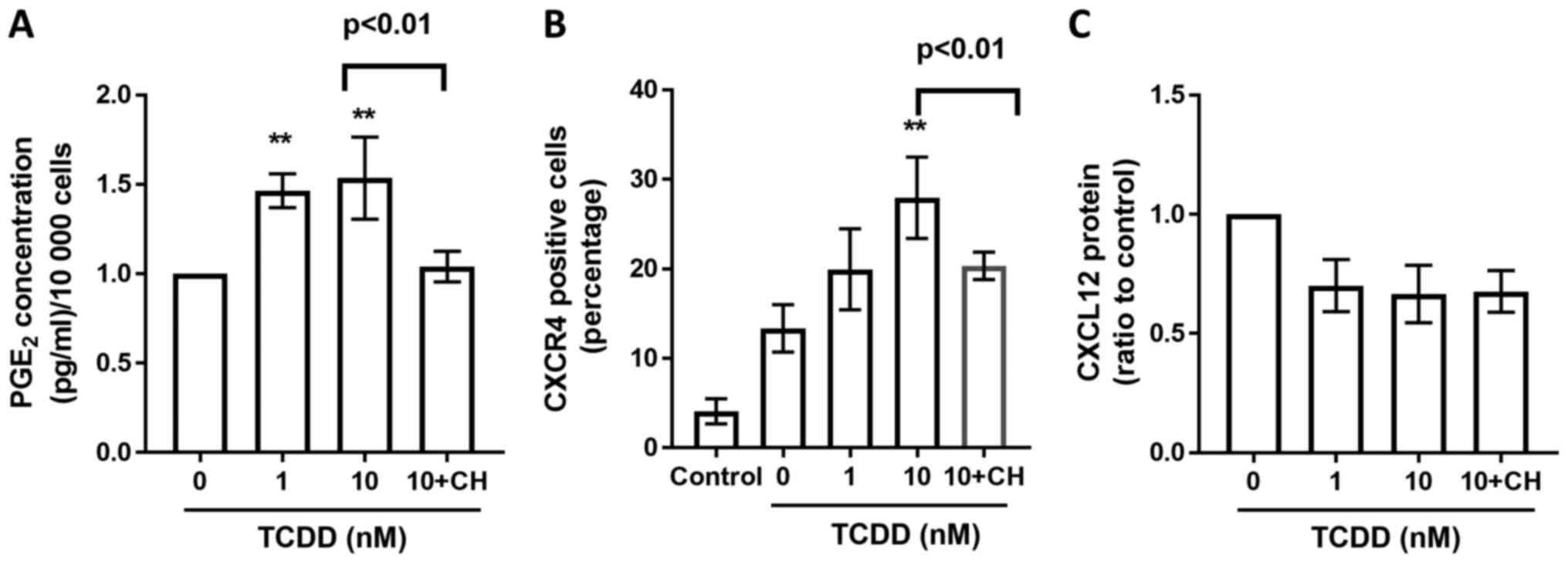 | Figure 7.PGE2, CXCR4, CXCL12 profiles derived
from MG-63 cells treated with AhR agonists. (A) Cells were treated
with toluene (vehicle control), 1 or 10 nM TCDD and 10 nM TCDD plus
CH-223191 for 72 h. PGE2 protein levels in the supernatants were
measured by ELISA. (B) CXCR4 receptor expressions were measured by
flow cytometry after toluene (vehicle control), 1 nM, 10 nM TCDD
and 10 nM TCDD plus CH-223191 treatment for 24 h. The number
represented the percentage of total cells. (C) CXCL12 protein
production in the supernatant after cells were treated with toluene
(vehicle control), 1 nM, 10 nM TCDD and 10 nM TCDD plus CH-223191
for 24 h was measured by ELISA. Data are presented as the mean ±
standard error of the mean (n=3). **P<0.01 vs. the
toluene-treated control (n=3). CH, 10 µM CH-223191; TCDD,
2,3,7,8-tetrachlorodibenzo-p-dioxin; AhR, aryl hydrocarbon
receptor; PGE2, prostaglandin E2. |
Discussion
The present study investigated how TCDD treatment
affected the activation of the osteosarcoma cell line, MG-63 cell.
Currently, there are only limited studies on the environmental
impact on osteosarcoma and on osteoblast functions. Previous
studies have pointed out differences in gene expression profiles of
osteosarcoma cell lines and normal osteoblasts (23,24).
However, among the currently available osteosarcoma cells lines,
Saos-2 cells have the most mature osteoblastic labeling profile,
while U-2 OS cells are negative for most of the investigated
osteoblastic markers (25). The MG-63
cells are the intermediate cell type in terms of differences in
extracellular matrix formation. Recently, Miki et al
(26) reported AhR influenced by
environmental compounds, such as 3-methylcholanthrene (3-MC) and
β-naphthoflavone (β-NF), could regulate estrogen synthesis and
metabolism in bone tissues through cytokine/aromatase signaling
based on a hFOB cell and MG-63 cell line model. These increments of
AhR-target genes in MG-63 cells were inhibited by co-treatment of
AhR antagonist, CH-223191. Compare to the study, we try to
investigate the effect of TCDD at a lower concentration which is
more physiological feasible. Our results showed that the
expressions of RANKL, COX-2, PGE2 and CXCR4 were upregulated under
TCDD treatment, and inhibited by co-treatment of CH-223191. Our
findings further expand the knowledge on genes mediated by AhR
pathway in MG-63 cell line even under low concentration of TCDD,
and possibly linked to the aggressiveness in osteosarcoma.
The AhR, a ligand-activated transcriptional
regulator, is ubiquitously expressed in most organs and can be
activated by a structurally diverse range of chemicals (27,28). The
best-characterized ones include a variety of environmental
contaminants. Gathered evidence point to the pro-inflammatory role
of TCDD via activation of the AhR pathway, and modulation of bone
microarchitecture and material properties (13). The TCDD are possible carcinogens but
the risks are not closely linked to dose exposure (29,30).
Evidence indicates that dioxins may interfere with differentiation
of osteoblasts and osteoclasts in TCDD concentrations as low as 100
fM (14). Thus, we propose that TCDD,
a ubiquitous environmental contaminant, may be associated with the
aggressiveness of osteosarcoma even under low concentrations of
exposure.
The TCDD concentrations used in the present study
were between 0–10 nM were based on the previous epidemiology study
investigated on the historical event that happened on 10 July 1976,
a chemical explosion in Seveso, Italy, that resulted in the highest
known residential exposure to TCDD (31). The median (interquartile range) serum
TCDD concentration soon after the explosion was 73.2 (33.1–193.0)
Parts Per Trillion (ppt) for all women, and roughly equals to 0.22
(0.10–0.60) nM. In our results, TCDD treatment may the upregulation
of RANKL under low concentrations (0.1 and 1 nM) at early time
point, which indicates that TCDD may be associated with
aggressiveness of MG-63 cells through the interaction with
osteoclasts under different concentrations of TCDD exposure
(32).
In our study, the amounts of Cyclooxygenase-2
(COX-2) protein are persistently elevated at 24 and 72 h after TCDD
treatment. Currently, COX-2 expression has also been established as
a marker in human osteosarcoma (33).
COX-2 may mediates increased PGE2 production in bone in response to
various stimuli including parathyroid hormone (PTH), mechanical
strain, or inflammatory cytokines (34). TCDD can stimulate COX-2 expression
through both a nongenomic pathway or activation of the AhR pathway
(35). The alterations to COX-2
expression and the abundance of its enzymatic product PGE2 have key
roles in influencing the development, progression, and metastasis
potentials of cancer (36).
PGE2 was essential both for expression of functional
CXCR4 and for production of its ligand CXCL12 in cancer-associated
MDSCs model (37). The increased
expression of CXCR4 may be a possible factor leading to the
migration and proliferation abilities in a variety of tumors
(38). Gathered evidence support that
that TCDD is involved in the regulation of CXCR4/CXCL12 axis. In a
comprehensive study, Hsu et al (39) reported that TCDD downregulates CXCR4
mRNA in five of 19 cell lines tested, but also upregulates CXCR4
mRNA in seven of the cell lines tested. Variations on the CXCL12
expressions on the cell lines are also seen (39). Using AhR antagonist, Yun et al
(40) reported that TCDD induced
CXCR4 in an AhR dependent manner. These findings indicate that
CXCR4/CXCL12 are different mediated according to cells and
microenvironments differences.
Results of our present analysis reveal that CXCR4
expression are increased after TCDD treatment in a dose response
manner. Levels of CXCL12 showed the trends toward decreasing after
TCDD treatment, but the differences are not statistically
different. While the TCDD induced expression of COX-2, PGE2 and
CXCR4 are blocked by the co-administration of AHR antagonist,
CH-223191, our findings revealed that AHR pathway are involved in
the regulation of COX-2, PGE2 and CXCR4.
Current evidences revealed that RANKL are considered
as a possibly hopeful therapeutic target molecule for osteosarcoma
patients. In a recent study, IHC results demonstrate RANKL
expression was observed in the tumor element in 68% of human
osteosarcoma (41). However, the
staining intensity was relatively low and only 37% of samples
exhibited equal or mayor 10% RANKL positive tumor cells; moreover,
RANK expression was not observed in OS tumor cells (41). The absence of RANK expression in
primary human OS cells suggests that an autocrine RANKL/RANK
signaling in human OS tumor cells is not operative. However, in a
whole-body Rankl deletion mouse model, Yan Chen et al
(42) provide a rationale to consider
RANKL blockade for the treatment and prevention of aggressive
RANKL-overexpressing OS. This finding indicates that RANKL is
associated with disease prognosis. Our results demonstrate that the
RANKL is increased initially at 24 h under low concentrations of
TCDD, but decreased at 72 h, and are increased under the AhR
antagonist CH-223191. These findings indicate that the effect of
TCDD on RANKL are complicated. More comprehensive studies are
needed to establish possible treatment considerations for patients
with osteosarcoma.
In conclusion, our results indicate that chronic
TCDD exposure on MG-63 cells may be associated with clinical
aggressiveness through the upregulation of COX-2, PGE2, and CXCR4.
However, the TCDD induced RANKL expressions need to be investigated
under different concentration and time points. Therefore, the
results of our present study did indicate that TCDD could possibly
disturb MG-63 cell homeostasis by disrupting the AhR pathway. Taken
together, these findings suggest that ‘AhR signal therapy’ should
be further explored as a therapeutic option in the treatment of
osteosarcoma.
Acknowledgements
The authors would like to acknowledge Mr. Ho Chung
Ju and Ms. Huang Tzu Ting (Department of Thoracic Medicine, Chang
Gung University, Taoyuan, Taiwan) for their technical assistance
with antibody preparations during the experiments.
Funding
The present study was supported by grants from the
E-Da Hospital and I-Shou University (grant no. EDAHP-103025) and
the Chang Gung Memorial Hospital (grant no. CMRPG3E0731).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SYH and PCC performed the experiments. PCC, SCY and
CHW were responsible for the acquisition and analysis of the data.
SCY, YKT and PCC conceived and designed the study. SYH and PCC
contributed reagents, materials and analysis tools. SCY and PCC
wrote the paper and gave final approval of the submitted
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Harada S and Rodan GA: Control of
osteoblast function and regulation of bone mass. Nature.
423:349–355. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Trieb K and Windhager R: Receptor
activator of nuclear factor κB expression is a prognostic factor in
human osteosarcoma. Oncol Lett. 10:1813–1815. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Caetano-Lopes J, Canhão H and Fonseca JE:
Osteoblasts and bone formation. Acta Reumatol Port. 32:103–110.
2007.PubMed/NCBI
|
|
4
|
Yellowley C: CXCL12/CXCR4 signaling and
other recruitment and homing pathways in fracture repair. Bonekey
Rep. 2:3002013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shahnazari M, Chu V, Wronski TJ, Nissenson
RA and Halloran BP: CXCL12/CXCR4 signaling in the osteoblast
regulates the mesenchymal stem cell and osteoclast lineage
populations. FASEB J. 27:3505–3513. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu C, Fuertes E, Flexeder C, Hofbauer LC,
Berdel D, Hoffmann B, Kratzsch J, von Berg A and Heinrich J:
GINIplus Study Group; LISAplus Study Group: Associations between
ambient air pollution and bone turnover markers in 10-year old
children: Results from the GINIplus and LISAplus studies. Int J Hyg
Environ Health. 218:58–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qi H, Aguiar DJ, Williams SM, La Pean A,
Pan W and Verfaillie CM: Identification of genes responsible for
osteoblast differentiation from human mesodermal progenitor cells.
Proc Natl Acad Sci USA. 100:3305–3310. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ray S and Swanson HI: Activation of the
aryl hydrocarbon receptor by TCDD inhibits senescence: A tumor
promoting event? Biochem Pharmacol. 77:681–688. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barouki R, Coumoul X and
Fernandez-Salguero PM: The aryl hydrocarbon receptor, more than a
xenobiotic-interacting protein. FEBS Lett. 581:3608–3615. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Veldhoen M, Hirota K, Westendorf AM, Buer
J, Dumoutier L, Renauld JC and Stockinger B: The aryl hydrocarbon
receptor links TH17-cell-mediated autoimmunity to environmental
toxins. Nature. 453:106–109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mimura J and Fujii-Kuriyama Y: Functional
role of AhR in the expression of toxic effects by TCDD. Biochim
Biophys Acta. 1619:263–268. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Harvey WA, Jurgensen K, Pu X, Lamb CL,
Cornell KA, Clark RJ, Klocke C and Mitchell KA: Exposure to
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) increases human hepatic
stellate cell activation. Toxicology. 344–346:26–33. 2016.
View Article : Google Scholar
|
|
13
|
Herlin M, Finnilä MA, Zioupos P, Aula A,
Risteli J, Miettinen HM, Jämsä T, Tuukkanen J, Korkalainen M,
Håkansson H, et al: New insights to the role of aryl hydrocarbon
receptor in bone phenotype and in dioxin-induced modulation of bone
microarchitecture and material properties. Toxicol Appl Pharmacol.
273:219–226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Korkalainen M, Kallio E, Olkku A, Nelo K,
Ilvesaro J, Tuukkanen J, Mahonen A and Viluksela M: Dioxins
interfere with differentiation of osteoblasts and osteoclasts.
Bone. 44:1134–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schwarz M and Appel KE: Carcinogenic risks
of dioxin: Mechanistic considerations. Regul Toxicol Pharmacol.
43:19–34. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Garcia-Moure M, Martinez-Vélez N,
Patiño-García A and Alonso MM: Oncolytic adenoviruses as a
therapeutic approach for osteosarcoma: A new hope. J Bone Oncol.
9:41–47. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kansara M, Teng MW, Smyth MJ and Thomas
DM: Translational biology of osteosarcoma. Nat Rev Cancer.
14:722–735. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dalla-Torre CA, de Toledo SRC, Yoshimoto
M, Petrilli AS, Andrade JA, Chilton-MacNeill S, Squire JA and
Zielenska M: Expression of major vault protein gene in osteosarcoma
patients. J Orthop Res. 25:958–963. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Le Vu B, de Vathaire F, Shamsaldin A,
Hawkins MM, Grimaud E, Hardiman C, Diallo I, Vassal G, Bessa E,
Campbell S, et al: Radiation dose, chemotherapy and risk of
osteosarcoma after solid tumours during childhood. Int J Cancer.
77:370–377. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang YY, Huang HL, Chen YC, Hsu JT, Shieh
TM and Tsai MT: Biological characteristics of the MG-63 human
osteosarcoma cells on composite tantalum carbide/amorphous carbon
films. PLoS One. 9:e955902014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Svec D, Tichopad A, Novosadova V, Pfaffl
MW and Kubista M: How good is a PCR efficiency estimate:
Recommendations for precise and robust qPCR efficiency assessments.
Biomol Detect Quantif. 3:9–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma Q and Baldwin KT:
2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced degradation of aryl
hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway.
Role of the transcription activaton and DNA binding of AhR. J Biol
Chem. 275:8432–8438. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Benayahu D, Shur I, Marom R, Meller I and
Issakov J: Cellular and molecular properties associated with
osteosarcoma cells. J Cell Biochem. 84:108–114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bilbe G, Roberts E, Birch M and Evans DB:
PCR phenotyping of cytokines, growth factors and their receptors
and bone matrix proteins in human osteoblast-like cell lines. Bone.
19:437–445. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pautke C, Schieker M, Tischer T, Kolk A,
Neth P, Mutschler W and Milz S: Characterization of osteosarcoma
cell lines MG-63, Saos-2 and U-2 OS in comparison to human
osteoblasts. Anticancer Res. 24:3743–3748. 2004.PubMed/NCBI
|
|
26
|
Miki Y, Hata S, Ono K, Suzuki T, Ito K,
Kumamoto H and Sasano H: Roles of Aryl hydrocarbon receptor in
aromatase-dependent cell proliferation in human osteoblasts. Int J
Mol Sci. 18:pii: E2159. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Denison MS and Nagy SR: Activation of the
aryl hydrocarbon receptor by structurally diverse exogenous and
endogenous chemicals. Annu Rev Pharmacol Toxicol. 43:309–334. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Denison MS, Soshilov AA, He G, DeGroot DE
and Zhao B: Exactly the same but different: Promiscuity and
diversity in the molecular mechanisms of action of the Aryl
hydrocarbon (Dioxin) receptor. Toxicol Sci. 124:1–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Walker NJ: Unraveling the complexities of
the mechanism of action of dioxins. Toxicol Sci. 95:297–299. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huff J, Lucier G and Tritscher A:
Carcinogenicity of TCDD: Experimental, mechanistic, and
epidemiologic evidence. Annu Rev Pharmacol Toxicol. 34:343–372.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eskenazi B, Warner M, Sirtori M, Fuerst T,
Rauch SA, Brambilla P, Mocarelli P and Rubinacci A: Serum dioxin
concentrations and bone density and structure in the Seveso Women's
Health Study. Environ Health Perspect. 122:51–57. 2014.PubMed/NCBI
|
|
32
|
Clohisy JC, Frazier E, Hirayama T and
Abu-Amer Y: RANKL is an essential cytokine mediator of
polymethylmethacrylate particle-induced osteoclastogenesis. J
Orthop Res. 21:202–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qu L and Liu B: Cyclooxygeanse-2 promotes
metastasis in osteosarcoma. Cancer Cell Int. 15:692015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rezzonico R, Schmid-Alliana A, Romey G,
Bourget-Ponzio I, Breuil V, Breittmayer V, Tartare-Deckert S, Rossi
B and Schmid-Antomarchi H: Prostaglandin E2 induces interaction
between hSlo potassium channel and Syk tyrosine kinase in
osteosarcoma cells. J Bone Miner Res. 17:869–878. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu Y, Liu Q, Guo S, Zhang Q, Tang J, Liu
G, Kong D, Li J, Yan S, Wang R, et al: 2, 3, 7,
8-Tetrachlorodibenzo-p-dioxin promotes endothelial cell apoptosis
through activation of EP3/p38MAPK/Bcl-2 pathway. J Cell Mol Med.
21:3540–3551. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Greenhough A, Smartt HJ, Moore AE, Roberts
HR, Williams AC, Paraskeva C and Kaidi A: The COX-2/PGE2 pathway:
Key roles in the hallmarks of cancer and adaptation to the tumour
microenvironment. Carcinogenesis. 30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Obermajer N, Muthuswamy R, Odunsi K,
Edwards RP and Kalinski P: PGE2-induced CXCL12 production and CXCR4
expression controls the accumulation of human MDSCs in ovarian
cancer environment. Cancer Res. 71:7463–7470. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu Y, Guan GF, Chen J, Hu B, Sun C, Ma Q,
Wen YH, Qiu XC and Zhou Y: Aberrant CXCR4 and β-catenin expression
in osteosarcoma correlates with patient survival. Oncol Lett.
10:2123–2129. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hsu EL, Yoon D, Choi HH, Wang F, Taylor
RT, Chen N, Zhang R and Hankinson O: A proposed mechanism for the
protective effect of dioxin against breast cancer. Toxicol Sci.
98:436–444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yun C, Katchko KM, Schallmo MS, Jeong S,
Yun J, Chen CH, Weiner JA, Park C, George A, Stupp SI, et al: Aryl
hydrocarbon receptor antagonists mitigate the effects of dioxin on
critical cellular functions in differentiating human
osteoblast-like cells. Int J Mol Sci. 19:pii: E225. 2018.
View Article : Google Scholar
|
|
41
|
Branstetter D, Rohrbach K, Huang LY,
Soriano R, Tometsko M, Blake M, Jacob AP and Dougall WC: RANK and
RANK ligand expression in primary human osteosarcoma. J Bone Oncol.
4:59–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen Y, Di Grappa MA, Molyneux SD, McKee
TD, Waterhouse P, Penninger JM and Khokha R: RANKL blockade
prevents and treats aggressive osteosarcomas. Sci Transl Med.
7:317ra1972015. View Article : Google Scholar : PubMed/NCBI
|