Introduction
Anaplastic thyroid carcinoma (ATC) is a highly
invasive, undifferentiated tumor characterized by rapid growth and
distant metastasis. ATC accounts for <2% of all types of thyroid
cancer, but is responsible for >50% of cases of thyroid
cancer-associated mortality (1–3). Half of
all cases of ATC-associated mortality are caused by upper airway
obstruction and asphyxia, whereas the remaining cases are
attributed to local or distant metastases and/or treatment
complications (4). The patient
survival time after diagnosis is ~16 weeks, and the 1 and 5 year
survival rates are 17 and 8%, respectively (5). In addition, the available treatments
for ATC, including surgery, radiotherapy and chemotherapy, have
poor results (6). Therefore, the
development of novel approaches is crucial, and may include novel
cytotoxic drugs, targeted molecular therapies and gene therapies,
in order to kill ATC cells (7–10).
Vanadium is a transition metal that belongs to the
group of micronutrients that are essential for normal metabolism.
Notably, sodium orthovanadate (SOV) is a vanadium compound and
phosphate analog, which possesses numerous biological activities,
including inhibition of nonselective protein tyrosine phosphatases,
alkaline phosphatases and ATPases, activation of tyrosine kinases,
promotion of mitogenesis and neuroprotection, and inhibition of
diabetic effects by an insulin-mimetic property (11,12). SOV
can also inhibit the growth of central nervous system tumors
(13), lung cancer (14), prostate cancer (15), bladder cancer (16) and liver cancer (17,18).
However, the effects of SOV on ATC cells have not yet been
reported.
The present study aimed to investigate the effects
of SOV on ATC in vitro and in vivo. To do so, the
influence of SOV on cell growth, cell cycle progression, apoptosis
and mitochondrial membrane potential (Δψm) were investigated.
Materials and methods
Cell culture, reagents and
antibodies
The human ATC cell line 8505C was kindly provided by
Dr Jihua Han (The Third Affiliated Hospital of Harbin Medical
University, Harbin, China). Cells were cultivated in RPMI 1640
medium supplemented with 10% fetal bovine serum and 1%
penicillin/streptomycin at 37°C in a humidified incubator
containing 5% CO2. SOV was purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). Antibodies against Ki-67 (catalog
no. 9449S) were provided by Cell Signaling Technology, Inc.
(Danvers, MA, USA).
Cell viability assay
Cell viability was assessed with the Cell Counting
kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). Briefly, cells were cultured in 96-well plates with RPMI
1640 medium overnight prior to treatment with SOV (0, 0.5, 1, 2, 4
and 8 µM) for 1–6 days and incubated at 37°C. The cells were seeded
between 500 and 3,000 cells/well, in 100 µl culture medium,
depending on the culture times tested (1–6 days). After the
appropriate culture time, 10 µl CCK-8 solution was added to each
well for 1 h, prior to measuring the absorbance at 450 nm with a
microplate reader. The half maximal inhibitory concentration
(IC50) values were calculated according to the
Reed-Muench method (19).
Experiments were performed three times.
Plate colony formation assay
Cells were seeded in 6-well plates at a density of
500 cells/well and incubated overnight at 37°C. They were then
treated with various concentrations of SOV (0, 0.5, 1, 2, 4 and 8
µM) for 14 days and incubated at 37°C. Each treatment was carried
out in triplicate. Plates were incubated for 14 days. Medium
containing the appropriate concentration of SOV was replaced twice
weekly. Eventually, plates were washed twice with cold PBS, fixed
with 4% paraformaldehyde for 10 min, and stained with 3% crystal
violet. Colonies containing ≥50 cells were scored.
Cell cycle analysis
Cell cycle progression of 8505C cells was analyzed
using a Cell Cycle kit (catalog no. 558662; BD Biosciences, San
Jose, CA, USA). Briefly, following treatment with various
concentrations of SOV (0, 0.5, 1, 2, 4 and 8 µM) at 37°C for 48 h,
cells were harvested. A total of 1×106 cells were
incubated with reagents A-C, according to the manufacturer's
protocol, and cells were subjected to flow cytometry. Experiments
were performed three times.
Apoptosis analysis
Cell apoptosis was analyzed with the fluorescein
isothiocyanate (FITC)/Annexin V Apoptosis Detection kit I (BD
Biosciences), according to the manufacturer's protocol. Briefly,
cells were treated with various concentrations of SOV (0, 2 and 4
µM) and incubated at 37°C for 48 h. Subsequently, cells were washed
twice with cold PBS, resuspended in 1X Binding Buffer at a
concentration of 1×106 cells/ml, and 100 µl of each cell
solution was transferred to individual 5 ml culture tubes. A volume
of 5 µl FITC-conjugated Annexin V and 5 µl propidium iodide were
added to the tubes, which were gently vortex-mixed, prior to 15 min
incubation in the dark at room temperature. A volume of 400 µl 1X
Binding Buffer was then added to each tube. The samples were
immediately analyzed by flow cytometry (<1 h). Experiments were
performed three times.
Δψm assessment. The lipophilic cationic dye
JC-1 was used to measure alterations in Δψm. The JC-1 probe was
provided by Beyotime Institute of Biotechnology (Haimen, China) and
the assay was performed according to the manufacturer's protocol.
Briefly, cells were incubated with SOV (0, 2 and 4 µM) in 6-well
plates at 37°C for 48 h, prior to adding 5 µg/ml JC-1 for 20 min at
37°C. After incubation with JC-1, cells were washed twice with PBS.
The Δψm was assessed by determining the dual emissions from
mitochondrial JC-1 monomers (green, 490 nm stimulated luminescence
and 530 nm emission light) and aggregates (red, 525 nm stimulated
luminescence and 590 nm emission light) using a confocal laser
scanning microscope. An increase in the green/red fluorescence
intensity ratio indicated mitochondrial depolarization. The
relative Δψm was calculated according to the following formula:
Experimental ratio value/Control ratio value ×100. Experiments were
performed three times.
Xenograft tumor model and
treatments
Female nude mice (5 weeks old; 16–18 g; Beijing
Vital River Laboratories Animal Technology Co., Ltd., Beijing,
China) were housed at a constant temperature (23°C) and relative
humidity (60%) with a fixed 12-h light-dark cycle, and free access
to food and water. After 1 week of feeding in the new environment,
mice were then used to generate a subcutaneous tumor model. All
surgical procedures and animal care protocols were approved by the
Ethics Committee of the First Affiliated Hospital of Harbin Medical
University (Harbin, China). The study also complied with
institutional guidelines for animal care. An 8505C cell suspension
(100 µl) in serum-free RPMI 1640 culture medium (1×106)
was subcutaneously injected into the flanks of nude mice. Tumor
formation and tumor size were monitored daily. When tumors were ~4
mm in diameter, animals were randomly divided into three groups,
according to their similar average tumor sizes (n=4 mice/group).
Mice received daily 200 µl intraperitoneal injections of either PBS
(control) or SOV (5 or 10 mg/kg) in PBS. The doses and methods were
based on our preliminary experiments and previous reports (17,20).
Tumor size was assessed twice weekly at the skin surface and
calculated as follows: Tumor volume=(width)2 ×
(length/2). After 4 weeks treatment, tumors were harvested, weighed
and their volume was measured.
Detection of 8505C cell proliferation
in vivo
Immuno histochemical analyses of the tumors were
performed using an anti-Ki-67 antibody, in order to detect cancer
cell proliferation. Briefly, tumor paraffin sections (4 µm) were
prepared by fixation with 10% formaldehyde solution for 12 h at
room temperature, washing with water for 24 h, dehydration,
transparency, embedding with paraffin, sectioning and baking for 30
min at 60°C. Following dewaxing, rehydration and antigen retrieval,
the tumor sections were blocked in 3% bovine serum albumin (catalog
no. 37520; Thermo Fisher Scientific, Inc.) at room temperature for
2 h, and incubated overnight at 4°C with primary antibody against
Ki-67 (1:200; catalog no. 8112S; Cell Signaling Technology, Inc.).
Sections were subsequently incubated at room temperature for 30 min
with SignalStain® Boost Detection Reagent (HRP, Mouse)
(catalog no. 8125S; Cell Signaling Technology, Inc.), then examined
by light microscopy (magnification, ×400).
Detection of apoptosis of 8505C cells
in vivo
Cell apoptosis was determined in situ with a
terminal deoxynucleotidyl transferase dUTP nick-end labeling
(TUNEL) cell apoptosis detection kit (Beyotime Institute of
Biotechnology), according to the manufacturer's protocol. Briefly,
4 µm tumor sections were dewaxed with xylene twice for 5 min, and
soaked in 100% ethanol for 5 min, 90% ethanol for 2 min and 70%
ethanol for 2 min. Sections were rinsed with distilled water for 2
min and incubated with 20 µg/ml proteinase K without DNase at 37°C
for 15 min. They were eventually washed three times with PBS, and
exposed to 50 µl TUNEL working fluid. A fluorescence microscope was
used to capture images of 400 high-power fields from the slides.
The apoptosis index (%) was calculated according to the following
formula: Apoptosis index=Number of apoptotic cells/Total number of
nucleated cells ×100. Experiments were performed three times.
Statistical analysis
GraphPad Prism 7.0 (GraphPad Software, Inc., La
Jolla, CA, USA) was used for statistical analysis. Data are
expressed as the means ± standard deviation. The differences among
samples were evaluated by one-way analysis of variance followed by
Dunnett's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Inhibitory effect of SOV on 8505C cell
viability
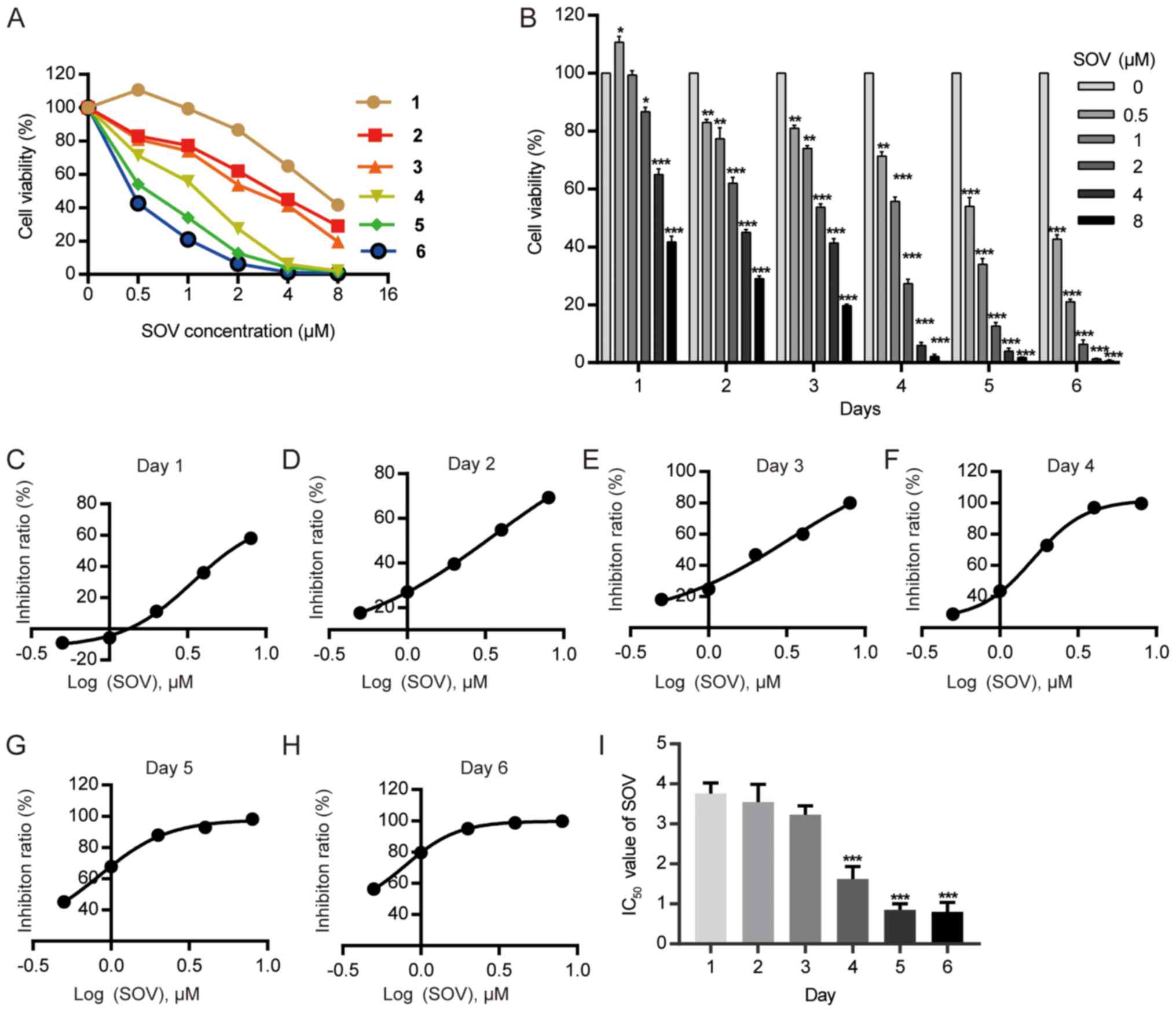
The 8505C cell line was cultured with various
concentrations of SOV (0.5, 1, 2, 4 and 8 µM) or without SOV
(control group) for 1–6 days. The cell survival rate was determined
using the CCK-8 kit (Fig. 1A). SOV
inhibited the viability of 8505C cells, and exhibited a stronger
effect at higher concentrations (Fig.
1B). The IC50 values of SOV for 8505C growth were
3.76, 3.55, 3.23, 1.62, 0.85 and 0.80 µM on days 1–6, respectively
(Fig. 1C-I). The mean
IC50 was 2.30 µM.
SOV inhibits the clonogenic survival
of 8505C cells
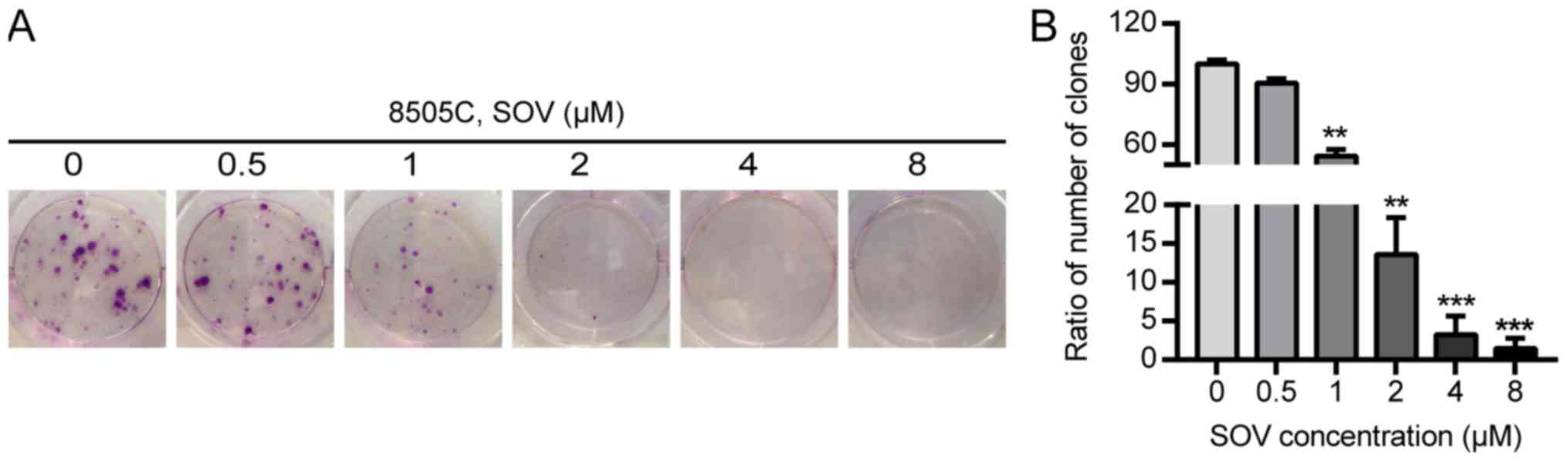
The effects of SOV on the clonogenic survival of
8505C cells were evaluated using colony formation assays. The 8505C
cells were exposed to increasing concentrations of SOV (0.5, 1, 2,
4 and 8 µM) or culture medium for 14 days. A decrease in the number
of ATC colonies following SOV treatment was observed in a
concentration-dependent manner (Fig. 2A
and B). Concentrations of SOV ≥1 µM inhibited >50% of 8505C
cell colony formation compared with in the control group
(P<0.01), and 8 µM SOV inhibited 98% of the colony formation
(P<0.001).
SOV induces G2/M cell cycle
arrest in 8505C cells
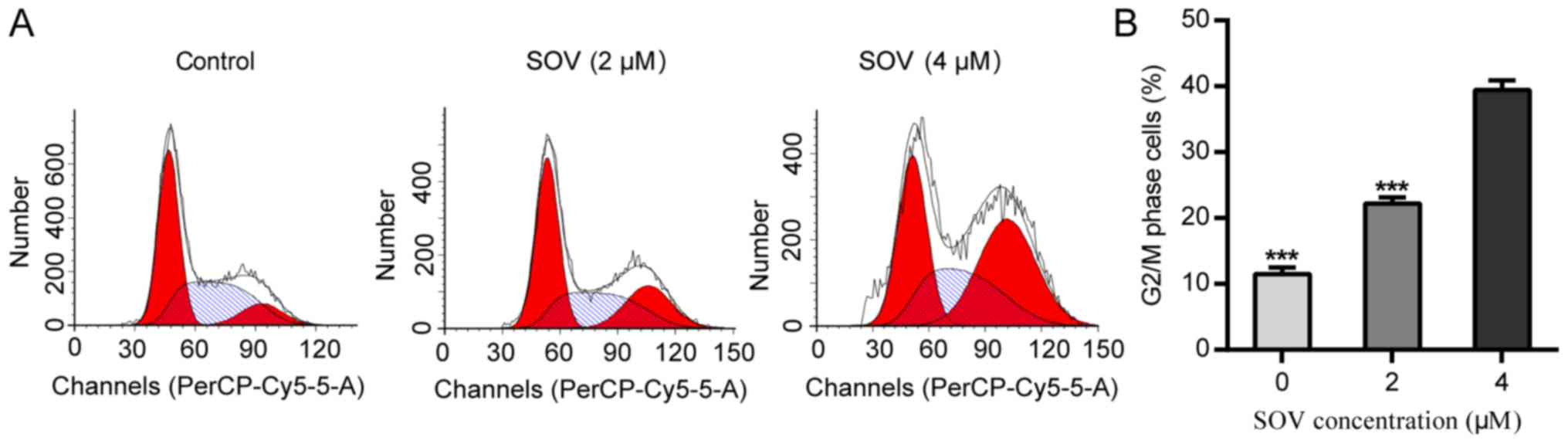
In order to explore the anti-proliferative mechanism
of SOV, 8505C cell cycle progression was assessed following
treatment with SOV. Briefly, 8505C cells were cultured in the
presence of 0, 2 or 4 µM SOV, according to the mean IC50
for 48 h. Flow cytometric analysis revealed that SOV blocked the
progression of 8505C cells beyond the G2/M phase
(Fig. 3A and B). Treatment with 4 µM
SOV resulted in the accumulation of 40% of cells in the
G2/M phase, whereas only 10% of cells in the control
group were in the G2/M phase (P<0.001). These data
suggested that SOV may lead to G2/M phase arrest in
8505C cells.
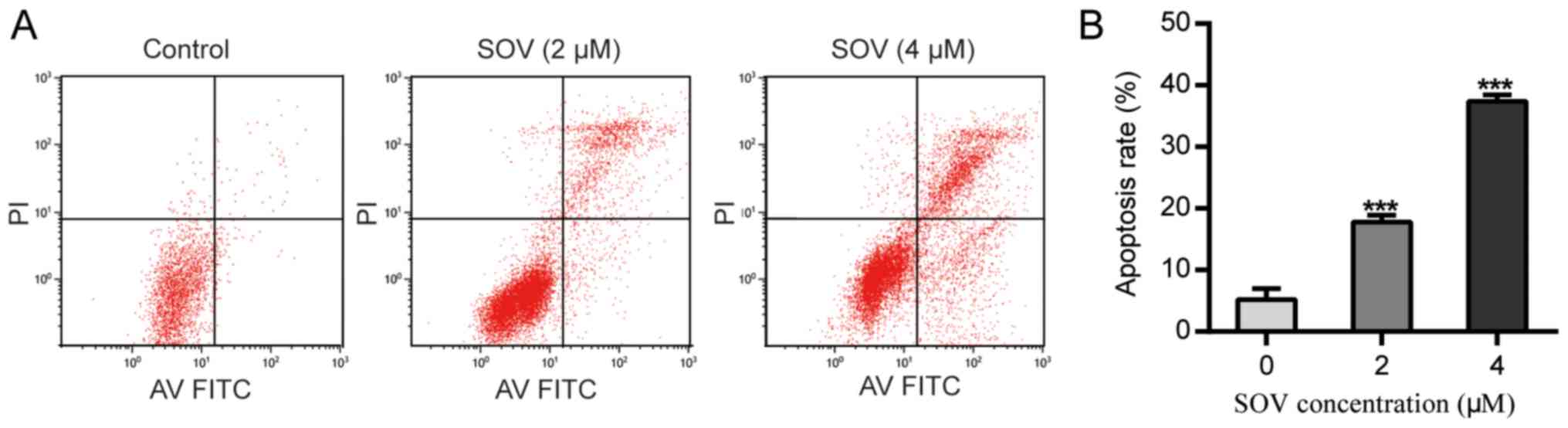
SOV induces apoptosis of 8505C
cells
Following treatment with SOV for 48 h, the apoptotic
rate of 8505C cells treated with SOV was significantly higher than
that of the control group (P<0.01, Fig. 4A and B). Treatment with 2 µM SOV for
48 h increased the percentage of apoptotic 8505C cells to 20%, and
4 µM SOV resulted in ~40% apoptotic cells. These data suggested
that SOV may induce 8505C cell apoptosis in a
concentration-dependent manner.
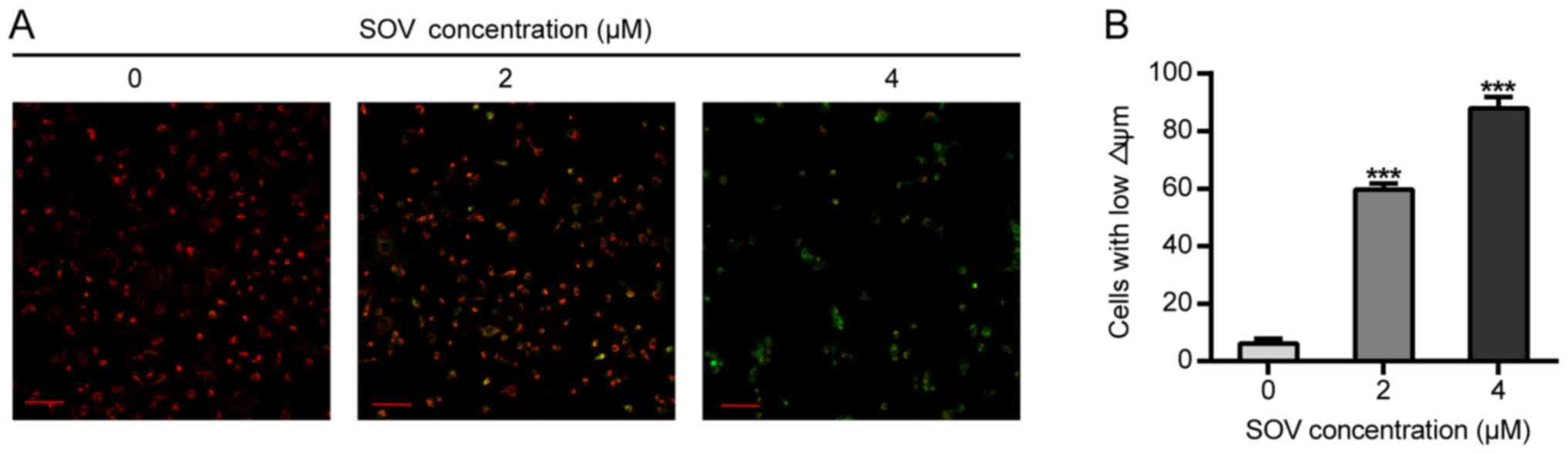
SOV diminishes the Δψm of 8505C
cells
The effects of SOV on Δψm were then assessed. The
8505C cells treated with 2 or 4 µM SOV for 48 h exhibited lower red
and higher green fluorescence than those of the control group, and
Δψm alterations were observed in a concentration-dependent manner
(Fig. 5A). In addition, 60 and 80%
of cells exhibited green fluorescence (early apoptotic) in the
groups treated with 2 and 4 µM, respectively (Fig. 5B).
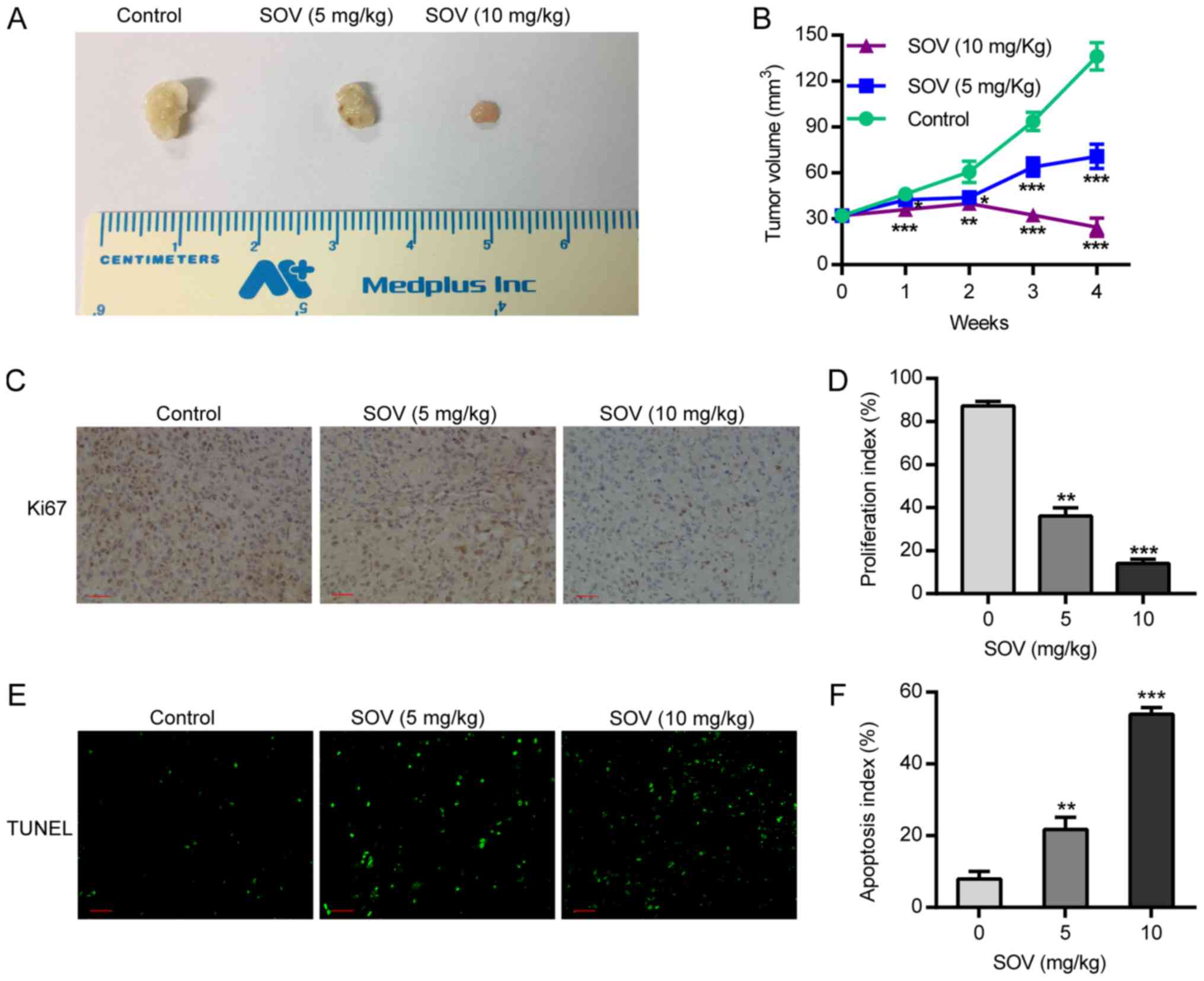
SOV inhibits xenograft tumor growth
and induces apoptosis in vivo
Following treatment of nude mice with SOV, or PBS as
a control, tumor volume was calculated by assessing tumor diameter
weekly. The results demonstrated that the tumor volume in the SOV
group was smaller than that in the control group from the first
week, and the difference in tumor volume between groups was
positively associated with treatment duration. In the two SOV
intervention groups, the tumor volume in the high-concentration
group (10 mg/kg) was smaller than that in the low-concentration
group (5 mg/kg; P<0.05, Fig. 6A and
B). This suggested that treatment with SOV significantly
inhibited tumor growth compared to that in the control group.
Therefore, SOV may inhibit the growth of ectopic ATC.
In order to detect 8505C cell proliferation after
SOV or PBS treatment, the expression of the proliferative biomarker
protein Ki-67 was detected by immunohistochemistry. The results
revealed that the expression of Ki-67 in the tumor tissues of the
SOV group was significantly lower than that in the control group.
In addition, Ki-67 expression in the high-SOV group (10 mg/kg) was
significantly lower than that in the low-SOV group (5 mg/kg;
P<0.05, Fig. 6C and D). In order
to assess the apoptosis of 8505C cells, an in situ TUNEL
assay was performed. The results demonstrated that the number of
apoptotic cells treated with SOV was higher than that in the
control group, and was positively associated with SOV concentration
(Fig. 6E and F). These results
revealed that SOV inhibited the proliferation and promoted the
apoptosis of 8505C cells in vivo.
Discussion
ATC is the most malignant type of thyroid cancer,
and is characterized by early metastasis and invasion of local
organs. Surgical treatment commonly fails as a radical treatment.
To the best of our knowledge, there is currently no efficient
chemotherapeutic drug able to control ATC development. It is
therefore crucial to develop novel treatments. In the present
study, SOV promoted the apoptosis of 8505C cells, leading to
G2/M phase cell cycle arrest. These results provided a
novel option for the treatment of undifferentiated thyroid
cancer.
Previous studies have revealed that vanadium salts
can inhibit the progression of various tumors and trigger apoptosis
of numerous cancer cells, including neuroblastoma (SH-SY5Y cells)
(13), lung cancer (A549 cells)
(14), renal cancer (HTB44 cells)
(14), prostate cancer (DU145 and
PC-3 cells) (14,15), bladder cancer (T24 cells) (16), liver cancer (HepG2, SK-Hep-1, Hep3B
and h35-19 cells) (17,18) and monocytic leukemia (U937 cells)
(21). Nevertheless, some studies
revealed that vanadium salts can promote tumor progression. For
instance, Afshari et al demonstrated that vanadium salts
cause Syrian hamster embryo cells to escape senescence and become
immortalized at a low frequency, eventually leading to the
occurrence of tumors (22). In
addition, Hwang et al reported that vanadium salts lead to
tumor formation by activating hypoxia-inducible factor 1 (23). The present results revealed that a
low concentration of SOV (0.5 µM) administered over a short period
of time (24 h) stimulated 8505C cell growth; however, increases in
the concentration and the treatment period inhibited 8505C cell
growth in a concentration- and time-dependent manner. The
IC50 values of SOV for 8505C growth were 3.76, 3.55,
3.23, 1.62, 0.85 and 0.80 µM on days 1–6, respectively. In
addition, Kordowiak et al indicated that vanadium salts at
low concentrations (0.5–1 µM) improve the morphology and viability
of H35-19 rat hepatoma cells, whereas higher concentrations (2.5
µM) of vanadium act as a cellular growth inhibitor (18). In addition to cell growth, cell clone
formation also decreased with increasing concentration. In the
present study, SOV had a dual effect on 8505C cells: Low
concentrations may promote cell growth and high concentrations
inhibited tumor cell growth. This area requires further
investigation, and lower SOV concentrations and treatments duration
should be tested.
Induction of cell cycle arrest can inhibit cell
growth. In the present study, SOV induced tumor cell cycle arrest,
resulting in the majority of cells being arrested in the
G2/M phase. Liu et al also reported that vanadium
salts induce G2/M-phase arrest by reactive oxygen
species-mediated degradation of cell division cycle 25C in PC-3
cells (15). Wu et al
reported that vanadium salts increase the phosphorylation of cyclin
B1 at Thr161 and reduce its phosphorylation at Tyr15, leading to
G2/M phase arrest in HepG2, SK-Hep-1 and Hep3B cells
(17). Nevertheless, Zhang et
al reported that vanadate is also able to induce S phase arrest
via p53- and p21-dependent pathways (24). In the present study, SOV induced
8505C cell cycle arrest in the G2/M phase in a
concentration-dependent manner. Treatment with 4 µM SOV resulted in
the accumulation of 40% of cells in G2/M phase, whereas only 10% of
cells in the control group were in G2/M phase.
Apoptosis induction is one of the main effects of
antitumor drugs. Numerous studies have demonstrated that vanadium
salts can regulate apoptosis in different ways. They can suppress
(25), enhance (26–28) or
induce (29) apoptosis. Morita et
al revealed that SOV inhibits p53-mediated apoptosis (30), whereas Günther et al reported
that SOV potentiates apoptosis induction, possibly via a
p53-independent mechanism (16). In
addition to p53, apoptosis is closely associated with protein
levels of B-cell lymphoma 2 (Bcl-2) and Bcl-extra large (Bcl-xl)
(16). Bcl-2 is located in
mitochondrial membranes and endoplasmic reticulum, and regulates
apoptosis. The present study demonstrated that SOV induced
apoptosis of 8505C cells in a concentration-dependent manner. In
addition, SOV reduced Δψm of 8505C cells, which suggested that SOV
induced cell apoptosis via the mitochondrial pathway (31).
Animal models are essential for studying the effects
of chemotherapeutic drugs, as their effects on complex systems,
including host metabolism, host defense and the endocrine system,
must be considered in order to evaluate their safety and efficacy
(32). Previous studies have
demonstrated that orthovanadate exerts significant anticancer
effects on tumor-bearing mice. Günther et al reported that
vanadium salts inhibit Ehrlich ascites carcinoma proliferation and
enhance mice survival, and observed that vanadium combined with
ascorbate at a pharmacological dose has an even better anticancer
effect (16). Wu et al
demonstrated that SOV treatment results in a clear suppression of
tumor growth in a mouse orthotopic transplantation model of
hepatocellular carcinoma (17). In
addition, it has been indicated that vanadium administration
suppresses colon carcinogenesis in rats (33). The antitumor effects of SOV on ATC
xenografts in vivo were investigated in the present study.
The results exhibited that SOV markedly suppressed tumor growth in
mice xenograft models of 8505C cells. In addition, tumor growth
inhibition was stronger at higher SOV concentrations (10 mg/kg)
than at lower concentrations (5 mg/kg). Furthermore, via the
detection of the biomarker Ki-67, results revealed that SOV
significantly inhibited tumor proliferation, and TUNEL assays
indicated that SOV also significantly induced tumor cell
apoptosis.
A previous study has revealed that SOV acts as an
antitumor agent and promotes cell death by inhibiting autophagy
(17). To the best of our knowledge,
the antitumor mechanism of SOV is currently not well understood,
and mostly involves protein phosphatase inhibition and increases in
phosphodiesterase and protein kinase activity, which are involved
in cell growth and development, DNA damage, regulation of genes and
proteins, and oxidative stress.
The present study has some limitations. It was not
possible to elucidate the underlying mechanism of the antitumor
effects of SOV, and the expression of apoptosis-associated proteins
induced by cell cycle arrest were not assessed. Furthermore, the
state of and changes in p53 in 8505C cells were not detected in the
pathogenesis of apoptosis. Subsequently, the association between
SOV-induced apoptosis and SOV-induced p53 regulation was not
investigated. Finally, no attempt was made to elucidate the reason
why low concentrations of SOV promoted 8505C cell growth. It may
therefore be important to further investigate this point by
decreasing duration and concentration of SOV treatment.
In conclusion, the present study revealed that SOV
had some important anticancer effects in ATC, including the
inhibition of tumor cell viability, induction of G2/M
cell cycle arrest and promotion of apoptosis. These results
provided a basis for further investigations on the development of
novel SOV-based chemotherapeutic drugs for the treatment of
undifferentiated thyroid cancer.
Acknowledgements
The authors would like to thank Dr Shangha Pan
(Central Laboratory, The First Affiliated Hospital of Harbin
Medical University, Harbin, China) for his assistance with the
experiments.
Funding
This study was supported by the China Medical Board
(grant no. 08-894).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QY, HJ and WD conceived and designed the study. QY,
WJ, DL, MG, KL, and LD performed the experiments. CW and QY
analyzed the data. QY wrote the manuscript.
Ethics approval and consent to
participate
All surgical procedures and animal care protocols
were approved by the Ethics Committee of the First Affiliated
Hospital of Harbin Medical University (Harbin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nagaiah G, Hossain A, Mooney CJ,
Parmentier J and Remick SC: Anaplastic thyroid cancer: A review of
epidemiology, pathogenesis, and treatment. J Oncol.
2011:5423582011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davies L and Welch HG: Increasing
incidence of thyroid cancer in the United States, 1973–2002. Jama.
295:2164–2167. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cornett WR, Sharma AK, Day TA, Richardson
MS, Hoda RS, van Heerden JA and Fernandes JK: Anaplastic thyroid
carcinoma: An overview. Curr Oncol Rep. 9:152–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Neill JP, O'Neill B, Condron C, Walsh M
and Bouchier-Hayes D: Anaplastic (undifferentiated) thyroid cancer:
Improved insight and therapeutic strategy into a highly aggressive
disease. J Laryngol Otol. 119:585–591. 2005.PubMed/NCBI
|
|
5
|
Paunovic IR, Sipetic SB, Zoric GV, Diklic
AD, Savic DV, Marinkovic J and Zivaljevic VR: Survival and
prognostic factors of anaplastic thyroid carcinoma. Acta Chir Belg.
115:62–72. 2015. View Article : Google Scholar
|
|
6
|
Perri F, Lorenzo GD, Scarpati GD and
Buonerba C: Anaplastic thyroid carcinoma: A comprehensive review of
current and future therapeutic options. World J Clin Oncol.
2:150–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim SH, Kang JG, Kim CS, Ihm SH, Choi MG,
Yoo HJ and Lee SJ: Apigenin induces c-Myc-mediated apoptosis in FRO
anaplastic thyroid carcinoma cells. Mol Cell Endocrinol.
369:130–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu XM, Jaskulasztul R, Ahmed K, Harrison
AD, Kunnimalaiyaan M and Chen H: Resveratrol induces
differentiation markers expression in anaplastic thyroid carcinoma
via activation of Notch1 signaling and suppresses cell growth. Mol
Cancer Ther. 12:1276–1287. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Che H, Guo H, Si X, You Q and Lou W:
Additive effect by combination of Akt inhibitor, MK-2206, and PDGFR
inhibitor, tyrphostin AG 1296, in suppressing anaplastic thyroid
carcinoma cell viability and motility. Onco Ther. 7:425–432.
2014.
|
|
10
|
Reeb AN, Li W, Sewell W, Marlow LA, Tun
HW, Smallridge RC, Copland JA, Spradling K, Chernock R and Lin RY:
S100A8 is a novel therapeutic target for anaplastic thyroid
carcinoma. J Clin Endocrinol Metab. 100:232–242. 2015. View Article : Google Scholar
|
|
11
|
Alonso A, Sasin J, Bottini N, Friedberg I,
Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J and Mustelin
T: Protein tyrosine phosphatases in the human genome. Cell.
117:699–711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Korbecki J, Baranowska-Bosiacka I,
Gutowska I and Chlubek D: Biochemical and medical importance of
vanadium compounds. Acta Biochim Pol. 59:195–200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tian X, Fan J, Hou W, Bai S, Ao Q and Tong
H: Sodium orthovanadate induces the apoptosis of SH-SY5Y cells by
inhibiting PIWIL2. Mol Med Rep. 13:874–880. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Klein A, Holko P, Ligeza J and Kordowiak
AM: Sodium orthovanadate affects growth of some human epithelial
cancer cells (A549, HTB44, DU145). Folia Biol (Krakow). 56:115–121.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu TT, Liu YJ, Wang Q, Yang XG and Wang
K: Reactive- oxygen-species-mediated Cdc25C degradation results in
differential antiproliferative activities of vanadate, tungstate,
and molybdate in the PC-3 human prostate cancer cell line. J Biol
Inorg Chem. 17:311–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Günther TM, Kviecinski MR, Baron CC,
Felipe KB, Farias MS, da Silva FO, Bücker NC, Pich CT, Ferreira EA,
Wilhelm Filho D, et al: Sodium orthovanadate associated with
pharmacological doses of ascorbate causes an increased generation
of ROS in tumor cells that inhibits proliferation and triggers
apoptosis. Biochem Biophys Res Commun. 430:883–888. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu Y, Ma Y, Xu Z, Wang D, Zhao B, Pan H,
Wang J, Xu D, Zhao X, Pan S, et al: Sodium orthovanadate inhibits
growth of human hepatocellular carcinoma cells in vitro and in an
orthotopic model in vivo. Cancer Lett. 351:108–116. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kordowiak AM, Klein A, Goc A and Dabroś W:
Comparison of the effect of VOSO4, Na3VO4 and NaVO3 on
proliferation, viability and morphology of H35-19 rat hepatoma cell
line. Pol J Pathol. 58:51–57. 2007.PubMed/NCBI
|
|
19
|
Reed LJ and Muench H: A simple method of
estimating fifty percent endpoints. Am J Hyg. 27:1938.
|
|
20
|
Ostrowski J, Woszczyński M, Kowalczyk P,
Trzeciak L, Hennig E and Bomsztyk K: Treatment of mice with EGF and
orthovanadate activates cytoplasmic and nuclear MAPK, p70S6k, and
p90rsk in the liver. J Hepatol. 32:965–974. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choi YJ, Lim SY, Woo JH, Kim YH, Kwon YK,
Suh SI, Lee SH, Choi WY, Kim JG, Lee IS, et al: Sodium
orthovanadate potentiates EGCG-induced apoptosis that is dependent
on the ERK pathway. Biochem Biophys Res Commun. 305:176–185. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Afshari CA, Kodama S, Bivins HM, Willard
TB, Fujiki H and Barrett JC: Induction of neoplastic progression in
Syrian hamster embryo cells treated with protein phosphatase
inhibitors. Cancer Res. 53:1777–1782. 1993.PubMed/NCBI
|
|
23
|
Hwang JT, Lee M, Jung SN, Lee HJ, Kang I,
Kim SS and Ha J: AMP-activated protein kinase activity is required
for vanadate-induced hypoxia-inducible factor 1alpha expression in
DU145 cells. Carcinogenesis. 25:24972004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Z, Huang C, Li J and Shi X:
Vanadate-induced cell growth arrest is p53-dependent through
activation of p21 in C141 cells. J Inorg Biochem. 89:142–148. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morita A, Zhu J, Suzuki N, Enomoto A,
Matsumoto Y, Tomita M, Suzuki T, Ohtomo K and Hosoi Y: Sodium
orthovanadate suppresses DNA damage-induced caspase activation and
apoptosis by inactivating p53. Cell Death Differ. 13:499–511. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gamero AM and Larner AC: Vanadate
facilitates interferon alpha-mediated apoptosis that is dependent
on the Jak/Stat pathway. J Bioll Chem. 276:13547–13553. 2001.
View Article : Google Scholar
|
|
27
|
Stewart C, Mihai R and Holly JM: Increased
tyrosine kinase activity but not calcium mobilization is required
for ceramide-induced apoptosis. Exp Cell Res. 250:329–338. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo YL, Baysal K, Kang B, Yang LJ and
Williamson JR: Correlation between sustained c-Jun N-terminal
protein Kinase activation and apoptosis induced by tumor necrosis
factor-α in rat mesangial cells. J Biol Chem. 273:4027–4034. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Figiel I and Kaczmarek L: Orthovanadate
induces cell death in rat dentate gyrus primary culture.
Neuroreport. 8:2465–2470. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morita A, Yamamoto S, Wang B, Tanaka K,
Suzuki N, Aoki S, Ito A, Nanao T, Ohya S, Yoshino M, et al: Sodium
orthovanadate inhibits p53-mediated apoptosis. Cancer Res.
70:257–265. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cossarizza A, Franceschi C, Monti D,
Salvioli S, Bellesia E, Rivabene R, Biondo L, Rainaldi G, Tinari A
and Malorni W: Protective effect of N-acetylcysteine in tumor
necrosis factor-alpha-induced apoptosis in U937 cells: The role of
mitochondria. Exp Cell Res. 220:232–240. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ray RS, Ghosh B, Rana A and Chatterjee M:
Suppression of cell proliferation, induction of apoptosis and cell
cycle arrest: Chemopreventive activity of vanadium in vivo and in
vitro. Int J Cancer. 120:13–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kanna P, Mahendrakumar CB, Chakraborty T,
Hemalatha P, Banerjee P and Chatterjee M: Effect of vanadium on
colonic aberrant crypt loci induced in rats by 1,2 dimethyl
hydrazine. World J Gastroenterol. 9:1020–1027. 2003. View Article : Google Scholar : PubMed/NCBI
|