Introduction
In the clinical setting, KRAS Proto-Oncogene, GTPase
(KRAS) genotyping is a powerful predictive tool used to stratify
patients with colorectal cancer (CRC) for anti-epidermal growth
factor receptor (EGFR) therapy (1).
KRAS is a molecular switch protein at the apex of multiple
signaling cascades that control diverse biological processes,
including cell proliferation, differentiation and apoptosis
(2). Activating KRAS
mutations, known to occur in 35–40% of colorectal tumors, result in
the expression of active oncoproteins that continuously transmit
positive growth signals to downstream effectors (3). As KRAS functions downstream of EGFR,
blocking the receptor provides little therapeutic benefit to
patients with activating KRAS mutations. KRAS testing
is therefore, imperative prior to anti-EGFR indication to avoid
administration of ineffective, potentially toxic, and highly
expensive therapy (4,5).
Activating KRAS mutations in CRC are single
point mutations found mostly in codons 12 or 13 of exon 2, and
codon 61 of exon 3 (6). These
missense mutations produce constitutively active protein products
capable of initiating cellular transformation in relevant cell line
models including primary mouse embryonic fibroblasts (MEFs)
(7), murine NIH3T3 (8) and rat Rat-1 fibroblasts (9). Although the morphological, functional,
and molecular features conferred by these mutations have already
been extensively studied, current KRAS genotyping guidelines
limited to testing only for these canonical mutants could present
possible complications (10,11). Firstly, occurrence of canonical
KRAS mutations only accounts for 30–40% of anti-EGFR
non-responders (4). Hence,
additional predictive markers, such as genetic lesions downstream
of KRAS, including B-Raf proto-oncogene, serine/threonine kinase,
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α,
phosphatase and tensin homolog or in the NRAS proto-oncogene GTPase
isoform, are currently being investigated as potential biomarkers
in association with anti-EGFR therapy, for patients with the
wild-type KRAS allele (5,12,13).
Another problem lies with the fact that the bulk of epidemiological
data used to establish the predominance of the canonical KRAS
mutations in CRC were collected from western countries and other
developed nations (14). In
addition, based on the varying epidemiological distribution of
mutations reported in different countries such as the U.K. (27.4%)
(14), Switzerland (37%) (15), Jordan (44%) (16) and China (42.2%) (17), KRAS mutation frequency and spectrum
in CRC are most likely dependent on the population under study
(14,18). In light of this paradigm,
investigations on the functional consequence of rare and novel KRAS
mutations across populations are crucial in improving CRC
diagnostics, prognostics, and therapeutics.
Between 2003 and 2010, a retrospective survey of
exon 2 KRAS mutations in 153 histopathologically verified
malignant colorectal tissue samples at the University of the
Philippines Manila-National Institutes of Health revealed novel
mutations in addition to the canonical mutations in codons 12 and
13 (unpublished data: Cutiongco-Dela Paz et al). One such
mutation was observed in KRAS exon 2, codon 31 (NM_004985.3:
c.93A>T). The significance of this mutation is currently
unknown. This missense mutation changes the amino acid at position
31 from glutamate to aspartate (p.31E>D; E31D). Another putative
activating KRAS mutation detected in CRC tissues was
reported for codon 63 in exon 2 (19). This mutation (NM_004985.3:
c.187G>A) changes a glutamic acid residue into lysine
(p.63E>K; E63K), strongly hinting at probable functional
alterations in the wild-type protein due to charge alteration at a
position critical in stabilizing the guanosine triphosphate (GTP)
hydrolysis reaction (20). Similar
to the novel mutation found in codon 31, functional
characterization studies of the codon 63 mutant, to the best of our
knowledge, have yet to be reported in the literature. In the
present study, the functional characterization of the E31D and E63K
mutations in KRAS were reported. Their effects on proliferative
rates, invasiveness, cytoskeletal organization and general
morphology were investigated.
Materials and methods
Cloning and site-directed mutagenesis
of wild-type and KRAS mutant variants
The wild-type KRAS isoform b precursor (NM_004985.3)
was re-amplified from a previous clone in pGem®-T Easy
(Promega Corporation, Madison, WI, USA) available in the
laboratory, using wild-type primers appended with restriction sites
for HindIII and NotI at the 5′ and 3′ ends,
respectively. The primers used to generate all KRAS variants used
in this study are as follows: KRAS, F
5′-GCCGTCCTGAAGCTTCGCCGGATGACTGAATATAAACTTG-3′, R
5′-GTCGTGCCGCGGCCGCGTGCCGTTACATAATTACACACTTTG-3′; KRAS E31D, R
5′-GTTGGATCATAA*TCGTCCAC-3′, F 5′-GTGGACGAT*TATGATCCAAC-3′; KRAS
E63K, R 5′-GCACTGTACTT*CTCTTGACC-3′, F 5′-GGTCAAGAGA*AGTACAGTGC-3′;
KRAS G12D, F 5′-GCCGTCCTGAAGCTTCGCCGGATGACTGAATAT;
AAACTTGTGGTAGTTGGAGCTGA*TGGCGTAGG-3′; KRAS G13D, F
5′-GCCGTCCTGAAGCTTCGCCGGATGACTGAATAT;AAACTTGTGGTAGTTGGAGCTGGTGA*CGTAGG-3′.
Site-directed mutagenesis by overlap extension PCR
was used to generate the mutants. The general PCR components and
conditions used in this study are as follows. Each reaction mixture
contained a final concentration of 1X PCR buffer
(Titanium® Taq PCR buffer, Clontech Laboratories, Inc.,
Mountain View, CA, USA), 0.125 mM of each deoxynucleoside
triphosphate (iNtRON Biotechnology, Inc., Sangdaewon-Dong,
Jungwon-Seongnam, South Korea), 2 mM each of the appropriate
forward and reverse primers, 1X Taq polymerase
(Titanium® Taq polymerase, Clontech Laboratories), and
50 ng of the cloned pTargeT™-KRAS WT template. The template DNA was
initially denatured at 94°C for 5 min. Twenty cycles of
denaturation at 94°C for 5 min, annealing at 55°C for 30 sec, and
extension at 72°C for 1 min were then performed using the C1000
Touch™ Thermal Cycler (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Two fragments of the same mutant were then fused
using overlap extension-PCR, where 25 ng of each fragment was used
as a template with KRAS-F and KRAS-R as primers. The same PCR
conditions stated above were used with the additions of a final
extension step at 72°C for 10 min after 20 cycles of amplification
to ensure the addition of A-overhangs for TA-cloning into the
pTargeT™ mammalian expression vector (Promega Corporation).
Amplified PCR products were cloned directly into pTargeT™ and
verified error-free and in the correct orientation by Sanger
sequencing.
Cell culture and transfection of
NIH3T3 cells
NIH3T3 cells were purchased from the American Type
Culture Collection (ATCC; Manassas, Virginia, USA; cat. no.
CRL-1658). The cells were maintained in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% bovine calf serum (BCS; Gibco; Thermo
Fisher Scientific, Inc.), 3 mg/ml sodium bicarbonate
(NaHCO3), 100 U/ml penicillin/streptomycin, and
incubated in a humidified atmosphere containing 5% CO2
at 37°C. Cells were seeded onto 12-well polypropylene culture
plates. At 80–90% confluency, the cells in each well were
transfected with 2 µg pTargeT™ construct, using 4 µl Lipofectamine™
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). A mammalian
expression vector containing a green fluorescent protein gene
reporter (pmR-ZsGreen1; Clontech Laboratories) was also transfected
vis-à-vis the KRAS expression constructs to approximate
transfection efficiency. For particular experiments, each pTargeT™
construct was co-transfected with pmR-ZsGreen1 in a 1:7 vector
ratio (pmiR-ZsGreen1: pTargeT) to fluorescently track cells that
were most likely transfected and are overexpressing the KRAS
variant. Transfection efficiency was assessed by fluorescence
microscopy between 24 and 72 h post-transfection, and the cells
were observed or harvested for subsequent morphological, functional
and molecular characterization experiments. Routinely and
periodically, all constructs were also tested for ability to
express the transcript and/or protein via RT-PCR and western blot,
respectively.
Morphological characterization
Morphological appearance, including size,
refringency, presence of filopodia, presence of lamellipodia, and
depolarization of transfected fibroblasts was examined under an
inverted brightfield microscope at 100× magnification 48 h
post-transfection. To quantitatively compare the transforming
effect on cellular morphology by the different forms of KRAS,
percentage of cells exhibiting transformed characteristics was
determined for each transfection setup. Each transfected well was
imaged in three random fields of view. Using the Fiji opensource
platform for biological image analysis (21), fibroblasts with aberrant morphology
were counted for each documented field. A total cell count per view
was also performed. The mean percentage of morphologically
transformed cells was then computed for all fields of view. The
values were then statistically compared among all
transfectants.
Monitoring changes in actin
cytoskeletal architecture
NIH3T3 cells were seeded into an 8-well glass
chamber slide (EMD Millipore, Burlington, MA, USA), and transfected
the next day as described previously. Forty-eight hours
post-transfection, the cells were fixed, permeabilized, and stained
with a phalloidin fluorescent conjugate. All steps were performed
at room temperature. Briefly, cells were washed twice with 1X PBS,
and fixed for 20 min using 4% paraformaldehyde. The fixative was
removed, cells were washed twice, and subsequently permeabilized
for 5 min using 0.1% Triton-X in 1X PBS. Cells were then blocked
with 1% bovine serum albumin (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) in 1X PBS for 20 min. After washing, cells were stained
for 30 min with Alexa Fluor™488 Phalloidin (Thermo Fisher
Scientific, Inc.), by adding 100 µl of the working stain solution
(0.165 µM) to each well in the dark. The cells were washed twice,
and counterstained with Hoechst 33342 for 15 min by adding 100 µl
of the working stain solution (1 µg/ml) to each well in the dark.
Stained cells were mounted in 1X PBS and sealed with a cover slip.
Slides were observed under a fluorescence microscope (Olympus
FSX100™; Olympus Corporation, Tokyo, Japan) at 400× magnification,
using the green fluorescent filter (λex/λem: 490/525 nm) to
visualize stained filamentous actin structures, and the blue
fluorescent filter (λex/λem: 355/465 nm) to visualize the
nuclei.
Scratch wound assay
Cells were seeded and transfected with a pTargeT™
KRAS construct (wild-type or mutant) in 12-well plates as
previously described. Upon reaching full confluence, the cell
monolayer was scratched with a sterile white pipette tip, washed
with 1X PBS, and fed with serum-depleted media (2.5% BCS). Bright
field and fluorescence images of the same gap area were captured in
4-h intervals starting immediately after the scratch was made. The
rate of wound closure was quantified over a 16-h interval, at which
point the gaps were still defined by drawing parallel lines along
the cell migration front and measuring the difference in gap
distance using imaging software (Olympus DP2-BSW; Olympus Soft
Imaging Solutions GmbH, Münster, Germany). Additional scratch wound
assay experiments were performed to visually track migrating cells.
In these parallel experiments, cells were cotransfected with each
pTargeT™ construct and pmR-ZsGreen1 in a 1:7 vector ratio
(pmiR-ZsGreen1: pTargeT) as previously described and wound gaps
were observed over a 40-h interval, to track the expression of the
fluorescent reporter.
Cell proliferation assay
CellTiter 96® AQueous One Solution Cell
Proliferation Assay (Promega Corporation) was used to measure the
rate of cell proliferation. Transfected cells in 12-well plates
were trypsinized at 37°C 24 h post-transfection. For each
transfection reaction, the detached cell suspension was split into
two aliquots, and cells were collected via centrifugation at 400 ×
g at room temperature for 2 min. One half of the cells was
resuspended in complete, serum-rich media (10% BCS), while the
other half was resuspended in serum-depleted media (2.5% BCS). The
cells in both suspensions were stained at room temperature with
trypan blue for 5 min and subsequently quantified using a
hemocytometer. The suspensions were diluted to a final
concentration of 2,500 cells/100 µl. An equal number of cells
(i.e., 2,500) were seeded in triplicate in two 96-well plates,
corresponding to two time points at which the number of viable
cells was measured (i.e., 48 and 72 h post-transfection). The
plates were incubated in a humidified atmosphere containing 5%
CO2 at 37°C and processed 48 and 72 h post-transfection.
To process a plate for spectrophotometric analysis, 10 µl of
CellTiter 96® AQueous One Solution was added to each
well followed by a 2 h incubation to facilitate color development.
Simultaneously, wells with predetermined cell counts were also
prepared to serve as standards. Absorbance value of each well was
measured at 490 nm by a colorimetric plate reader (FLUOstar Omega
Microplate Reader, BMG LABTECH, Cary, NC, USA). A standard curve
showing the linear relationship between absorbance value and cell
count was generated and used to quantify the number of viable cells
in each well. The cell counts of all transfectants were compared at
both time points and statistically analyzed.
ELK1, ETS transcription
factor-transactivation domain (ELK-TAD) luciferase reporter
assay
The 293 cells with an ELK-TAD luciferase reporter
assay system (Signosis Inc., Silicon Valley, San Francisco, USA;
cat. no. SL-0040-FP) was used to measure the ability of the KRAS
mutants to activate the mitogen-activated protein kinase (MAPK)
signaling pathway. The cell line is stably integrated with a fusion
protein of the ELK-transactivation domain and Gal4. Activation of
ELK-TAD through the MAPK pathway drives the expression of firefly
luciferase, which is regulated by Gal4-UAS. Cells were maintained
in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.) and 100 U/ml
penicillin/streptomycin, then incubated in a humidified atmosphere
containing 5% CO2 at 37°C. For the luciferase assay,
10,000 cells were seeded into each well of a 96-well plate. Cells
were then transfected with 200 ng of the pTargeT-KRAS constructs
per well using Lipofectamine™ 2000. Twenty-four hours
post-transfection, the media was changed to DMEM supplemented with
4% FBS to serum deprive the cells, which were then allowed to grow
a further 24 h prior to the assay. Cells were lysed by adding 20 µl
of Passive Lysis Buffer (cat. no. E1941; Promega Corporation) into
each well, followed by a 20-min incubation at room temperature. The
cell lysates were transferred into an opaque 96-well white plate,
and 100 µl of the Luciferase Assay Reagent (Promega Corporation;
cat. no. E1483) was added into each well. The plate was read using
a luminometric plate reader (FLUOstar Omega Microplate Reader, BMG
LABTECH) under the pre-set Firefly luciferase settings. Readings
were normalized against the relative cell count measured through
fluorescent staining of the cells' DNA using the CyQUANT NF Cell
Proliferation Assay kit (Thermo Fisher Scientific Inc.; cat. no.
C35007), according to the manufacturer's protocols, on transfected
cells in parallel wells. Cells in these wells had their media
removed, and 100 µl of the CyQuant NF reagent was added per well.
The plate was then incubated at 37°C for 1 h. Fluorescence
measurements were performed using a plate reader (FLUOstar Omega
Microplate Reader, BMG LABTECH) with an excitation wavelength of
485 nm, and emission detection at 530 nm.
Bioinformatics-based prediction of
potential functional impact of mutations
The functional impact of KRAS E31D and E63K
mutations was assessed using three sequence-based prediction
platforms: Polymorphism Phenotyping (POLYPHEN-2; version 2)
(22), Sorting Intolerant From
Tolerant (SIFT; version 5.2.2) (23)
and Mutation Assessor (release 2) (24).
The impact of the amino acid changes on KRAS protein
structure was evaluated by building homology models of the mutants
based on the solved 3D structure of human KRAS (PDB: 3GFT). As 3GFT
harbors the Q61H mutation, the wild-type structure was initially
prepared by reverting this activating mutation in silico.
The mutants were built in Accelrys Discovery Studio Client 2.5
(Dassault Systèmes BIOVIA, San Diego, CA, USA) using the MODELER
protocol, which scores and optimizes the conformation of a mutated
residue and its neighbors within a defined structure (15). The global main chain root-mean-square
distance (RMSD) of each mutant model was then computed based on
sequence alignment with the wild-type.
The putative effect of the mutants on GTP binding
was assessed by simulating ligand-receptor interactions. First, the
GTP ligand was prepared by generating multiple docking poses based
on optimum conformations, tautomers, and isomers at the
physiological pH. Similarly, the mutated KRAS structures were
prepared using a general protocol that cleans the protein
structure, removes water molecules, and optimizes the side-chain
conformation for residues within the binding site (25). A minimization step was also performed
to the cleaned protein models in order to minimize the energy of
the structure, relax the protein conformation, and remove steric
overlaps that may produce inaccurate interactions. Using the
binding site coordinates defined by the reference solved crystal
structure, the generated ligand poses were docked within the
binding sphere of each KRAS protein variant. The CDOCKER algorithm
was used to generate the docked conformations of the ligand
(26). The calculated docking scores
were compared among the different forms of KRAS.
Statistical analysis
All quantitative data were presented as mean ±
standard error. One-way analysis of variance with post hoc Tukey's
Honest Significant Differences was performed to compare and
evaluate the quantitative results of the cellular assays. A P-value
of 0.05 was used for all assays.
Results
Rare mutants E31D and E63K induce cell
rounding, refringency, and cytoplasmic shrinkage
The flattened phenotype of normal non-transformed
NIH3T3 fibroblasts was predominant in cells transfected with the
empty expression vector. Non-transformed cells are reasoned to be
flat and well-spread because of the appropriate and well
established adhesion of cells onto the culture substrate (27). On the other hand, transformed NIH3T3
cells are characteristically smaller, rounder, and highly
refractile compared with their non-transformed counterparts
(28,29). Transformed cells also possess
pronounced pseudopods and cellular protrusions (8). These characteristics likely reflect
cytoskeletal reorganization resulting in poor surface adhesion and
promotion of cellular motility.
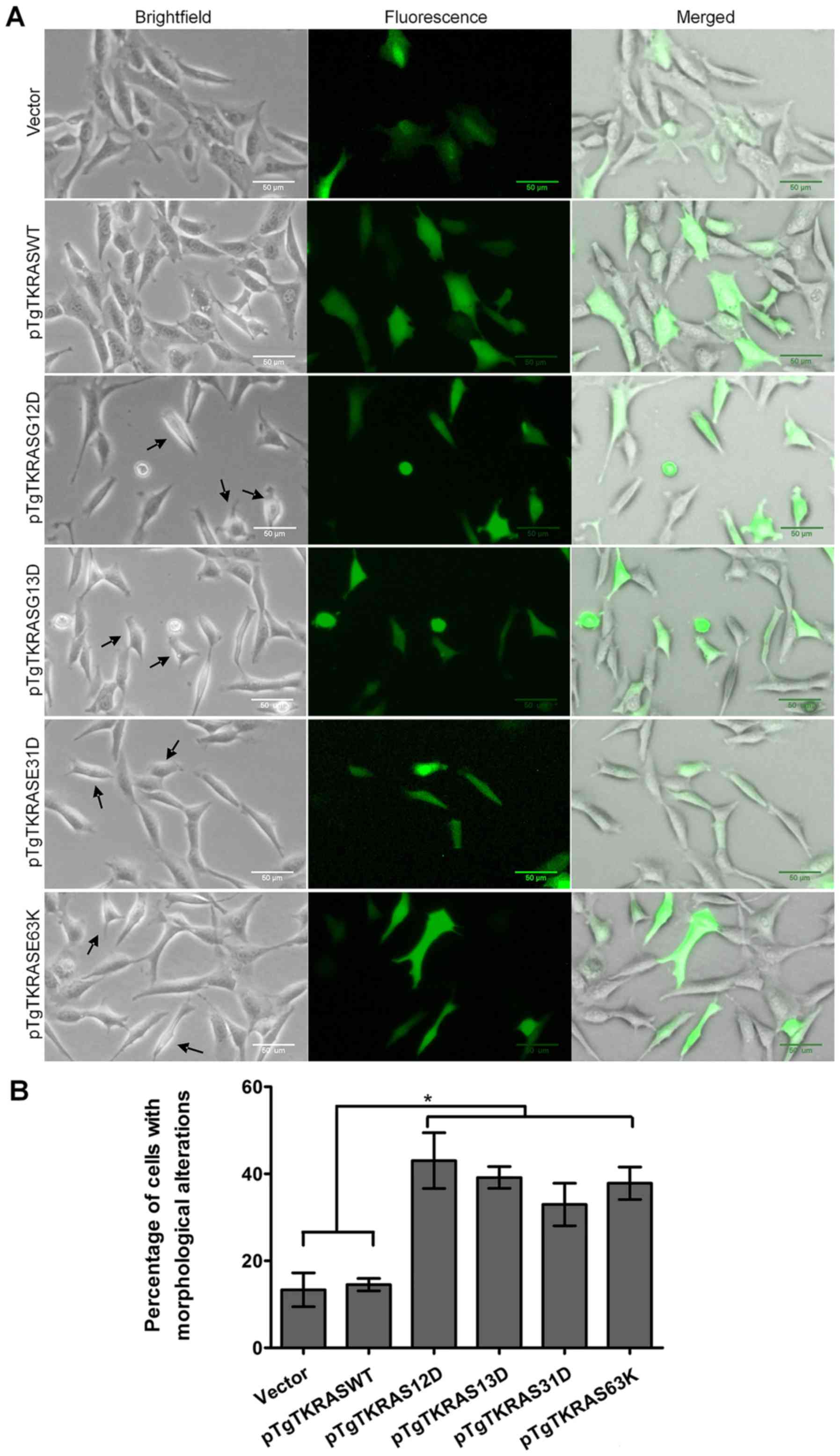
Compared with the vector control, no significant
morphological changes were seen in cells overexpressing the
wild-type protein (Fig. 1). This
observation is corroborated by previous reports that amplified
expression of normal RAS is usually not sufficient for cellular
transformation and therefore produces no discernible morphological
changes in NIH3T3 cells (28,29).
As expected, a significant fraction of cells
transfected with constitutively active forms of KRAS, including
KRAS G12D and KRAS G13D, showed morphological features that mirror
those of transformed fibroblasts, as the cells became smaller,
rounder, and more refringent (Fig.
1). Cytoplasmic shrinkage was also apparent in the transfected
fibroblasts. A significant percentage (P<0.05) of cells
transfected with either the KRAS E31D (33%) or KRAS E63K (38%)
mutant also share the morphology typical of transformed cells,
hinting at the transformation potential of the rare mutations.
While equal cell densities among the treated cells could not be
guaranteed at the observation time-point, considering the combined
cytotoxic and proliferative effects of transfection and KRAS
overexpression, respectively, the trend of increased morphological
aberration in KRAS-mutant overexpressing cells was highly
reproducible. In all our experiments it was also ensured that
morphology assessment was performed before cells reached full
confluence and that any perceived differences in cell densities can
be attributed to the aforementioned morphological changes.
Cytochemical staining of filamentous
actin in cells overexpressing KRAS mutants reveals altered
cytoskeletal organization
Activating RAS mutations are known to affect
actin organization, consequently facilitating the onset of cellular
transformation (30). To determine
if the rare KRAS mutations under study have the same
deleterious impact on cytoskeletal dynamics similar to canonical
mutants, filamentous actin of transfected cells was visualized via
fluorescence staining. Transfected cells cultured in 8-well chamber
slides were washed, fixed and permeabilized prior to staining.
High-affinity binding of phalloidin to actin was used to visualize
the actin cytoskeleton. Hoechst was used to counter-stain the
nuclei.
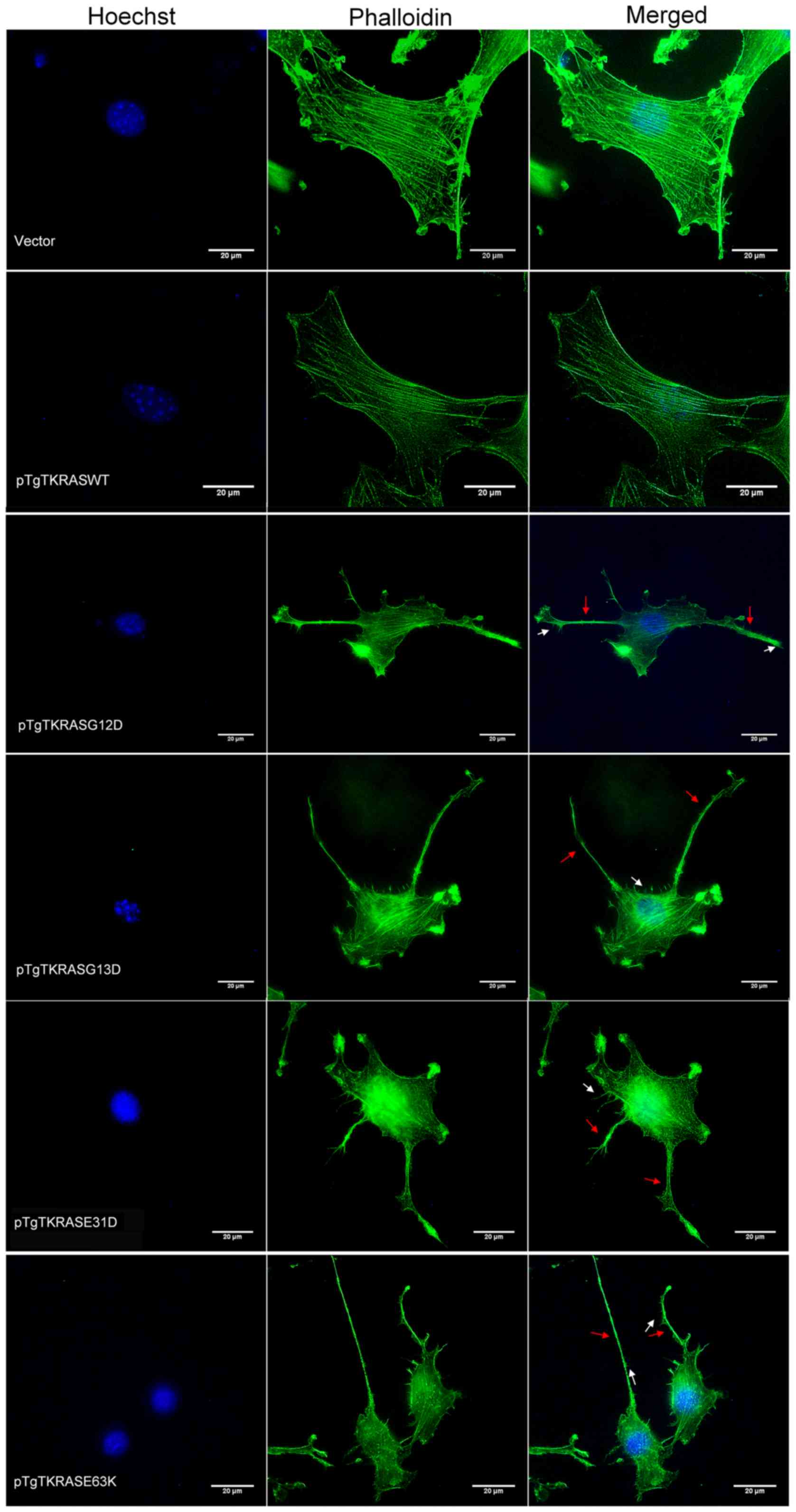
Whereas the vector control- and wild-type-expressing
cells exhibited prominent and well-oriented stress fibers, all
canonical- and rare mutant-expressing cells developed poorly
defined and thin stress fibers (Fig.
2). In KRAS E31D- and KRAS E63K-transfected cells, as well as
in the positive controls, cell depolarization was apparent with
extensive membrane ruffling and lamellipodia formation around the
cell periphery. Formation of filopodia and pseudopodia were also
increased. Altogether, these cytoskeletal features are typical of
contractile and motile cells with highly dynamic actin networks,
suggesting the effect of the rare KRAS mutants on cellular
migration.
KRAS E31D and E63K mutations confer
increased proliferative capacity
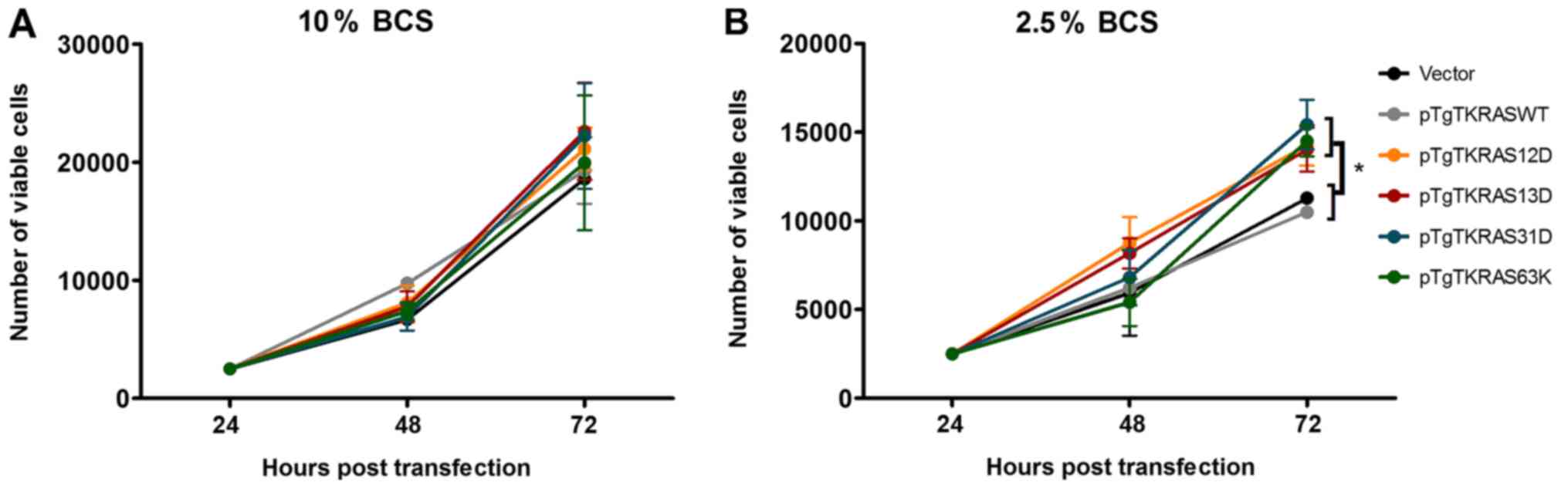
The proliferation rates of cells transfected with
the KRAS E31D and KRAS E63K mutants were assessed and compared with
the vector, wild-type, and canonical mutant controls (Fig. 3). This was done by seeding an equal
number of cells that have been transfected for 24 h onto 96-well
plates in triplicates and quantifying the number of viable cells
via a colorimetric reagent 48 and 72 h post-transfection. At high
serum concentration (10%), all transfectants showed comparable
number of viable cells at the observed time points (Fig. 3A). In contrast, in serum-depleted
(2.5%) conditions, cells expressing KRAS G12D, KRAS G13D, KRAS
E31D, and KRAS E63K all showed a significant increase in cell count
72 h post-transfection (Fig. 3B).
The assays also indicated that the absolute number of viable cells
was, as expected, dependent on serum concentration.
Cells expressing canonical and rare
KRAS mutants show increased cell migration rates in scratch wound
assays
Given the impact of KRAS E31D and KRAS E63K
overexpression on cellular morphology and cytoskeletal actin,
scratch wound assays were performed to investigate cell motility
and its association with the expression of the rare mutants. The
transfected cell monolayers were grown to full confluence, upon
which a scratch was introduced using a sterile pipette tip. The
cultures were then shifted to serum-depleted media (2.5%) and the
wound fields were observed in a time-course manner. In some trials,
pmR-ZsGreen1 was co-transfected with the expression vectors to
ascertain, through expression of the fluorescent protein, whether
migrating cells were positively transfected or otherwise.
Wound gaps were routinely observed at 4-h intervals
until the confluent monolayer for each transfected culture was
restored. As it became difficult to delineate the opposing
migrating fronts beyond a certain time point, as cells started to
extensively populate the gap wound from either side, the rate of
wound closure was calculated 16 h post-scratch (Fig. 4A). The gap closure rates in cells
expressing KRAS E31D and KRAS E63K mutants were significantly
higher (P<0.05) compared with the vector and wild-type controls,
and comparable with cells expressing the constitutively active
forms of KRAS (Fig. 4B). Further, in
co-transfected setups, fluorescent cells were particularly
prominent midway the closing gap for cultures transfected with
either KRAS E31D or KRAS E63K (Fig.
4C). The same was observed for cultures transfected with either
canonical mutant. Although non-fluorescent cells were also present
in the gap it is highly likely that these cells harbor the KRAS
construct regardless, since cells were transfected with a 1:7
vector ratio (pmiR-ZsGreen1: pTargeT). Altogether, these results
imply that positively transfected cells were more likely to migrate
into the gap, suggesting the functional consequence of the
mutations on cell motility.
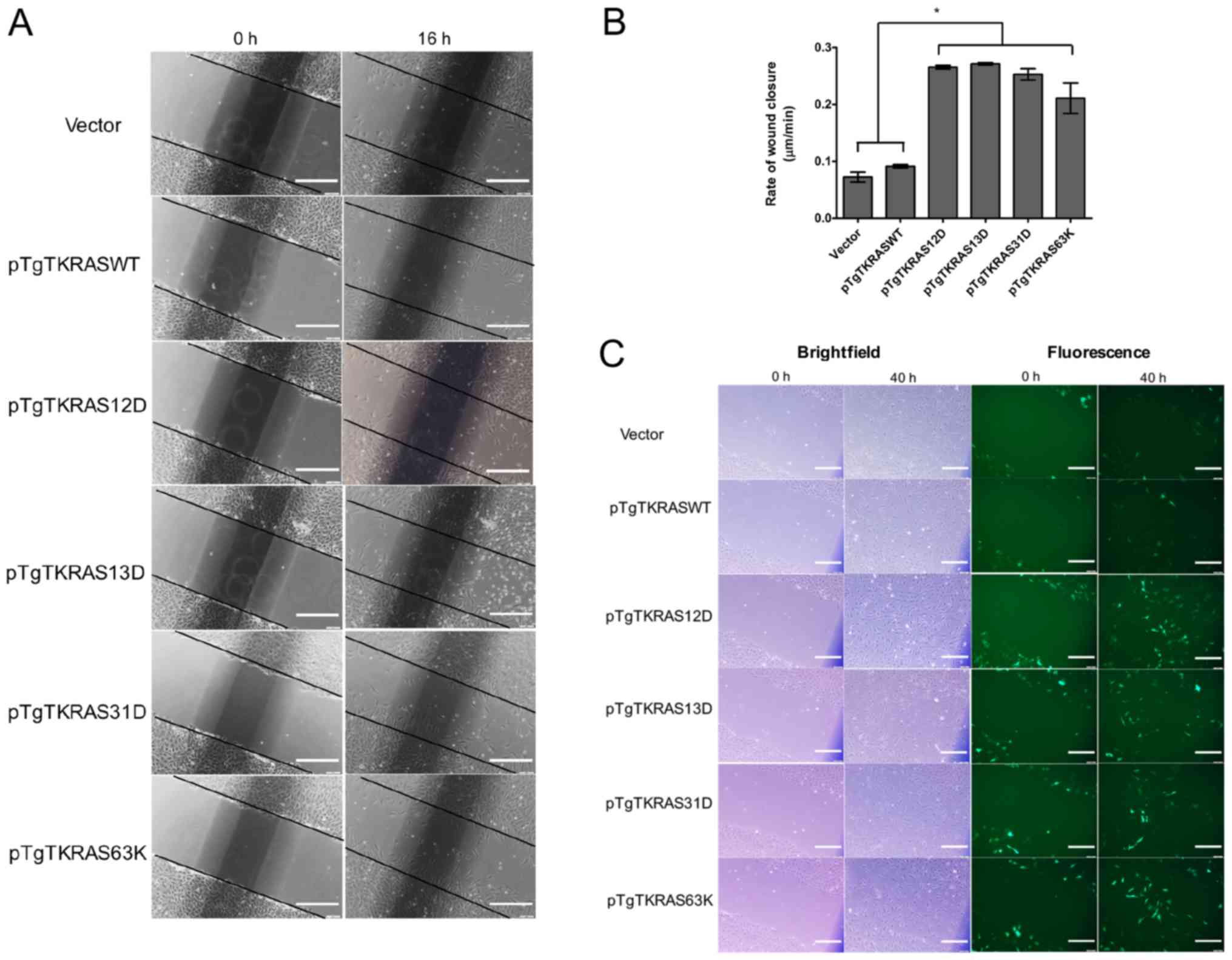 | Figure 4.Overexpression of canonical and novel
KRAS mutants resulted in increased cell migration in vitro.
Scratch wound assays were conducted using transfected NIH3T3 cells
grown in 2.5% serum. (A) Micrographs of wound fields directly after
scratching the monolayer (0 h), and 16 h post-scratch. Compared
with the controls (i.e., vector, pTgTKRASWT), narrower scratch gaps
were noticeable for cells overexpressing KRAS mutant variants.
Scale bars: 100 µm. Representative of three trials. (B) The
distance between lines approximating the cell migration front was
measured for each setup at time points 0 and 16 h, and the rate of
front migration was calculated. Wound closure was significantly
faster for cells overexpressing the mutant protein. *P<0.01. (C)
NIH3T3 cells positively transfected with mutant KRAS constructs
have an increased tendency to migrate into wound gaps. Cells used
for scratch wound assays were co-transfected with a transfection
marker, pmiR-ZsGreen1. Migrating fibroblasts were tracked directly
after scratching the monolayer (0 h), and 40 h post-scratch to
verify that cells facilitating the gap closure are positively
transfected. Scale bars: 100 µm. WT, wild-type; KRAS, KRAS
Proto-Oncogene, GTPase. |
In contrast, at 40 h following wounding, even fewer
fluorescent cells were spotted halfway into the scratch wound of
cells transfected with either the empty vector or wild-type KRAS,
and a conspicuous gap could still be seen under the green
fluorescent filter. Because transfection efficiency was consistent
among treatments based on fluorescent quantification using
fluorescence microscopy (data not shown), this underscores the
comparable tendency for migration of empty vector- and
KRAS-WT-transfected cells, adding evidence to the limited
transforming effect of wild-type KRAS overexpression. For the empty
vector and wild-type KRAS-transfected cultures, non-fluorescent
cells were able to migrate into the gaps, due to the relatively
long observation period. These cells most likely reached their
terminal positions at reduced velocities, as indicated by the data
on wound closure rates.
Effect of KRAS mutations on ELK
transactivation
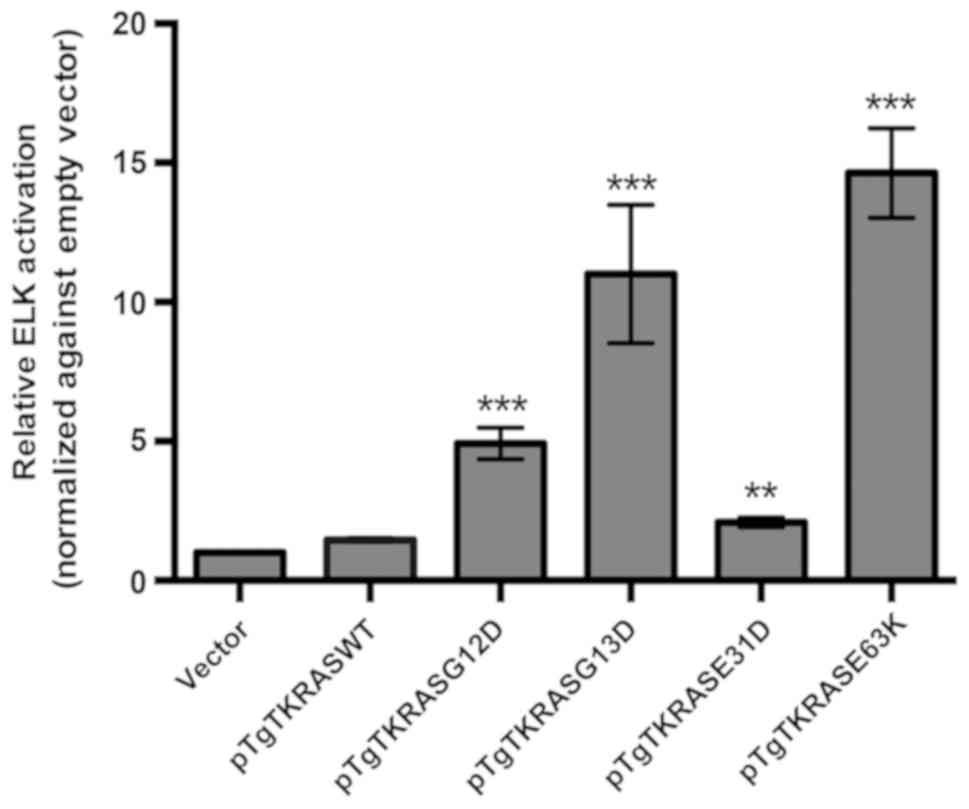
To determine whether KRAS E31D and KRAS E63K have an
effect on downstream targets, an ELK-TAD luciferase reporter assay
was employed. ELK-1 is an established nuclear target and one of the
best characterized substrates of ERK, a downstream effector of KRAS
(31,32). The different KRAS constructs were
transfected into a 293 cell line harboring a stably integrated
ELK/luc reporter to monitor their effects on ELK-1 transactivation.
As shown in Fig. 5, the canonical
mutants KRAS G12D and KRAS G13D, as well as the novel mutant KRAS
E63K, consistently stimulated ELK-1-mediated transcriptional
response. KRAS E31D, on the other hand, only showed stimulation of
ELK-1 in two out of three trials and only achieved significance in
one trial (P<0.05). As discussed below, in silico
analyses suggested that the effects of this mutation may be benign.
However, given the consistent and comparably aggressive phenotypic
readout of this mutant compared with KRAS G12D and KRAS G13D in the
different cancer hallmark assays, this may mean that KRAS E31D
mediates its effects not through ELK-1, but via another downstream
target.
Functional consequences of KRAS E31D
and KRAS E63K mutations as predicted through in silico protein
modelling and docking simulations
The effect of E31D and E63K mutations on KRAS GTPase
function was predicted using three bioinformatics platforms:
PolyPhen-2 (22), which utilizes a
physical and comparative approach in relating structure to
function; SIFT (23), which yields
predictions based on sequence homology and physical properties of
pertinent amino acids; and Mutation Assessor (24), which relies on the evolutionary
conservation of the mutated amino acid in protein homologs. In all
platforms, E31D was predicted to have null to minimal effect on
protein function, whereas E63K scored high ratings of potential
functional impact in PolyPhen-2 and SIFT (Table I). However, these predictions only
analyze the protein in isolation, precluding the possible effects
of a mutation on the binding of regulators, including adaptors,
scaffolds, and other proteins in a signalosome, in addition to
effectors.
 | Table I.Functional effects of KRAS E31D and
E63K as predicted by multiple bioinformatics platforms. |
Table I.
Functional effects of KRAS E31D and
E63K as predicted by multiple bioinformatics platforms.
| Mutation | Polyphen-2
(score) | SIFT (score) | Mutation assessor
(score) |
|---|
| E31D | Benign (0.000) | Tolerated
(0.06) | Neutral (0.49) |
| E63K | Probably
damaginga (0.985) | Affect protein
function (0.00) | Low (1.485) |
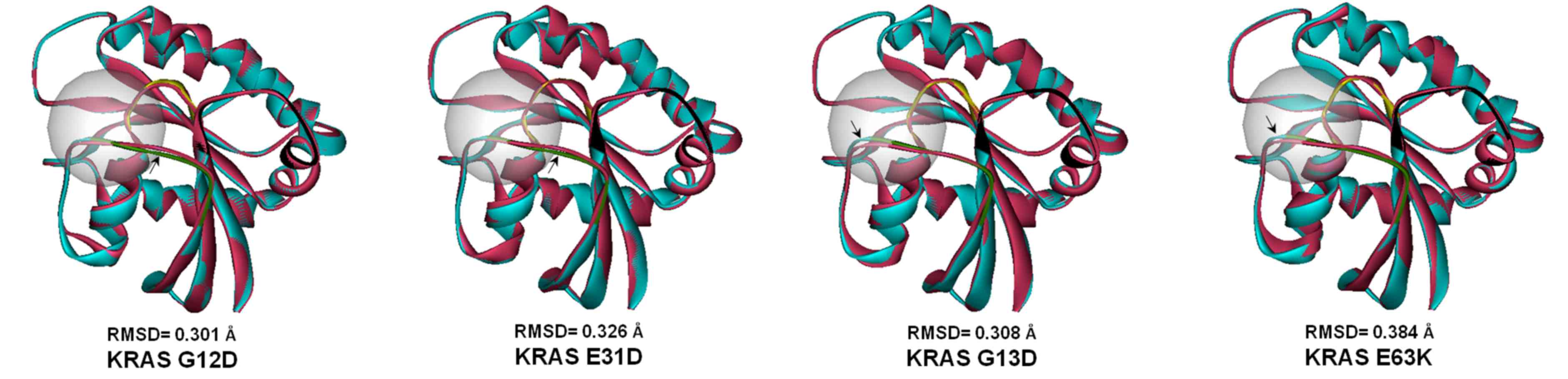
Similar to the canonical mutants KRAS G12D and KRAS
G13D, both KRAS E31D and KRAS E63K have minimal effect on the
overall structure of KRAS, based on the calculated RMSD values
(Fig. 6). However, all mutants
resulted in apparent conformational changes in a region critical to
the GTPase activity of the protein, the mobile loop switch I, whose
relative orientation dictates the activity status of KRAS. In both
KRAS E31D and KRAS E63K, noticeable repositioning of the switch I
loop was observed. The canonical mutants KRAS G12D and KRAS G13D
resulted in a reorientation of the same loop. Perturbations caused
by the canonical mutants are corroborated by crystal structures
solved previously (33).
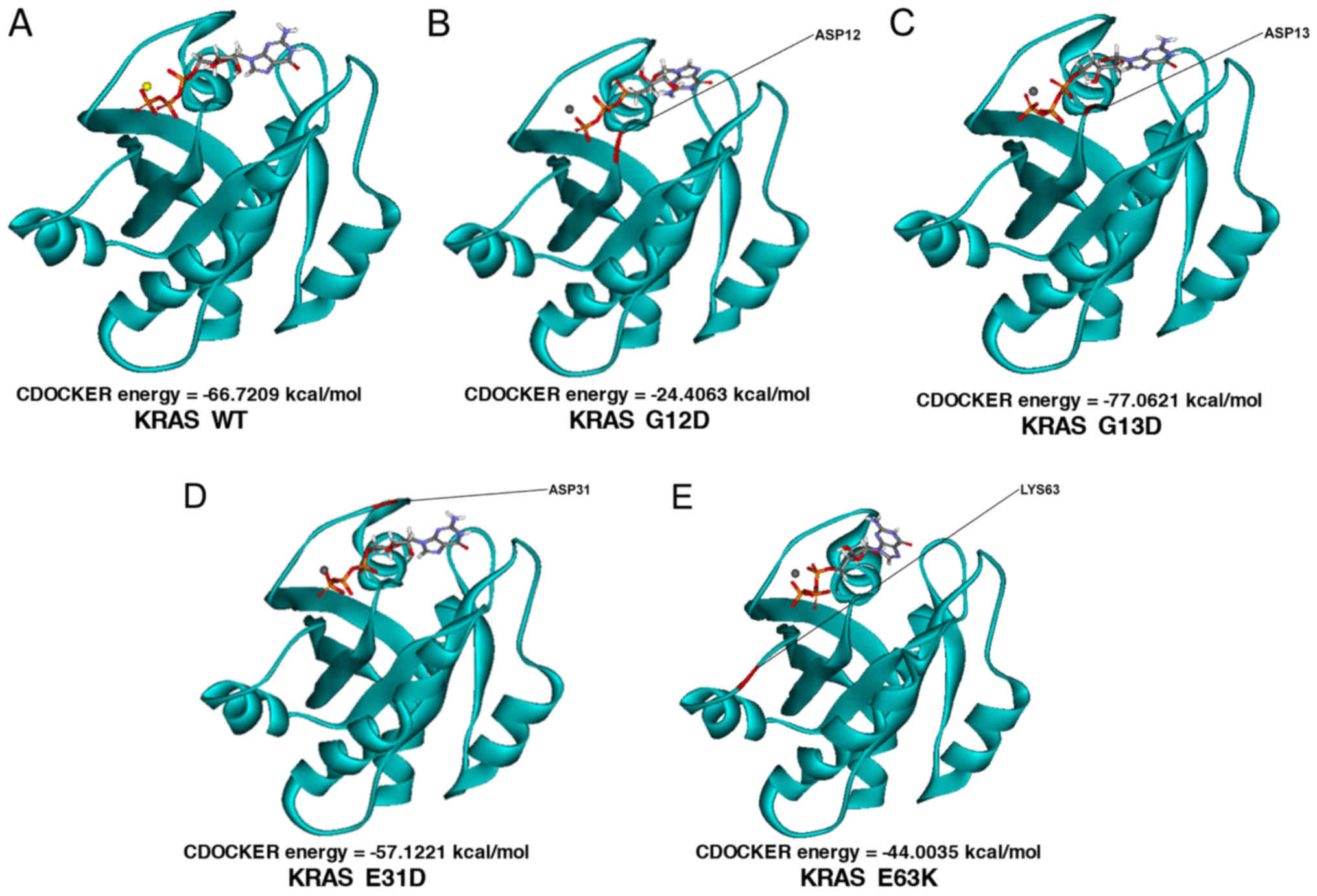
Docking simulations using CDOCKER were performed to
determine the effect of mutations on ligand binding. CDOCKER
functions on an algorithm wherein the receptor is held rigid while
the ligand is allowed to flex during pose refinement. The homology
mutant models of KRAS E31D, E63K, G12D, and G13D were prepared and
minimized prior to docking. A total of 64 poses of the pH-optimized
GTP ligand were generated for the simulations, with considerations
to possible conformations, tautomers, and isomers. For each variant
of KRAS, 192 GTP-receptor complex poses were scored. As shown by
the minimum docking scores, KRAS G13D had the most favorable
interaction with GTP, followed by the wild-type, E31D, E63K, and
finally, the G12D mutant (Fig. 7).
The exergonic CDOCKER energy values for all KRAS variants predicted
unimpeded and highly favorable ligand binding. In all cases, the
highly negative triphosphate moiety of GTP was correctly oriented
toward the positively charged metal cofactor within the binding
pocket.
Discussion
Based on the varying epidemiological distribution of
mutations reported in different countries, KRAS mutation
frequency and spectrum are most likely dependent on the population
under study (14,18). Even with the 2–4-fold increase in the
incidence of the disease in Asian countries, molecular statistics
considering the regional population is lacking (18). It is therefore paramount to
investigate rare and novel KRAS mutations detected in the
local population in association with their impact on CRC
diagnostics, prognostics, and therapeutics.
Another reason for examining the functional
consequences of novel KRAS mutations is intratumor
heterogeneity. This suggests that cancer cell clones coexisting
within a tumor present a divergent set of genetic aberrations
(34). Cells with similar phenotypes
may also segregate and enrich a particular region of the tumor,
leading to sampling bias and subsequent underestimation of
important mutations and biomarkers (35). The genetic variation within a tumor
has also been cited as a cause of targeted drug resistance:
Low-frequency cells carrying resistance-conferring mutations
eventually outgrow the dominant clones susceptible to the therapy
(34,36). Taking the aforementioned into
consideration, the presence of rare mutations could ultimately be
predictive of treatment outcome.
Further underscoring the significance of
low-frequency KRAS mutations in CRC is a recent report on
the non-responsiveness of patients harboring mutations beyond the
canonical hotspots to cetuximab treatment (37,38).
Although a definitive correlation could not be established between
the occurrence of the reported rare mutations and refractory cancer
due to low patient numbers, there is sufficient evidence to suggest
that these mutations are significant in predicting tumor
susceptibility to anti-EGFR treatment (39,40).
The functional impact of two previously reported
KRAS mutations was assessed in this study. The first
mutation, E31D in exon 2 (NM_004985.3: c.93A>T; p.31E>D), was
surveyed from a genetic screen of histopathologically positive CRC
tissues from Filipino patients at the Philippine General Hospital.
Alternative codon 31 mutations in KRAS have also previously
been reported in CRC (41),
papillary thyroid carcinoma (42),
and adrenal tumors (43). To the
best of our knowledge, the functional effects of these mutations
have yet to be investigated. The other mutation, E63K in exon 3
(NM_004985.3: c.187G>A; p.63E>K), is curated in the COSMIC
database and has been reported to occur in lesions of the large
intestine, genital tract, thyroid, lymphoid tissue, and medulla
(19). Eight separate E63K mutations
were cited in COSMIC, and three were identified in CRC (19). The results obtained in the current
study demonstrated that, the E63K mutation scored high ratings of
potential functional impact from two predictive amino acid
substitution tools (i.e., PolyPhen-2 and SIFT). Despite the rarity
of these KRAS mutations, the aforementioned reports suggest the
significance of amino acid positions 31 and 63 in the normal
cycling of the protein, and the potential transformative effects
brought about by mutations in the two loci.
In the present study, the transforming potential of
the rare KRAS mutations E31D and E63K were revealed based on
morphological and functional characterization of NIH3T3 cells
overexpressing the mutant oncoproteins. The murine embryonic
fibroblast NIH3T3 were selected as the heterologous host, due to
its well documented phenotypic response upon overexpression of
activating RAS genes (44–48). Unlike other common CRC lines
including HCT-116 (G13D), DLD-1 (G13D), SW480 (G12V), and SW948
(Q61L), NIH3T3 has a wild-type KRAS background, which is necessary
for the evaluation of putative activating mutants. Finally, unlike
a number of cell lines and primary cultures that require
complementation of cooperative oncogenes, NIH3T3 cells readily
exhibit oncogenic phenotypes upon lone expression of a transforming
RAS variant (49). A caveat
in our approach to functionalize the novel mutants is the use of a
transient overexpression system in the cellular assays. To mitigate
the potential confounding effects of inconsistent transfection
efficiencies, the robustness of the procedure was ensured by
co-transfection with pmiR-ZsGreen1, which encodes a green
fluorescent protein marker, followed by assessment by fluorescence
microscopy.
Cells overexpressing the rare mutants were indicated
to share phenotypic features with G12D or G13D-transfected cells in
terms of morphology, actin cytoskeleton organization, proliferative
capacity, and migration rate, validating the hypothesis that the
rare mutants could induce cellular transformation. The
non-transforming effect of wild-type KRAS transient overexpression
was also observed in the cellular assays, which was in accordance
with previous reports (28,29).
Enhanced cell motility by E31D and E63K
overexpressing cells was only observed in serum-depleted
conditions. In serum-rich media, all transfected cells showed
comparable rates of wound gap closure. Mitogens, cytokines, and
growth factors present in the serum may have saturated cell surface
receptors, which were significant to effector pathways associated
with cell motility and proliferation, including members of the ErbB
family of receptors (50). On the
other hand, utilizing serum-depleted media allows cells to
synchronize and enter the quiescent G0/G1
phase, preventing the mistaken collection of wound gap closure data
on actively proliferating cells (51). Working with a serum-depleted
background ensures differential pressures of the wild-type and
mutant KRAS oncoproteins on associated signaling pathways, without
the unwanted contribution of excess growth factors.
The majority of viable cells observed for both E31D
and E63K transfectants was further evidence of the transforming
potential of the rare mutants. This increase in proliferative
capacity may be attributed to the activation of the Raf-MEK-ERK
signaling pathway resulting from ectopic expression of either KRAS
E31D or E63K. Parallel experiments performed in high-serum
conditions yielded comparable cell counts across all transfectants
at the same time points, underlining the importance of the
background medium in obtaining biologically significant readout
from such sensitive assays. Notable differences in cell counts were
observed only upon reaching the 72-h post-transfection time point,
suggesting the time required to accelerate the mobilization of
molecular and cellular components necessary for cell proliferation
that are contingent on KRAS activation.
The ability of both canonical and novel KRAS mutants
to activate ELK-1, a downstream nuclear target and
well-characterized substrate of ERK, was also assessed through the
ELK-TAD luciferase reporter assay. Similar to the canonical mutants
KRAS G12D and KRAS G13D, the novel mutant KRAS E63K showed
activation of ELK, compared with KRAS WT and vector-only control.
In contrast, KRAS E31D only showed activation of ELK in two of
three trials and achieved significance only in one. While this may
be consistent with our in silico predictions of a benign
phenotype for E31D, the results of cellular assays as described
above prove otherwise. Therefore, it is reasonable to speculate
that KRAS E31D may be mediating its effects via a different
downstream effector. ERK has a plethora of downstream targets,
including transcription factors c-Fos (52), c-Jun (53), cytoskeletal elements (54) and intercellular domains of membrane
receptors (55). Therefore, it may
be more instructive to assess the effect of the novel KRAS mutants
on the more upstream effectors MEK and ERK, through phosphorylation
assays. Given that KRAS also signals via at least two other
pathways implicated in colorectal cancer, the effects of the
mutants on the PI3K-AKT-mTOR and RAL-GEF pathways should also be
assessed (56,57).
The present study examined the possibility of
structural changes elicited by the non-hotspot mutants on the KRAS
protein by performing mutation modeling and simulating
protein-ligand docking. Based on the generated global RMSD values,
incorporation of either E31D or E63K mutation elicits minimal
changes on the overall structure of KRAS. The constancy of the
phosphate binding loop structure also indicates intact GTP-binding
capability of the mutants. However, in silico analysis
revealed perturbations introduced by the rare mutations in the
switch I loop. The spatial influence of E31D on switch I may stem
from its position near the N-terminal flank of the loop, while the
positively charged amine side chain of E63K, as part of the switch
II loop, could introduce a change in the local electrostatic
environment that may affect the conformation of switch I
indirectly. It is speculated that the altered conformation in such
close proximity to the GTP-binding cleft interferes with GTP
hydrolysis via steric effects.
The unique range of docking energy values generated
upon simulation of GTP binding to the active site of the wild-type
and mutant KRAS also hint at perturbation of protein structure. The
negative CDOCKER energy values point to the spontaneous binding of
GTP to the active site of wild-type and mutant KRAS. In general,
the following trend in terms of favorability of binding (i.e., most
negative docking score) was observed:
G12D<E63K<E31D<wild-type<G13D. The trend
G12D<wild-type<G13D was previously reported using the
iGEMDOCK algorithm (58). These
observations indicate that the oncogenic potential of a certain
KRAS mutant does not solely rely on the strength of nucleotide
binding. Although loss-of-function mutations can be identified
through binding studies, the more important and more informative
parameter in considering an activating mutation in KRAS, is its
effect on the rate of GTP hydrolysis (33). Future docking simulations of
representative GTPase-activating proteins which facilitate GTP
hydrolysis, guanine nucleotide exchange factors which modulate
nucleotide exchange from inactive RAS-GDP to active RAS-GTP, and
other signal mediators to the KRAS mutants would provide additional
insight with respect to the biochemical interactions perturbed by
the amino acid alterations that ultimately lead to the observed
transformed-like phenotypes in NIH3T3 cells. These aforementioned
binding studies would be particularly interesting for the rare
mutants, considering that amino acid positions 31 and 63 are in
close proximity with the switch loops critical to GTP
hydrolysis.
Scaffold proteins also spatially regulate the
activation of RAS effectors by providing platforms for assembly,
and localizing signaling molecules to specific sites in the cell
(59,60). A number of these proteins also
protect activated signaling molecules from inactivation. For
example, RAS-dependent activation of Raf in the ERK module has been
shown to be accelerated, due to the binding of a scaffold protein,
Shoc2/SUR-8, to RAS-GTP, reserving it for subsequent Raf
interaction (61). These
interactions emphasize the complex molecular mechanisms underlying
signal transduction through RAS. In silico modelling of the
ternary structures involving the ligand, receptor, regulators,
and/or scaffolds is therefore recommended. The complete and more
definitive assessment of the structural impact of the mutants can
only be achieved through x-ray crystallography and nuclear magnetic
resonance techniques. It is crucial that computational and
predictive analyses be complemented by cellular assays, as
performed in the present study, to elucidate the functional
consequences of a mutation within a proper physiological
context.
Lastly, the oncogenic capacity of the novel KRAS
mutants E31D and E63K requires further validation through in
vivo animal studies and its potential association with clinical
outcomes determined, in order for them to be useful as predictive
biomarkers for non-responsiveness to anti-EGFR therapy.
Acknowledgements
The authors thank Dr Junie Billones (Department of
Physical Sciences and Mathematics, University of the Philippines,
Manila, Philippines) for advice on protein modeling, Mr. Joshua
Malapit (Disease Molecular Biology and Epigenetics Laboratory,
National Institute of Molecular Biology and Biotechnology,
University of the Philippines, Diliman, Philippines) for helpful
discussions on protein-ligand docking, and Ms. Isabelle Viola
(Disease Molecular Biology and Epigenetics Laboratory, National
Institute of Molecular Biology and Biotechnology, University of the
Philippines, Diliman, Philippines) for assistance with preparation
of figures.
Funding
This work was supported by grants from the
University of the Philippines System (grant no. 06-008) and the
Philippine Council for Health Research and Development (grant no.
FP150025).
Availability of data and materials
The data presented in this study, including those
for additional trials, are available from the corresponding author
on reasonable request.
Authors' contributions
RLG and EMCDP conceived project and wrote the
project proposal. AKJA and RTDY performed experiments. AKJA, RTDY
and RLG analyzed and organized the data. AKJA and RLG wrote
manuscript. All authors have read and approved of the final version
of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing financial
interest.
References
|
1
|
Porru M, Pompili L, Caruso C, Biroccio A
and Leonetti C: Targeting KRAS in metastatic colorectal cancer:
Current strategies and emerging opportunities. J Exp Clin Cancer
Res. 37:572018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Horsch M, Recktenwald CV, Schädler S,
Hrabé de Angelis M, Seliger B and Beckers J: Overexpressed vs
mutated Kras in murine fibroblasts: A molecular phenotyping study.
Br J Cancer. 100:656–662. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heinemann V, Stintzing S, Kirchner T,
Boeck S and Jung A: Clinical relevance of EGFR- and KRAS-status in
colorectal cancer patients treated with monoclonal antibodies
directed against the EGFR. Cancer Treat Rev. 35:262–271. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Di Nicolantonio F, Martini M, Molinari F,
Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L,
Frattini M, Siena S and Bardelli A: Wild-type BRAF is required for
response to panitumumab or cetuximab in metastatic colorectal
cancer. J Clin Oncol. 26:5705–5712. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baas JM, Krens LL, Guchelaar HJ, Morreau H
and Gelderblom H: Concordance of predictive markers for EGFR
inhibitors in primary tumors and metastases in colorectal cancer: A
review. Oncologist. 16:1239–1249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prior IA, Lewis PD and Mattos C: A
comprehensive survey of Ras mutations in cancer. Cancer Res.
72:2457–2467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stengel KR and Zheng Y: Essential role of
Cdc42 in Ras-induced transformation revealed by gene targeting.
PLoS One. 7:e373172012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guerrero S, Casanova I, Farré L, Mazo A,
Capellà G and Mangues R: K-ras codon 12 mutation induces higher
level of resistance to apoptosis and predisposition to
anchorage-independent growth than codon 13 mutation or
proto-oncogene overexpression. Cancer Res. 60:6750–6756.
2000.PubMed/NCBI
|
|
9
|
Seeburg PH, Colby WW, Capon DJ, Goeddel DV
and Levinson AD: Biological properties of human c-Ha-ras1 genes
mutated at codon 12. Nature. 312:71–75. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shankaran V, Obel J and Benson AB III:
Predicting response to EGFR inhibitors in metastatic colorectal
cancer: Current practice and future directions. Oncologist.
15:157–167. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao B, Wang L, Qiu H, Zhang M, Sun L,
Peng P, Yu Q and Yuan X: Mechanisms of resistance to anti-EGFR
therapy in colorectal cancer. Oncotarget. 8:3980–4000.
2017.PubMed/NCBI
|
|
12
|
van Krieken JH, Jung A, Kirchner T,
Carneiro F, Seruca R, Bosman FT, Quirke P, Fléjou JF, Plato Hansen
T, de Hertogh G, et al: KRAS mutation testing for predicting
response to anti-EGFR therapy for colorectal carcinoma: Proposal
for an European quality assurance program. Virchows Arch.
453:417–431. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Therkildsen C, Bergmann TK,
Henrichsen-Schnack T, Ladelund S and Nilbert M: The predictive
value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment
in metastatic colorectal cancer: A systematic review and
meta-analysis. Acta Oncol. 53:852–864. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zulhabri O, Rahman J, Ismail S, Isa MR and
Wan Zurinah WN: Predominance of G to A codon 12 mutation K-ras gene
in Dukes' B colorectal cancer. Singapore Med J. 53:26–31.
2012.PubMed/NCBI
|
|
15
|
Frattini M, Saletti P, Romagnani E, Martin
V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F and
Mazzucchelli L: PTEN loss of expression predicts cetuximab efficacy
in metastatic colorectal cancer patients. Br J Cancer.
97:1139–1145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Elbjeirami WM and Sughayer MA: KRAS
mutations and subtyping in colorectal cancer in Jordanian patients.
Oncol Lett. 4:705–710. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo F, Gong H, Zhao H, Chen J, Zhang Y,
Zhang L, Shi X, Zhang A, Jin H, Zhang J and He Y: Mutation status
and prognostic values of KRAS, NRAS, BRAF and PIK3CA in 353 Chinese
colorectal cancer patients. Sci Rep. 8:60762018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tong JH, Lung RW, Sin FM, Law PP, Kang W,
Chan AW, Ma BB, Mak TW, Ng SS and To KF: Characterization of rare
transforming KRAS mutations in sporadic colorectal cancer. Cancer
Biol Ther. 15:768–776. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bamford S, Dawson E, Forbes S, Clements J,
Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR
and Wooster R: The COSMIC (Catalogue of Somatic Mutations in
Cancer) database and website. Br J Cancer. 91:355–358. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pai EF, Krengel U, Petsko GA, Goody RS,
Kabsch W and Wittinghofer A: Refined crystal structure of the
triphosphate conformation of H-ras p21 at 1.35 A resolution:
Implications for the mechanism of GTP hydrolysis. EMBO J.
9:2351–2359. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schindelin J, Arganda-Carreras I, Frise E,
Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S,
Schmid B, et al: Fiji: An open-source platform for biological-image
analysis. Nat Methods. 9:676–682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reva B, Antipin Y and Sander C: Predicting
the functional impact of protein mutations: Application to cancer
genomics. Nucleic Acids Res. 39:e1182011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Billones JB, Carrillo MC, Organo VG, Sy
JB, Clavio NA, Macalino SJ, Emnacen IA, Lee AP, Ko PK and
Concepcion GP: In silico discovery and in vitro activity of
inhibitors against Mycobacterium tuberculosis 7,8-diaminopelargonic
acid synthase (Mtb BioA). Drug Des Devel Ther. 11:563–574. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu G, Robertson DH, Brooks CL III and
Vieth M: Detailed analysis of grid-based molecular docking: A case
study of CDOCKER-A CHARMm-based MD docking algorithm. J Comput
Chem. 24:1549–1562. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Der CJ: The ras family of oncogenes.
Cancer Treat Res. 47:73–119. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fernández-Medarde A and Santos E: Ras in
cancer and developmental diseases. Genes Cancer. 2:344–358. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tojkander S, Gateva G and Lappalainen P:
Actin stress fibers-assembly, dynamics and biological roles. J Cell
Sci. 125:1855–1864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang SH, Yates PR, Whitmarsh AJ, Davis RJ
and Sharrocks AD: The Elk-1 ETS-domain transcription factor
contains a mitogen-activated protein kinase targeting motif. Mol
Cell Biol. 18:710–720. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cruzalegui FH, Cano E and Treisman R: ERK
activation induces phosphorylation of Elk-1 at multiple S/T-P
motifs to high stoichiometry. Oncogene. 18:7948–7957. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hunter JC, Manandhar A, Carrasco MA,
Gurbani D, Gondi S and Westover KD: Biochemical and structural
analysis of common cancer-associated KRAS mutations. Mol Cancer
Res. 13:1325–1335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dagogo-Jack I and Shaw AT: Tumour
heterogeneity and resistance to cancer therapies. Nat Rev Clin
Oncol. 15:81–94. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yap TA, Gerlinger M, Futreal PA, Pusztai L
and Swanton C: Intratumor heterogeneity: Seeing the wood for the
trees. Sci Transl Med. 4:127ps102012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McGranahan N and Swanton C: Biological and
therapeutic impact of intratumor heterogeneity in cancer evolution.
Cancer Cell. 27:15–26. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Janakiraman M, Vakiani E, Zeng Z, Pratilas
CA, Taylor BS, Chitale D, Halilovic E, Wilson M, Huberman K,
Ricarte Filho JC, et al: Genomic and biological characterization of
exon 4 KRAS mutations in human cancer. Cancer Res. 70:5901–5911.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Smith G, Bounds R, Wolf H, Steele RJ,
Carey FA and Wolf CR: Activating K-Ras mutations outwith ‘hotspot’
codons in sporadic colorectal tumours-implications for personalised
cancer medicine. Br J Cancer. 102:693–703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tejpar S, Lenz HJ, Köhne CH, Heinemann V,
Ciardiello F, Beier RE, Stroh C, Duecker K and Bokemeyer C: Effect
of KRAS and NRAS mutations on treatment outcomes in patients with
metastatic colorectal cancer (mCRC) treated first-line with
cetuximab plus FOLFOX4: New results from the OPUS study. J Clin
Oncol. 32:LBA4442017. View Article : Google Scholar
|
|
40
|
Misale S, Di Nicolantonio F,
Sartore-Bianchi A, Siena S and Bardelli A: Resistance to anti-EGFR
therapy in colorectal cancer: From heterogeneity to convergent
evolution. Cancer Discov. 4:1269–1280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Murtaza BN, Bibi A, Nadeem MS, Chaudri MS
and Shakoori A: Identification of a novel mutation in codon 31 of
Kirstein rat sarcoma viral oncogene homologue in colon cancer:
Another evidence of non-canonical mutational pathway. Pakistan J
Zool. 44:1671–1676. 2012.
|
|
42
|
Cyniak-Magierska A, Brzeziańska E,
Januszkiewicz-Caulier J, Jarzab B and Lewinski A: Prevalence of RAS
point mutations in papillary thyroid carcinoma; a novel mutation at
codon 31 of K-RAS. Exp Clin Endocrinol Diabetes. 115:594–599. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lin SR, Tsai JH, Yang YC and Lee SC:
Mutations of K-ras oncogene in human adrenal tumours in Taiwan. Br
J Cancer. 77:1060–1065. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Willumsen BM, Norris K, Papageorge AG,
Hubbert NL and Lowy DR: Harvey murine sarcoma virus p21 ras
protein: biological and biochemical significance of the cysteine
nearest the carboxy terminus. EMBO J. 3:2581–2585. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thorgeirsson UP, Turpeenniemi-Hujanen T,
Williams JE, Westin EH, Heilman CA, Talmadge JE and Liotta LA:
NIH/3T3 cells transfected with human tumor DNA containing activated
ras oncogenes express the metastatic phenotype in nude mice. Mol
Cell Biol. 5:259–262. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Papageorge AG, Willumsen BM, Johnsen M,
Kung HF, Stacey DW, Vass WC and Lowy DR: A transforming ras gene
can provide an essential function ordinarily supplied by an
endogenous ras gene. Mol Cell Biol. 6:1843–1846. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
DeFeo-Jones D, Tatchell K, Robinson LC,
Sigal IS, Vass WC, Lowy DR and Scolnick EM: Mammalian and yeast ras
gene products: Biological function in their heterologous systems.
Science. 228:179–184. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cheng CM, Li H, Gasman S, Huang J, Schiff
R and Chang EC: Compartmentalized Ras proteins transform NIH 3T3
cells with different efficiencies. Mol Cell Biol. 31:983–997. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Garrett CT and Sell S: Cellular cancer
markers. Humana Press. (Totowa, NJ). 12. 455–477. 1995.
|
|
50
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pirkmajer S and Chibalin AV: Serum
starvation: Caveat emptor. Am J Physiol Cell Physiol.
301:C272–C279. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen RH, Abate C and Blenis J:
Phosphorylation of the c-Fos transrepression domain by
mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase.
Proc Natl Acad Sci USA. 90:10952–10956. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Morton S, Davis RJ, McLaren A and Cohen P:
A reinvestigation of the multisite phosphorylation of the
transcription factor c-Jun. EMBO J. 22:3876–3886. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Reszka AA, Seger R, Diltz CD, Krebs EG and
Fischer EH: Association of mitogen-activated protein kinase with
the microtubule cytoskeleton. Proc Natl Acad Sci USA. 92:8881–8885.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Northwood IC, Gonzalez FA, Wartmann M,
Raden DL and Davis RJ: Isolation and characterization of two growth
factor-stimulated protein kinases that phosphorylate the epidermal
growth factor receptor at threonine 669. J Biol Chem.
266:15266–15276. 1991.PubMed/NCBI
|
|
56
|
Hong S, Kim S, Kim HY, Kang M, Jang HH and
Lee WS: Targeting the PI3K signaling pathway in KRAS mutant colon
cancer. Cancer Med. 5:248–255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Martin TD, Samuel JC, Routh ED, Der CJ and
Yeh JJ: Activation and involvement of Ral GTPases in colorectal
cancer. Cancer Res. 71:206–215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen CC, Er TK, Liu YY, Hwang JK, Barrio
MJ, Rodrigo M, Garcia-Toro E and Herreros-Villanueva M:
Computational analysis of KRAS mutations: Implications for
different effects on the KRAS p.G12D and p.G13D mutations. PLoS
One. 8:e557932013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jaffe AB, Aspenstrom P and Hall A: Human
CNK1 acts as a scaffold protein, linking Rho and Ras signal
transduction pathways. Mol Cell Biol. 24:1736–1746. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nguyen A, Burack WR, Stock JL, Kortum R,
Chaika OV, Afkarian M, Muller WJ, Murphy KM, Morrison DK, Lewis RE,
et al: Kinase suppressor of Ras (KSR) is a scaffold which
facilitates mitogen-activated protein kinase activation in vivo.
Mol Cell Biol. 22:3035–3045. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Matsunaga-Udagawa R, Fujita Y, Yoshiki S,
Terai K, Kamioka Y, Kiyokawa E, Yugi K, Aoki K and Matsuda M: The
scaffold protein Shoc2/SUR-8 accelerates the interaction of Ras and
Raf. J Biol Chem. 285:7818–7826. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Adzhubei I, Jordan DM and Sunyaev SR:
Predicting functional effect of human missense mutations using
PolyPhen-2. Curr Protoc Hum Genet. Chapter 7: Unit7.20 2013 doi:
10.1002/0471142905.hg0720s76. PubMed/NCBI
|