Introduction
Felty’s syndrome (FS), which was first described in
1924, is a specific subcategory of rheumatoid arthritis (RA)
characterized by the triad of RA, severe extra-articular disease
and unexplained neutropenia (1). FS
usually develops after a >10-year course of RA and accounts for
<1% of RA patients (2). The
exact cause is unknown, however, several risk factors have been
proposed, including autoimmunity. The active extra-joint clinical
features can be misleading in FS and certain clinicians focus on
the severe extra-articular disease and neutropenia, which may lead
to subsequent infections. Thus, the correct diagnosis is
occasionally challenging.
The present study describes a case of FS with an
atypical arthritis presentation. The patient presented with a
fever, enlarged lymph nodes and hepatosplenomegaly, which can
easily be misdiagnosed as a hematological neoplasm. Written
informed consent was obtained from the patient.
Case report
A 17-year-old female with a 2-year history of
erosive nodular seropositive RA followed Chinese herbal treatment
for symptoms of repeated arthralgia in 2011. One year later, the
patient developed jaundice, hepatosplenomegaly and enlarged lymph
nodes. A physical examination revealed deformity in the proximal
interphalangeal joints, metacarpophalangeal joints, wrists and
ankles. Lymph nodes at the posterior neck, axillary fossa and
inguinal area were enlarged with a diameter of 0.5–1 cm.
In August 2012, the patient presented with a fever
>39°C. Blood tests revealed a white blood cell count of
4.12×109/l with an absolute neutrophil count of
3.15×109/l, hemoglobin levels of 80 g/l and a platelet
count of 159×109/l. A bone marrow biopsy showed
normocellular and maturing trilineage hematopoiesis. Glutamic
oxaloacetic transaminase levels were 352 U/l, while glutamate
pyruvate transaminase levels were 136 U/l. Bilirubin levels were
112.8 μmol/l and lactate dehydrogenase levels were 548 U/l. A rapid
erythrocyte sedimentation rate (35 mm/h) and high C-reactive
protein levels (60.2 mg/l) were revealed. Cultures of blood and
secreta showed no infection by microorganisms. Tests for human
immuodeficiency virus, syphilis, hepatitis B, hepatitis C and
autoimmune hepatitis-associated antibodies, and a purified protein
derivative skin test produced negative findings. The antinuclear
antibody titer was 1:320, while results for anti-dsDNA, anti-Sm and
ant-Rnp antibodies were negative. Levels of complement C3 and C4
were normal. Rheumatoid factor, antibodies to cyclic citrullinated
peptides, anti-keratin antibodies and anti-perinuclear factor
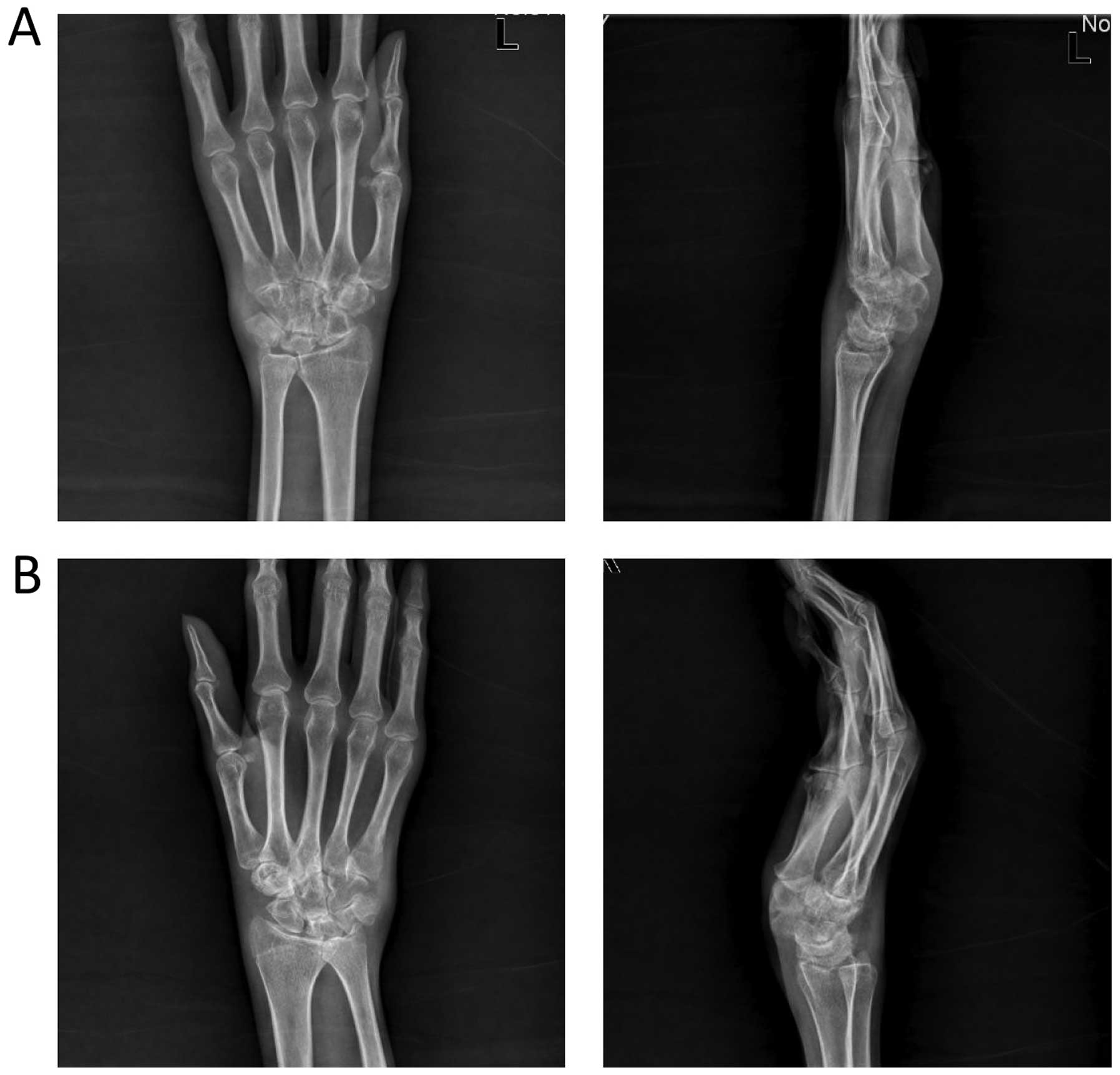
autoantibodies were negative. X-rays of the joints, including the
wrists and hands, showed soft-tissue swelling, bone erosion and
narrowing of the joint cavity (Fig.
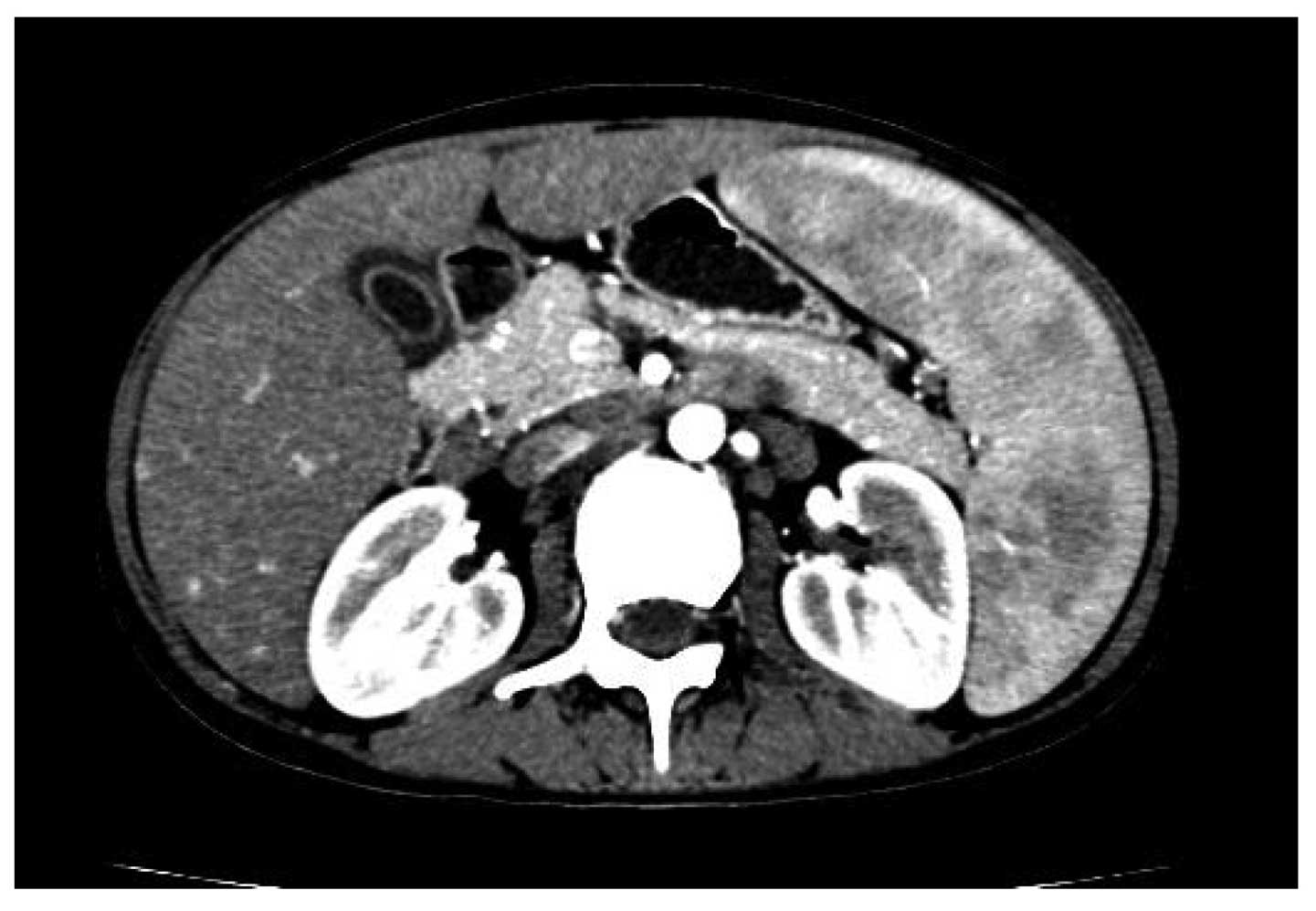
1). Splenomegaly (54×124 mm) with uniform density and multiple
enlarged lymph nodes distributed along the retroperitoneal space,
omental bursa, mesentery root and surrounding hepatic hilar was
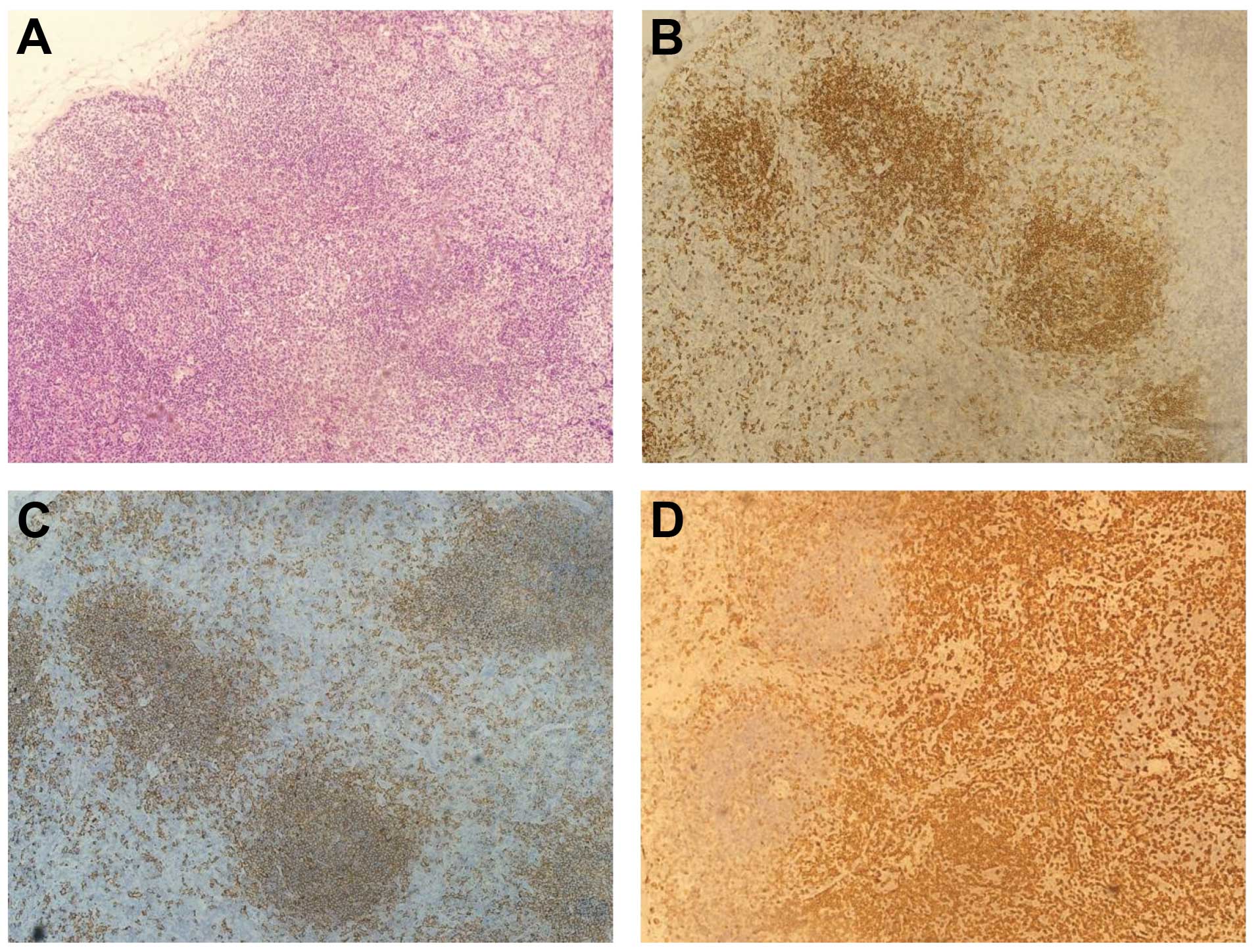
shown by abdominal computed tomography (CT) (Fig. 2). A lymph node biopsy from the right
cervical node revealed chronic inflammation, with positive results
for cluster of differentiation (CD)79α and CD20 in interfollicular
regions, and for CD5 in the paracortical area (Fig. 3). Fluorodeoxyglucose (FDG)-positron
emission tomography revealed lymphadenectasis of the bilateral
submandibular and superficial anterior cervical lymph node,
axillary fossa, retroperitoneal space, pelvic wall and inguinal
area without FDG uptake. The proliferation of hypermetabolic
lesions was observed in the shoulder, hip, knee, ankle and
interphalangeal joints, as well as knee joint-effusion, which was
the manifestation of RA. FDG uptake in the liver and spleen
occurred with a maximal standardized uptake value of 1.4 and 2.0,
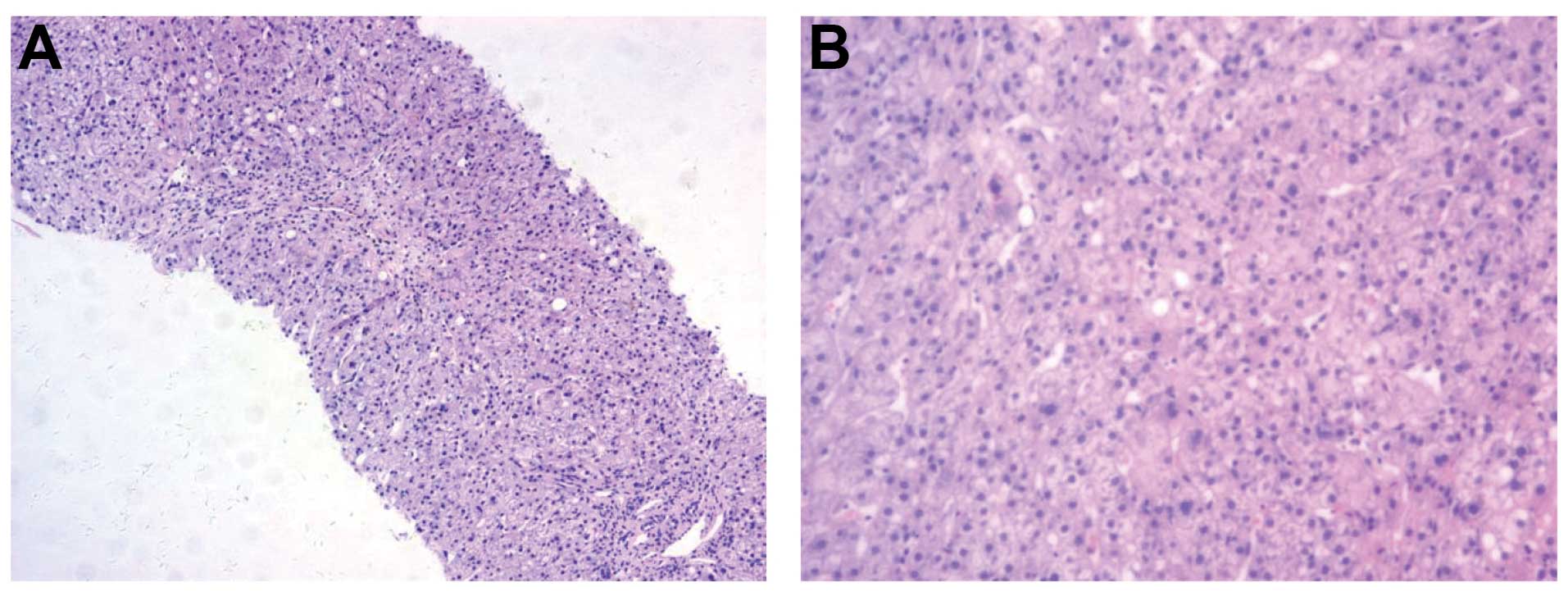
respectively. The pathological evaluation of liver tissue showed
inflammatory infiltration containing swelling and spotty or
fragmented necrosis in the hepatocytes (Fig. 4). Based on these findings, the
provisional diagnosis was of FS.
Treatment was initiated with methotrexate (7.5
mg/week), hydroxychloroquine (400 mg/day) and methylprednisolone
(16 mg/day). After tolerating overall treatment, the patient did no
longer had a fever. The levels of liver-associated enzymes and
bilirubin, and the erythrocyte sedimentation rate were decreased.
This decrease is usually associated with liver and spleen
shrinkage. Long-term follow-up is currently being performed.
Discussion
Severe joint destruction contrasting with moderate
or absent joint inflammation and severe extra-articular disease,
including, hepatopathy, lymphadenopathy, vasculitis, leg ulcers,
abnormal skin pigmentation and a high frequency of rheumatoid
nodules, are the clinical characteristics of FS. Due to severe
neutropenia, the affected individuals are susceptible to skin ulcer
formation and sepsis (2). There is
no specific diagnostic criterion for FS. FS is a clinical diagnosis
in patients with RA with unexplained neutropenia and splenomegaly.
Occasionally, the inactive joint symptoms lead clinicians to focus
on the severe extra-articular disease and neutropenia, subsequently
causing recurrent and even fatal infections. Therefore, the correct
diagnosis is challenging, and FS can easily be misdiagnosed as a
hematological neoplasm.
The hematological manifestations of RA can be
conveniently categorized into the following broad areas: Anemia
related to non-steroidal anitflammatory drugs or chronic disease,
FS or large granular lymphocyte syndrome-induced neutropenia and
hematological malignancy. Hepatosplenomegaly, neutropenia and
thrombocytopenia, often indicative of the diffuse spread of a
malignancy, may occasionally signify an underlying benign
inflammatory process in association with localized lymphoma.
Patients with long-standing FS disorders are considered at higher
risk of developing lymphoma (3). An
association exists between large granular lymphocyte (LGL) leukemia
and monoclonal expansion of LGLs and infiltration of the bone
marrow and spleen, which has a similar clinical presentation to FS;
this has also been termed ‘pseudo-Felty’ (4). The pivotal T cells involved in RA
pathogenesis have biological properties that are remarkably similar
to those in LGL leukemia (5). Liu
and Loughran (6) found that there
is an expansion of LGLs in 30–40% of patients with FS, and
indicated that FS and LGL leukemia with RA may be part of the same
disease spectrum, due to the prevalence of the immunogenic marker
HLA-DR4 in the two diseases. RA-associated LGL leukemia, lymphoma
and clinical manifestations of typical FS are occasionally not
distinguishable; therefore, the correct diagnosis of FS is
particularly important.
Neutropenia is the most common finding in FS. A
previous study showed that after mice were injected with the serum
from patients with FS, the number of circulating polymorphonuclear
cells decreased and human immune complexes were deposited in the
vascular bed of the lungs, which accounted for the pathogenesis of
the neutropenia in FS (7). Dwivedi
et al (8) proposed that FS
autoantibodies bind to deiminated histones and neutrophil
extracellular chromatin traps, which further stimulates
neutrophils, thus completing a self-sustaining cycle that drives
the depletion of mature neutrophils. Moreover, a long course of
erosive RA has also been indicated to contribute to FS neutropenia
as a consequence of immune complex-driven neutrophil depletion
(9). In the present case, the
patient had only a two-year history of RA, therefore the number of
neutrophils was normal.
The majority of experts recommend that the initial
choice of treatment for FS with increased infection rate should be
low-dose methotrexate (MTX) (1).
Other disease-modifying anti-rheumatic drugs (DMARDs), including
hydroxychloroquine, cyclosporin A, sulfasalazine, leflunomide,
azathioprine and cyclophosphamide, have also been used in the
treatment of FS (10). Rituximab is
used as a second-line therapy in patients with refractory FS who
are unresponsive to DMARDs (11,12).
Other anti-TNFα agents, including etanercept, infliximab and
adalimumab, have been reported as new methods to treat FS, while
anti-TNFα in combination with colony-stimulating factors
(granulocyte colony-stimulating factor) may be used with a
potential reduction of the infectious risk. In the present case,
the leukocyte and neutrophilic granulocyte counts were normal. By
two weeks of treatment with MTX, hydroxychloroquine and
methylprednisolone, the patient was in symptomatic remission and
long-term follow-up is currently being performed.
References
|
1
|
Felty AR: Chronic arthritis in the adult
associated with splenomegaly and leucopenia. Bull Johns Hopkins
Hosp. 35:16–20. 1924.
|
|
2
|
Balint GP and Balint PV: Felty’s syndrome.
Best Pract Res Clin Rheumatol. 18:631–645. 2004.
|
|
3
|
Nassif PS and Feldon SE: Orbital lymphoma
in a patient with Felty’s syndrome. Br J Ophthalmol. 76:173–174.
1992.PubMed/NCBI
|
|
4
|
Lamy T and Loughran TP Jr: How I treat LGL
leukemia. Blood. 117:2764–2774. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lois Iglesias A, Sifuentes Giraldo WA,
Bachiller Corral J, et al: Large granular lymphocyte leukemia as a
complication of rheumatoid arthritis. Reumatol Clin. 8:365–367.
2012.PubMed/NCBI
|
|
6
|
Liu X and Loughran TP Jr: The spectrum of
large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin
Hematol. 18:254–259. 2011.
|
|
7
|
Breedveld FC, Lafeber GJ, de Vries E, et
al: Immune complexes and the pathogenesis of neutropenia in Felty’s
syndrome. Ann Rheum Dis. 45:696–702. 1986.
|
|
8
|
Dwivedi N, Upadhyay J, Neeli I, et al:
Felty’s syndrome autoantibodies bind to deiminated histones and
neutrophil extracellular chromatin traps. Arthritis Rheum.
64:982–992. 2012.
|
|
9
|
Dwivedi N and Radic M: Neutrophil
activation and B-cell stimulation in the pathogenesis of Felty’s
syndrome. Pol Arch Med Wewn. 122:374–379. 2012.PubMed/NCBI
|
|
10
|
Mahévas M, Audia S, De Lastours V, et al:
Neutropenia in Felty’s syndrome successfully treated with
hydroxychloroquine. Haematologica. 92:e78–e79. 2007.
|
|
11
|
Narváez J, Domingo-Domenech E,
Gómez-Vaquero C, et al: Biological agents in the management of
Felty’s syndrome: a systematic review. Semin Arthritis Rheum.
41:658–668. 2012.
|
|
12
|
Shipley E, Héraud A, Hennette A and
Vernhes JP: Efficacy of rituximab in Felty’s syndrome. Joint Bone
Spine. 5:621–622. 2008.
|