Introduction
Glioblastoma multiforme (GBM), a grade IV
astrocytoma, is the most frequent and aggressive primary brain
tumor. Despite the increased understanding of the oncological
mechanisms underlying GBM pathophysiology, and treatment advances,
only a marginal improvement in overall survival has been seen
during the first decade of the 21st century (1). Abnormalities in multiple genetic
pathways have been identified including amplification of epidermal
growth factor (EGFR), loss of chromosome 10q, mutation of
phosphatase and tensin homolog (PTEN), mutation of p53, and
concomitant loss of p16 and p18 (2–4).
Investigation into similarities between growth factor signaling
elements implicated in GBM progression, and those that control
crucial stages in neural development has provided evidence
signifying neural stem and/or progenitor cells as the cell type of
origin for GBM (5–7). These cancer stem cells (CSCs) carry
three distinct properties: self-renewal, ability to differentiate
into multiple lineages and extensive proliferation. Their presence
in tumors has been demonstrated through the identification of
specific antigenic markers, and use of co-culture conditions that
were originally developed for normal neural stem cells (6,8,9).
Aberrant signaling pathways contributing to abnormal
cell migration, invasion, proliferation, as well as CSC maintenance
are responsible for the aggressive nature of GBM (10). Many of the identified mutations
result in activation of the lipid kinase PI3K and its downstream
target, the plekstrin-homology-domain serine threonine kinase AKT.
AKT has over 40 downstream targets (11). Prominent among these is mechanistic
target of rapamycin (mTOR; AKA mammalian target of rapamycin) and
recent reports have linked PI3K/Akt/mTOR activity directly to CSC
expansion and maintenance (10,12).
mTOR is an atypical serine/threonine kinase that
regulates several processes including autophagy, ribosome
biogenesis, and metabolism by integrating signals from growth
factors, nutrients, oxygen and energy status (13). Recent studies have indentified two
structurally and functionally distinct mTOR-containing multiprotein
complexes, mTOR complex 1 (mTORC1) and complex 2 (mTORC2). The two
complexes consist of unique mTOR-interacting proteins, which
determine their substrate specificity. mTORC1 acts as a downstream
effector of PI3K/Akt signaling, promoting cell growth,
proliferation, and CAP-dependent protein translation, alongside
inhibiting autophagy. mTOR2 phosphorylates AKT at Ser473, and as an
upstream activator regulates serum/glucocorticoid regulated kinase
(SGK) as well as PKC. mTORC2 also modifies cytoskeletal elements
and as a result is involved in controlling cellular migration. mTOR
has been implicated in promoting the maintenance of stem cells and
CSCs indicating its inhibition as an approach to prevent
self-renewal of stem cells (10,12,14).
An alternate proposed mechanism to target the CSCs
of GBM is to promote differentiation and therefore increase
sensitivity to therapy. All-trans retinoic acid (ATRA), a
derivative of retinoid, is capable of differentiating a variety of
stem cells, as well as normal neural progenitor cells (15). Retinoic acid and its derivatives
activate retinoic acid receptors and retinoid × receptor (RAR-RXR)
complexes and induce differentiation of neural stem cells (16). A combination of ATRA and interferon
γ (IFNγ) has shown the ability to induce both differentiation and
apoptosis of GBM cells independent of their PTEN status (17–19),
and clinically the use of ATRA with cytosine arabinoside resulted
in stability of recurrent GBM (20).
This investigation addresses means to target GBM
CSCs using the combination of differentiating agent ATRA with
inhibitors of the PI3K/AKT/mTOR pathways, and thus provide a novel
therapeutic approach.
Materials and methods
Cell line and reagents
The human glioblastoma cell lines U87 and LN18 were
used. These are commercially available neurosphere forming primary
GBM cell lines (ATCC, Manassas, VA, USA). U87 (21) cell line exhibits p16INK loss, PTEN
loss and wild-type p53. LN18 (22)
cell line exhibits intact PTEN and mutant p53 at codon 238. Cells
were maintained in Dulbecco’s modified Eagle’s medium (DMED;
Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS and 1%
penicillin/streptomycin/amphotericin in a humidified incubator with
5% CO2 at 37°C. Cells were made quiescent by serum
deprivation 24 h prior to treatment. Treatment included the mTORC1
inhibitor rapamycin (RAPA, 100 nM; EMD Chemicals, Gibbstown, NJ,
USA), PI3K inhibitor LY294002 (LY, 10 μM; EMD Chemicals), MAP
kinase 1 and 2 (MEK1/2) inhibitor U0126 (10 μM; Sigma-Aldrich, St.
Louis, MO, USA), and all-trans retinoic acid (ATRA, 10 μM;
Sigma-Aldrich). All reagents were attained from Sigma-Aldrich.
Isolation of protein
Protein extraction was performed with whole cell
lysis buffer containing 1% Triton X-100, 10 mM Tris-HCl, pH 7.5,
150 mM NaCl and 5 mM EDTA containing phosphatase and protease
inhibitors (Sigma-Aldrich). Protein concentrations were determined
by the modified Lowry method (Bio-Rad Laboratories, Hercules, CA,
USA).
Western blot analysis
Equal amounts of protein were resolved on a 10%
SDS-PAGE gel and then electrotransferred onto nitrocellulose
membrane. Membranes were processed according to the manufacturer’s
instructions (Santa Cruz Biotechnology, Santa Cruz, CA, USA; Cell
Signaling Technology, Danvers, MA, USA). A routine procedure
utilized primary antibodies for Nestin and activated extracellular
regulated kinase 1 and 2 (ERK, pERK1/2Thr202/Tyr204), at
1:1000 dilutions (Santa Cruz Biotechnology), followed by detection
by chemiluminescence (Millipore, Billerica, MA, USA). Blots were
stripped (Pierce Protein Biology Products, Rockford, IL, USA) and
re-probed with actin or respective total antibodies to ensure equal
loading. Densiometric analysis was performed using ImageJ (NIH,
Bethesda, MD, USA). Experiments were conducted at least three
times.
Fluorescence immunohistochemistry
Cells were treated with ATRA or RAPA for 24 h. After
treatments cells were fixed in 4% PFA, blocked with 10% goat serum
in PBS/0.1% Triton X-100, and stained with Nestin (1:500; Santa
Cruz Biotechnology) according to the manufacturer’s instructions.
FITC-Green or Rhodamine-Red secondary was used (Jackson
ImmunoResearch, West Grove, PA, USA) with
4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstaining. Cells
were visualized using imaging system of Zeiss Axiovert 100M
microscope (Thornwood, NY, USA).
Neurosphere assays
Serum-starved neurospheres were treated with RAPA,
LY, or ATRA for up to 3 days. Serial images of treated cells were
taken at ×10 using an Axiovert 100M (Zeiss) where field coordinates
were maintained. Neurosphere diameters were measured (AxioVision
software; Zeiss) across all visibly identifiable neurospheres by a
single blinded observer, while diameters across ellipsoid
neurospheres were measured diagonally across all treatments to
ensure uniformity. Neurosphere diameters were normalized to day
zero for each treatment. Effects of combination therapy were also
assessed with ATRA and RAPA and LY.
Scratch/wound healing migration
assay
The scratch wound migration technique was used to
determine the motility of GBM cells following treatment. LN18 cells
were grown to confluent monolayer, and when approaching 100% cell
confluence scratching the surface as uniformly as possible with a
pipette tip formed a wound. This initial wounding and the movement
of the cells in the scratched area were photographically monitored
using the Axiovert Zeiss 200 microscope (Carl Zeiss) with ×10
magnification (NA 0.25). The migration rate was expressed as a
percentage of the control, and it was calculated as the proportion
of the mean distance between both borderlines caused by scratching,
to the distance that remained cell-free after migration. Two
independent series of experiments were performed in
quadruplicate.
Statistical analysis
Values are presented as the means. One-way ANOVA was
carried out for multiple comparisons and two-tailed t-tests were
used for single comparisons. A P-value of <0.05 was considered
significant.
Results
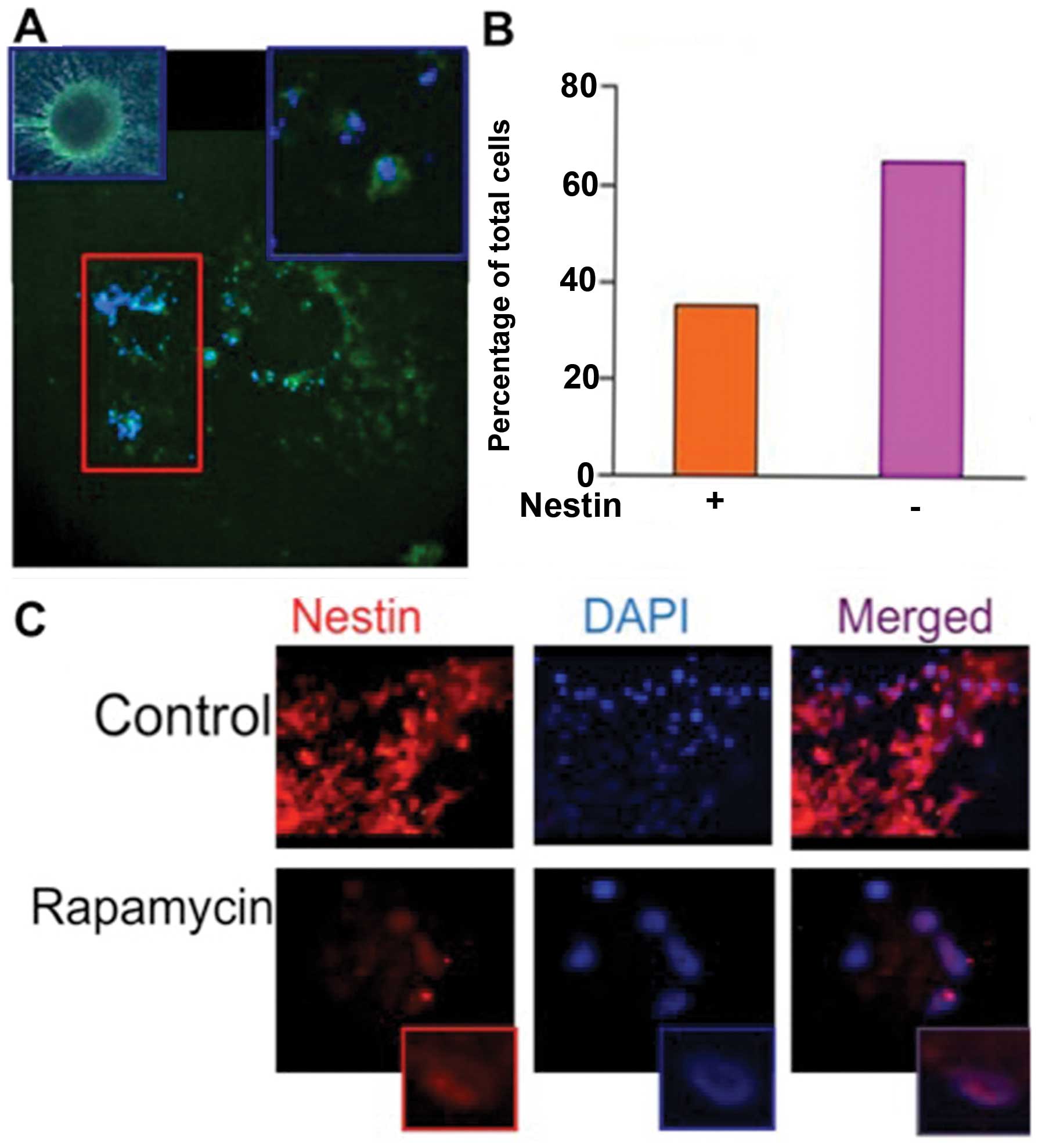
ATRA induces differentiation of CSCs,
evidenced by depletion of stem cell marker Nestin
In order to establish that ATRA induced
differentiation of CSCs, stem cells were treated with ATRA (10 μM)
for 24 h followed by immunofluorescence analysis of Nestin
expression (Fig. 1A). A quantified
comparison of cells expressing Nestin was performed after
counterstaining with DAPI. All non-treated CSCs demonstrated Nestin
expression, where only 40% of the cells expressed Nestin following
treatment with ATRA (Fig. 1B). This
indicates that treatment with ATRA was able to cause a significant
degree of differentiation among CSCs.
mTOR inhibition alters cellular
localization of Nestin
Cells treated with the mTORC1 inhibitor, RAPA (100
nM) for 24 h were subjected to immunofluorescence analysis
(Fig. 1C). The CSCs expressed
Nestin ubiquitously and was found primarily in the cytoplasm, as
evident by the absence of Nestin co-localization with DAPI in the
merged image (Fig. 1C, upper
panel). Following treatment with RAPA cytoplasmic expression of
Nestin was lost, though strong nuclear expression was evident
(Fig. 1C, lower panel).
Alterations in Nestin and ERK expression
following treatment with ATRA and PI3K/mTOR inhibitors
For further analysis of the effect of ATRA on
differentiation, CSCs were treated with ATRA (10 μM) for either 2
or 6 h and western blot analysis with densitometry analysis was
performed. A significant decrease in Nestin was seen after ATRA
treatment as compared to control. This reduction in Nestin
expression was greater at 6 than at 2 h suggesting a time-dependent
effect. Nestin expression was examined in CSCs treated with
combinations of ATRA and LY (10 μM), RAPA, (100 nM) or U0126 (10
μM) for 6 h. The results illustrated a decrease in Nestin
expression comparable to that of ATRA alone (Fig. 2A). The bar graph represents the
arbitrary value of Nestin in comparison to actin, and demonstrates
further Nestin inhibition with the use of pathway inhibitors.
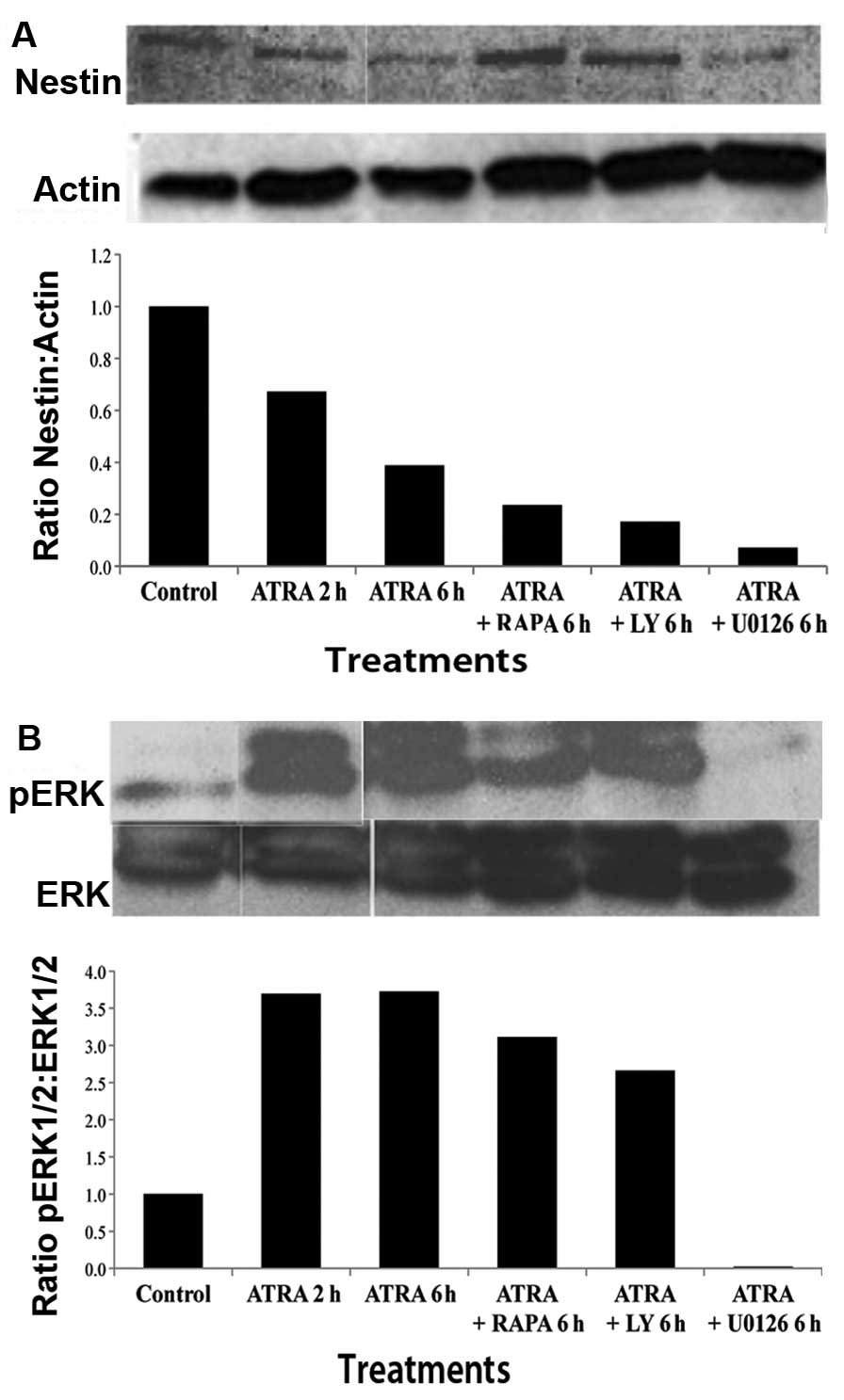 | Figure 2Western blot analysis of stem cell
marker Nestin and activated ERK. (A) Western blot analysis and
densitometry analysis represented as Nestin to actin ratio, shows a
time-dependent decrease in Nestin expression after treatment with
ATRA at 2 and 6 h. Combination treatment with ATRA and PI3K
inhibitor, LY, or mTOR inhibitor RAPA, or MEK1/2 inhibitor U0126,
showed a decrease in Nestin compared to control. (B) Western blot
analysis and densitometry analysis, expressed as a ratio of pERK1/2
to ERK1/2, shows a time-dependent increase in extracellular-related
kinase (ERK) following treatment with ATRA at 2 and 6 h. Cells
treated with ATRA in combination with LY or RAPA demonstrated a
slight increase in ERK1/2 expression as compared to control.
However, treatment with ATRA and MEK1/2 inhibitor, U0126, showed no
increase in ERK expression, suggesting that U0126 was able to
abrogate the effect of ATRA on cell differentiation. |
In order to investigate the mechanism of
differentiation induced by ATRA, we examined activated ERK1/2
(pERK1/2) expression in CSCs following ATRA treatment in the
presence or absence of mTOR/PI3K/MAPK inhibitors (Fig. 2B). pERK1/2 expression was found to
be increased following treatment with ATRA at 2 and 6 h in a
time-dependent manner. pERK1/2 expression was also found to be
increased following treatment with LY and RAPA in combination with
ATRA. Treatment with MEK1/2 inhibitor, U0126 (10 μM), in
combination with ATRA resulted in no alteration in ERK1/2
expression, suggesting that U0126 was able to abrogate the
activation caused by ATRA.
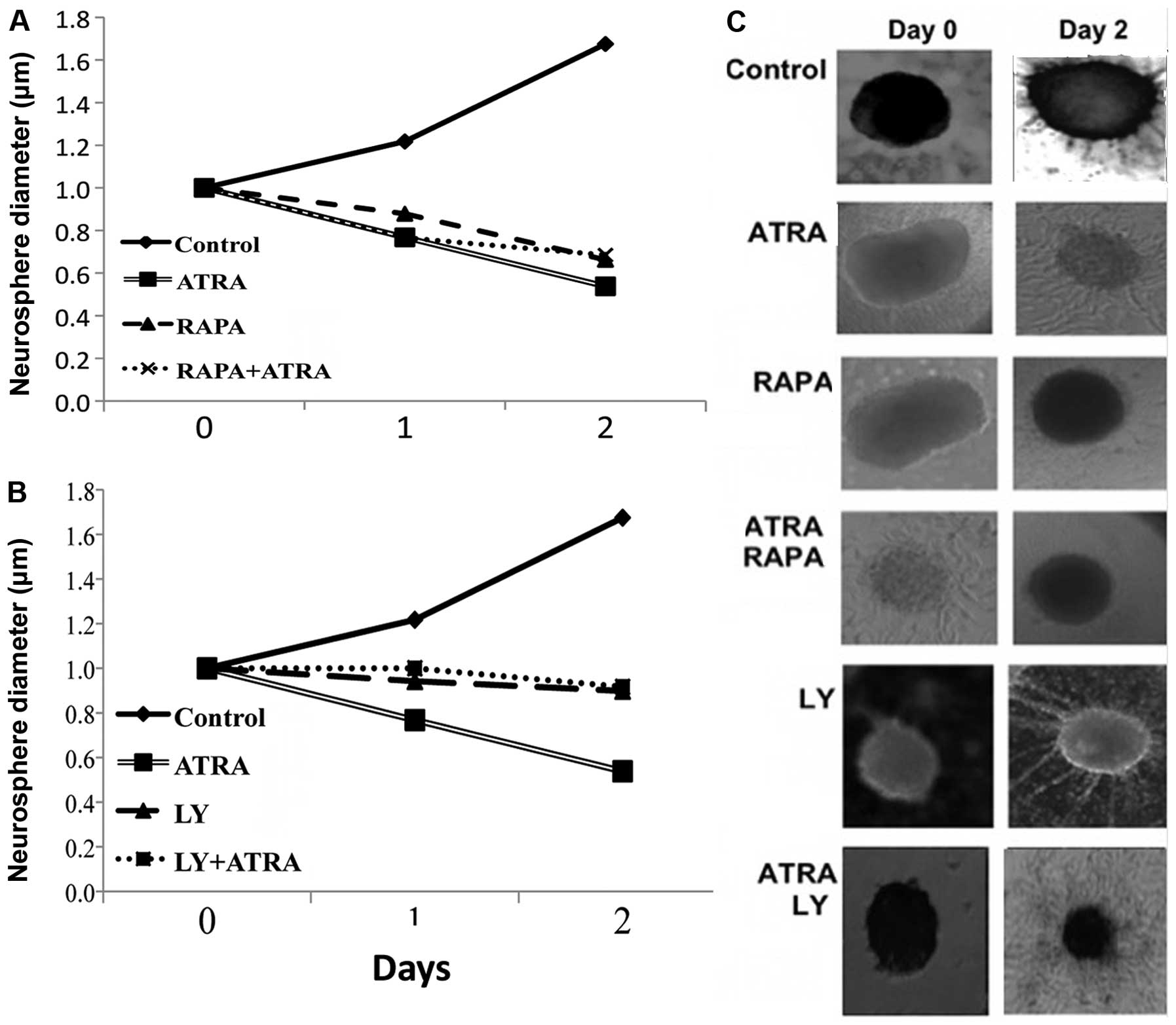
The effects of PI3K/mTOR inhibition in
combination with differentiating agents on CSC proliferation
The proliferation of CSCs was assessed by a
neurosphere assay determining the size changes in neurospheres. As
expected with time there was an exponential increase in cell
proliferation in controls (Fig. 3).
Following treatment with ATRA, and in the presence of mTOR or PI3K
inhibitors CSC proliferation was suppressed.
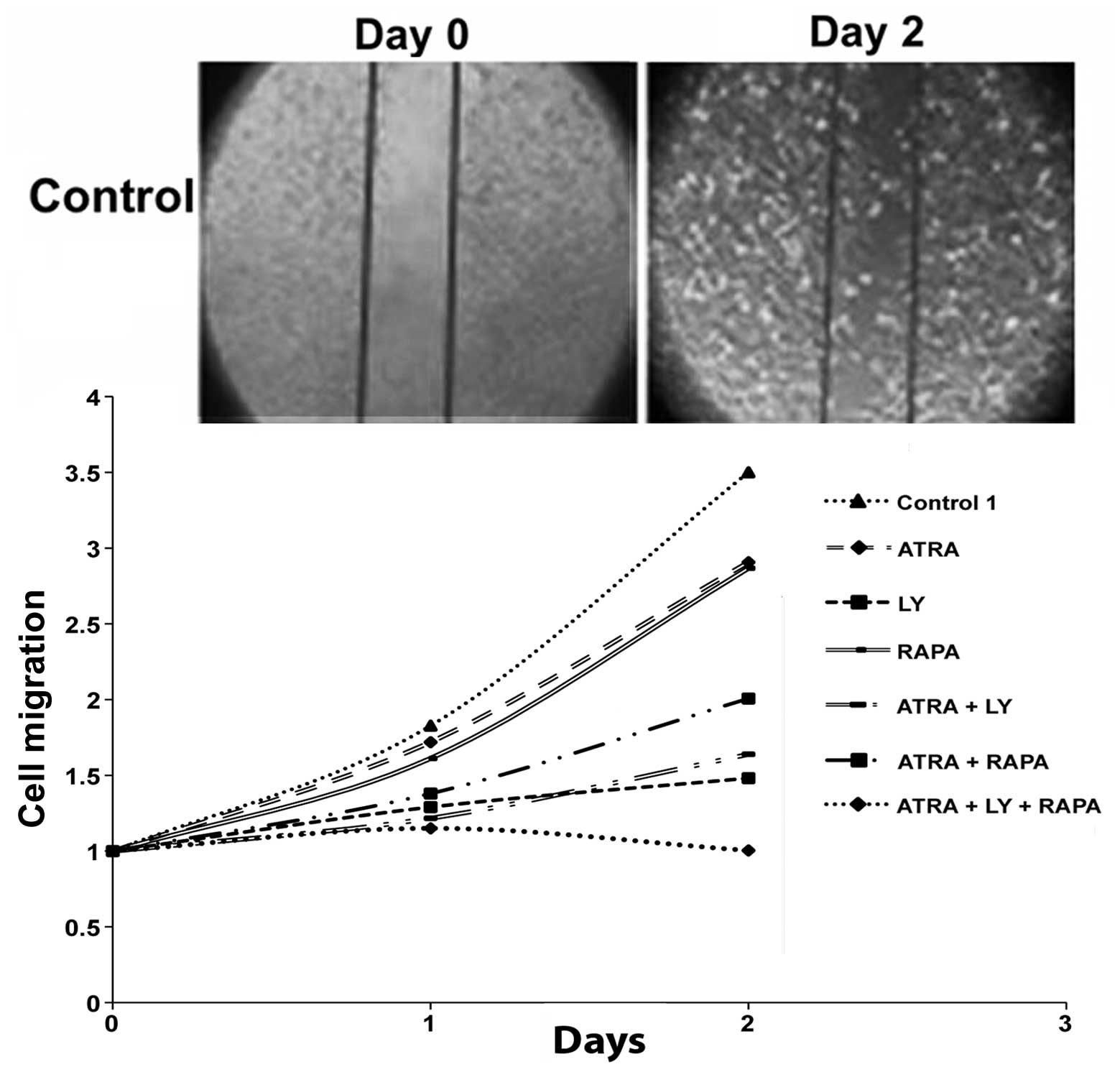
The effects of PI3K/mTOR inhibition in
combination with differentiating agents on cell migration
GBM cells treated with RAPA (100 nM), LY (10 μM) and
ATRA (10 μM) as single agents all demonstrated decreased cellular
motility as compared with control. Among these agents, LY exhibited
the highest degree of suppression. Treatment with LY and ATRA in
combination had a greater suppressive effect than compared to RAPA
and ATRA. Interestingly, the combination of all three agents; LY,
RAPA and ATRA, had a synergistic effect causing a complete halt in
GBM cell migration (Fig. 4).
Discussion
The findings of the present study provide further
evidence on the targeting of CSCs of GBM by differentiating agent
ATRA in combination with PI3K/mTOR inhibitors. The central role of
these CSCs in the recurrence and aggressive nature of this disease
has made them a key target in its management (5–7). There
is abundant evidence of the presence of CSCs within GBM, it has
been shown that the expression of stem cell markers (e.g. Nestin
and JNK), in tumor and peri-tumor areas can serve as a prognostic
indicator (23).
Nestin, a cytoskeletal intermediate filament has
long been described as a marker for neural stem cells (24). The confirmation of Nestin expression
in GBM neurospheres by immunofluoresence suggests further evidence
for the presence of stem cells in GBM. In addition, the loss of
Nestin expression following treatment with ATRA is indicative of
cellular differentiation occurring in a time-dependent manner. In
comparison, upon treatment with the mTORC1 inhibitor RAPA, there
was a marked translocation of Nestin expression from the cytoplasm
to the nucleus, suggesting that the cells underwent other molecular
alterations over differentiation.
Similar results were seen on western blot analysis
with a reduction in Nestin expression following ATRA treatment. The
combination of ATRA with PI3k inhibitor LY, mTORC1 inhibitor RAPA,
and the MEK1/2 inhibitor U0126 also caused a suppression of Nestin.
When investigating the mechanism underlying this phenomenon it
became suggestive that ATRA-induced differentiation was mediated by
the activation of ERK1/2 (pERK1/2). This was demonstrated by the
fact that after treatment of cells with ATRA an increase in pERK1/2
expression was observed (Fig. 2B).
ERK1/2 is a downstream signaling molecule of the Ras-Raf-MEK1/2
signaling pathway that has been found implicated in many cancers
including GBM (10,25). Studies have also shown that MEK1/2,
ERK1/2, and mTOR pathways are involved in a mutual negative
feedback loop mediated via p70-S6K, in which each pathway can
inhibit or activate the other affecting cellular proliferation or
differentiation (26,27). This has also led to previous
findings by our group that GBM CSCs treated with both MEK1/2,
ERK1/2 inhibitors and PI3K/mTOR inhibitors are more effective than
either inhibition alone (10).
Alternatively ERK1/2 inactivation with U0126 promoted
differentiation while inhibiting proliferation (28). Our findings suggest that ATRA
results in differentiation through the activation of ERK1/2, and
that this effect can be prevented with U0126 treatment. These
observations have pertinent clinical relevance since temozolamide
resistance in GBM has been found to be mediated by dysregulation of
MEK1/2, and ERK1/2 pathways and the inhibition of ERK1/2 pathway
may sensitize cells to chemotherapy treatment (29). One notable factor in our results is
that treatment with ATRA and U0126 in combination reversed the
activation of ERK 1/2 resulting from ATRA treatment alone, the
combined treatment did not completely halt differentiation as there
was still a significant decrease in Nestin expression with
combination treatment. This suggests the possibility of other
mechanisms by which ATRA is able to induce differentiation.
Other important results of this investigation
include a functional assessment of CSCs. Our results confirm
previous findings that inhibition of mTOR or PI3K leads to the
reduction of cellular proliferation and migration (30). We have confirmed a similar effect on
cell proliferation in GBM CSC populations by demonstrating that
mTORC1 inhibitor RAPA, and PI3K inhibitor LY suppressed neurosphere
growth, independent of the use of ATRA. The lack of additional
suppression in the presence of inhibitors suggests that the dose
and time of treatment was optimally achieved, and further decline
in proliferation with the addition of secondary agents was
unlikely. In addition, we examined the effect of PI3K/mTOR
inhibition on GBM cellular migration. We illustrated that
combination of PI3K inhibition, mTOR inhibition and treatment with
differentiating agent ATRA had a synergistic effect resulting in
the least amount of cellular migration. This may be mediated
through mechanisms involving the balance between ATRA-induced
differentiation and PI3K/mTOR inhibition regulating proliferation
and migration.
CSC proliferation and migration have important
clinical implications because these properties relate directly to
the invasive nature of GBM. Thus, evidence that inhibitors of mTOR
and differentiating agents can negatively affect proliferation and
migration suggest their potential therapeutic use. The finding that
the use of a differentiating agent in combination with mTOR pathway
inhibitors had a synergistic effect in preventing cell migration is
of particular significance. This indicates the possibility of
targeting CSCs by inducing differentiation, followed by treatment
with mTOR inhibitors. This is echoed by clinical findings in other
cancer types that are already examining inhibition of ERK1/2 and
mTOR pathways using existing inhibitors. A phase I trial looked at
236 patients with advanced colorectal cancer treated with a PI3K
inhibitor, a MAPK inhibitor, or a combination of the two (31). The results illustrated that dual
inhibition was superior in efficacy as compared to inhibition of a
single pathway. Therefore, elucidating the mechanism of only a
marginal benefit in an early phase trial of mTOR inhibitors
(temsirolimus) alone as treatment modality for recurrent GBM
(32).
In conclusion, this investigation demonstrates that
ATRA-induced differentiation is another mechanism of targeting the
MAPK/ERK1/2 pathway, and that treatment of cells with ATRA in
combination with PI3K and/or mTOR inhibition was an effective
strategy for decreasing CSC proliferation and migration. Thus,
using differentiating agents along with inhibitors of mTOR pathways
provides a molecular basis for the treatment of GBM.
Acknowledgements
We gratefully acknowledge the American Research
Foundation for their support, and the work done by Michael
Pendergast in the administration and maintenance of the
laboratory.
References
|
1
|
Lawrence YR, Mishra MV, Werner-Wasik M,
Andrews DW, Showalter TN, Glass J, et al: Improving prognosis of
glioblastoma in the 21st century: who has benefited most? Cancer.
118:4228–4234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wiedemeyer R, Brennan C, Heffernan TP,
Xiao Y, Mahoney J, Protopopov A, et al: Feedback circuit among INK4
tumor suppressors constrains human glioblastoma development. Cancer
Cell. 13:355–364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ohgaki H and Kleihues P: Genetic pathways
to primary and secondary glioblastoma. Am J Pathol. 170:1445–1453.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Endersby R and Baker SJ: PTEN signaling in
brain: neuropathology and tumorigenesis. Oncogene. 27:5416–5430.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Holland EC, Hively WP, DePinho RA and
Varmus HE: A constitutively active epidermal growth factor receptor
cooperates with disruption of G1 cell-cycle arrest pathways to
induce glioma-like lesions in mice. Genes Dev. 12:3675–3685. 1998.
View Article : Google Scholar
|
|
6
|
Galli R, Binda E, Orfanelli U, Cipelletti
B, Gritti A, De Vitis S, et al: Isolation and characterization of
tumorigenic, stem-like neural precursors from human glioblastoma.
Cancer Res. 64:7011–7021. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sanai N, Alvarez-Buylla A and Berger MS:
Neural stem cells and the origin of gliomas. N Engl J Med.
353:811–822. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, et al: Identification of human brain tumour
initiating cells. Nature. 432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ignatova TN, Kukekov VG, Laywell ED,
Suslov ON, Vrionis FD and Steindler DA: Human cortical glial tumors
contain neural stem-like cells expressing astroglial and neuronal
markers in vitro. Glia. 39:193–206. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jhanwar-Uniyal M, Albert L, McKenna E,
Karsy M, Rajdev P, Braun A and Murali R: Deciphering the signaling
pathways of cancer stem cells of glioblastoma multiforme: role of
Akt/mTOR and MAPK pathways. Adv Enzyme Regul. 51:164–170. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Manning BD and Cantley LC: AKT/PKB
signaling: navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jhanwar-Uniyal M, Jeevan D, Neil J,
Shannon C, Albert L and Murali R: Deconstructing mTOR complexes in
regulation of glioblastoma multiforme and its stem cells. Adv Biol
Regul. 53:202–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sarbassov DD, Ali SM and Sabatini DM:
Growing roles for the mTOR pathway. Curr Opin Cell Biol.
17:596–603. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gan B, Sahin E, Jiang S, Sanchez-Aguilera
A, Scott KL, Chin L, et al: mTORC1-dependent and -independent
regulation of stem cell renewal, differentiation, and mobilization.
Proc Natl Acad Sci USA. 105:19384–19389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Das A, Banik NL and Ray SK: Retinoids
induced astrocytic differentiation with down regulation of
telomerase activity and enhanced sensitivity to taxol for apoptosis
in human glioblastoma T98G and U87MG cells. J Neurooncol. 87:9–22.
2008. View Article : Google Scholar
|
|
16
|
Guan K, Chang H, Rolletschek A and Wobus
AM: Embryonic stem cell-derived neurogenesis. Retinoic acid
induction and lineage selection of neuronal cells. Cell Tissue Res.
305:171–176. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Das A, Banik NL and Ray SK: Molecular
mechanisms of the combination of retinoid and interferon-gamma for
inducing differentiation and increasing apoptosis in human
glioblastoma T98G and U87MG cells. Neurochem Res. 34:87–101. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang R, Banik NL and Ray SK: Combination
of all-trans retinoic acid and interferon-gamma upregulated
p27(kip1) and down regulated CDK2 to cause cell cycle
arrest leading to differentiation and apoptosis in human
glioblastoma LN18 (pten-proficient) and U87MG (pten-deficient)
cells. Cancer Chemother Pharmacol. 62:407–416. 2008.
|
|
19
|
Haque A, Das A, Hajiaghamohseni LM,
Younger A, Banik NL and Ray SK: Induction of apoptosis and immune
response by all-trans retinoic acid plus interferon-gamma in
human malignant glioblastoma T98G and U87MG cells. Cancer Immunol
Immunother. 56:615–625. 2007.PubMed/NCBI
|
|
20
|
Defer GL, Adle-Biassette H, Ricolfi F,
Martin L, Authier FJ, Chomienne C, et al: All-trans retinoic
acid in relapsing malignant gliomas: clinical and radiological
stabilization associated with the appearance of intratumoral
calcifications. J Neurooncol. 34:169–177. 1997. View Article : Google Scholar
|
|
21
|
Clark MJ, Homer N, O’Connor BD, Chen Z,
Eskin A, Lee H, et al: U87MG decoded: the genomic sequence of a
cytogenetically aberrant human cancer cell line. PLoS Genet.
6:e10008322010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Diserens AC, de Tribolet N, Martin-Achard
A, Gaide AC, Schnegg JF and Carrel S: Characterization of an
established human malignant glioma cell line: LN-18. Acta
Neuropathol. 53:21–28. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mangiola A, Lama G, Giannitelli C, De
Bonis P, Anile C, Lauriola L, et al: Stem cell marker nestin and
c-jun NH2-terminal kinases in tumor and peritumor areas of
glioblastoma multiforme: possible prognostic implications. Clin
Cancer Res. 13:6970–6977. 2007. View Article : Google Scholar
|
|
24
|
Lendahl U, Zimmerman LB and McKay RD: CNS
stem cells express a new class of intermediate filament protein.
Cell. 60:585–595. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roskoski R: ERK1/2 MAP kinases: structure,
function, and regulation. Pharmacol Res. 66:105–143. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Albert L, Karsy M, Murali R and
Jhanwar-Uniyal M: Inhibition of mTOR activates the MAPK pathway in
glioblastoma multiforme. Cancer Genomics Proteomics. 6:255–261.
2009.PubMed/NCBI
|
|
27
|
Sunayama J, Matsuda K, Sato A, Tachibana
K, Suzuki K, Narita Y, et al: Crosstalk between the PI3K/mTOR and
MEK/ERK pathways involved in the maintenance of self-renewal and
tumorigenicity of glioblastoma stem-like cells. Stem Cells.
28:1930–1939. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang B, Gao Y, Xiao Z, Chen B, Han J,
Zhang J, et al: Erk1/2 promotes proliferation and inhibits neuronal
differentiation of neural stem cells. Neurosci Lett. 461:252–257.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sato A, Sunayama J, Matsuda K, Seino S,
Suzuki K, Watanabe E, et al: MEK-ERK signaling dictates DNA-repair
gene MGMT expression and temozolomide resistance of stem-like
glioblastoma cells via the MDM2-p53 axis. Stem Cells. 29:1942–1951.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gulati N, Karsy M, Albert L, Murali R and
Jhanwar-Uniyal M: Involvement of mTORC1 and mTORC2 in regulation of
glioblastoma multiforme growth and motility. Int J Oncol.
35:731–740. 2009.PubMed/NCBI
|
|
31
|
Shimizu T, Tolcher AW, Papadopoulos KP,
Beeram M, Rasco DW, Smith LS, et al: The clinical effect of the
dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK
pathways in patients with advanced cancer. Clin Cancer Res.
18:2316–2325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Galanis E, Buckner JC, Maurer MJ,
Kreisberg JI, Ballman K, Boni J, et al: Phase II trial of
temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a
north central cancer treatment group study. J Clin Oncol.
23:5294–5304. 2005. View Article : Google Scholar : PubMed/NCBI
|