1. Introduction
Hepatocellular carcinoma (HCC) is a global health
problem. HCC is one of the most common forms of cancer and the
third leading reason for cancer-related death (1). Unfortunately, there are no typical
clinical manifestations of early-stage HCC. Therefore, most
patients are identified at later stages of the disease, and are
often treated by therapies including radical liver resection and
liver transplantation. The long-term outcome of current palliative
treatments for HCC such as transcatheter arterial chemoembolization
(TACE) and radiofrequency ablation (RFA) is not satisfactory
(2,3). Thereby, there is an urgent need to
investigate the underlying molecular pathogenesis in order to
develop novel therapies for HCC.
Hedgehog (Hh) signaling pathways are objects of
intense investigation in HCC studies. Hh signaling contributes to
cell differentiation, organ formation, carcinogenesis and cancer
metastasis. Studies indicate that Hh signaling is aberrantly
activated to promote proliferation, viability, migration and
invasion of HCC cells (4–6). In this review, we summarize basic
components of the Hh pathway and its role in HCC pathogenesis.
2. Characterization of Hh signaling
Hh signaling was first identified by the Nobel
laureates Wieschaus and Nusslein-Volhard by mutagenesis screening
assays in Drosophila(7).
They found that Hh mutations lead to abnormal hedgehog-like
denticle formation in flies. This signaling pathway is conserved
from flies to vertebrates, and includes 7 main components: Hedgehog
(Hh), Patched (PTCH), Smoothened (Smo), GLIs, kinesin-like protein
Costal 2 (Cos 2), fused (Fu) and suppressor of fused (SuFu).
There are 3 types of Hh protein found in mammalians
including Sonic hedgehog (Shh), Indian hedgehog (Ihh) and Desert
hedgehog (Dhh) (8). The Hh protein
precursor consists of a C-terminal protease domain and an
N-terminal signaling unit. After undergoing a series of
modifications, the N-terminus of the Hh protein is modified with
palmitates (9), and its C-terminus
is bound to a cholesterol moiety (10), which facilitates Hh protein to be
secreted from cells.
Hg targets the PTCH receptor, which is a 12-span
transmembrane protein. PTCH acts as a negative regulator of the Hh
pathway by inhibiting the G protein-coupled receptor Smo in the
absence of Hh protein. After binding with Hh protein, PTCH allows
Smo to release GLIs from a multiple protein complex. These GLIs
then enter the cell nucleus to regulate transcription of target
genes. There are three GLI proteins found in humans including GLI1,
GLI2 and GLI3. The GLI1 gene was initially identified as being
amplified in human glioma (11–13).
GLI1, GLI2 and GLI3 share a highly similar protein
structure consisting of 5 conserved tandem zinc fingers, a relative
conserved N-terminal domain, several possible PKA sites and other
small conserved domains in the C-terminus. They also have a common
DNA-binding domain which can target the DNA sequence GACCACCCA
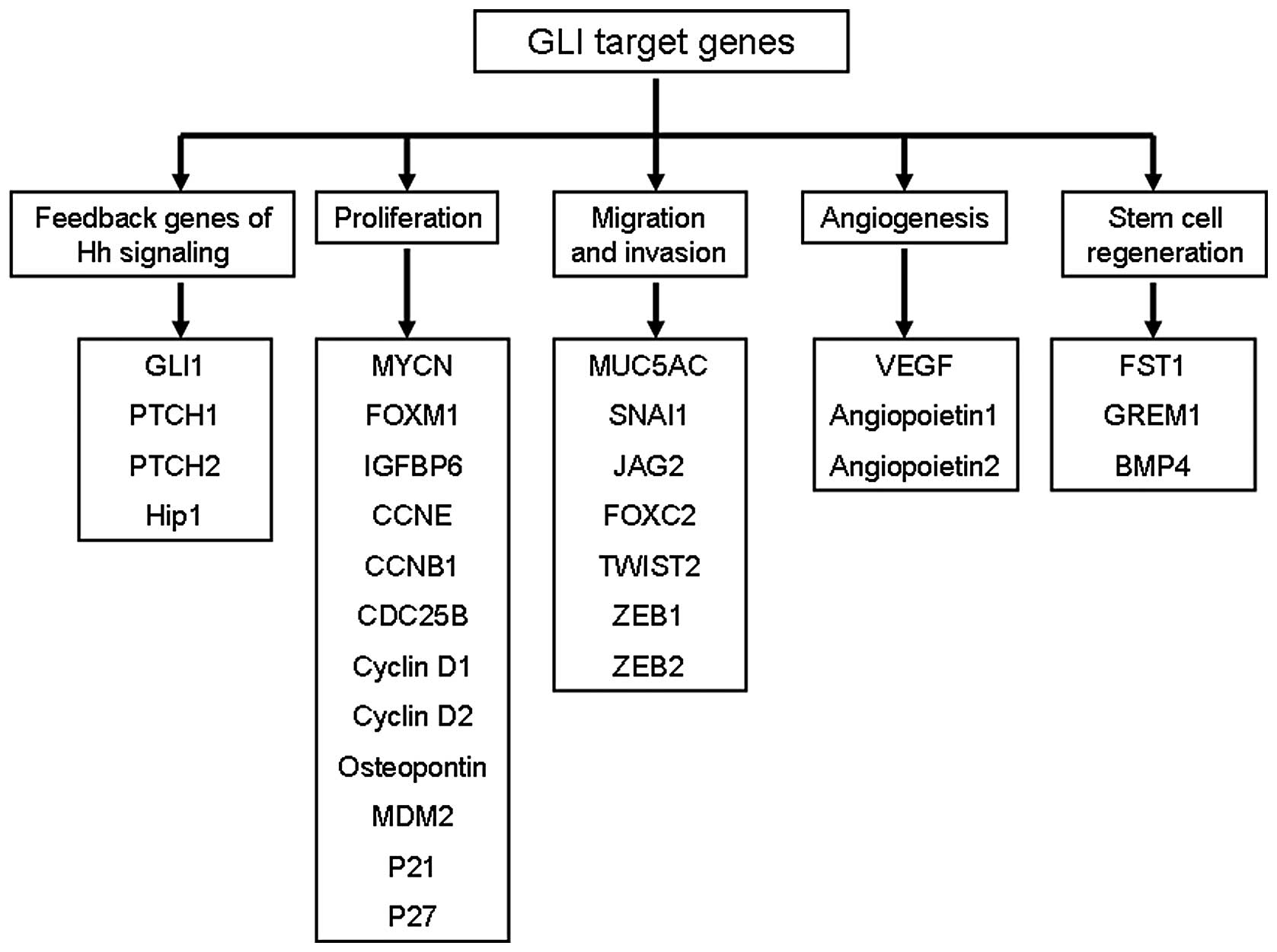
(14). As shown in Fig. 1, several GLI target genes have been
identified which affect cell proliferation [MYCN (15), CCND1 (16), CCND2 (17), FOXM1 (18)], stem regeneration [JAG2 (16), FST (19)], cell survival [BCL2 (16), CFLAR (20)], and EMT [FOXC2 (21), SNAI1 (4), TWIST2 (22)].
GLI proteins associate with Cos 2, Fu and SuFu to
join the GLI-Cos 2-Fu-SuFu complex in the cytoplasm in the absence
of Hh (23). While associated with
the cytoplasmic complex GLI proteins are sequestered from the
nucleus and target genes are not activated. In addition, protein
kinases including PKA, GSK3 and CK1 can promote phosphorylation of
GLIs to suppress there transcriptional activity. These
phosphorylated GLIs enter the cell nucleus to inhibit the
regulatory effect of Hh signaling on target genes by SIN3-HDAC or
SKI-HDAC dependent mechanisms (24).
When Hh protein binds with PTCH, Smo is activated
and translocates to primary cilia. Consequently, GLI proteins are
cleaved into activated forms, released from the cytoplasmic GLI-Cos
2-Fu-SuFu complex, and bind to promoter regions to regulate the
transcription of downstream genes. However, recent studies indicate
that GLI activation can also be controlled by Hh-independent
mechanisms. For example, the RAS-MEK/AKT pathway can modify the Hh
pathway in cancer cells (25).
Other studies suggest that TGFβ1 can induce GLI1 upregulation in
HCC (4).
3. Aberrant activation of Hh signaling in
cancers
Components of the Hh pathway are frequently mutated
in cancer. There have been three main events by which the Hh
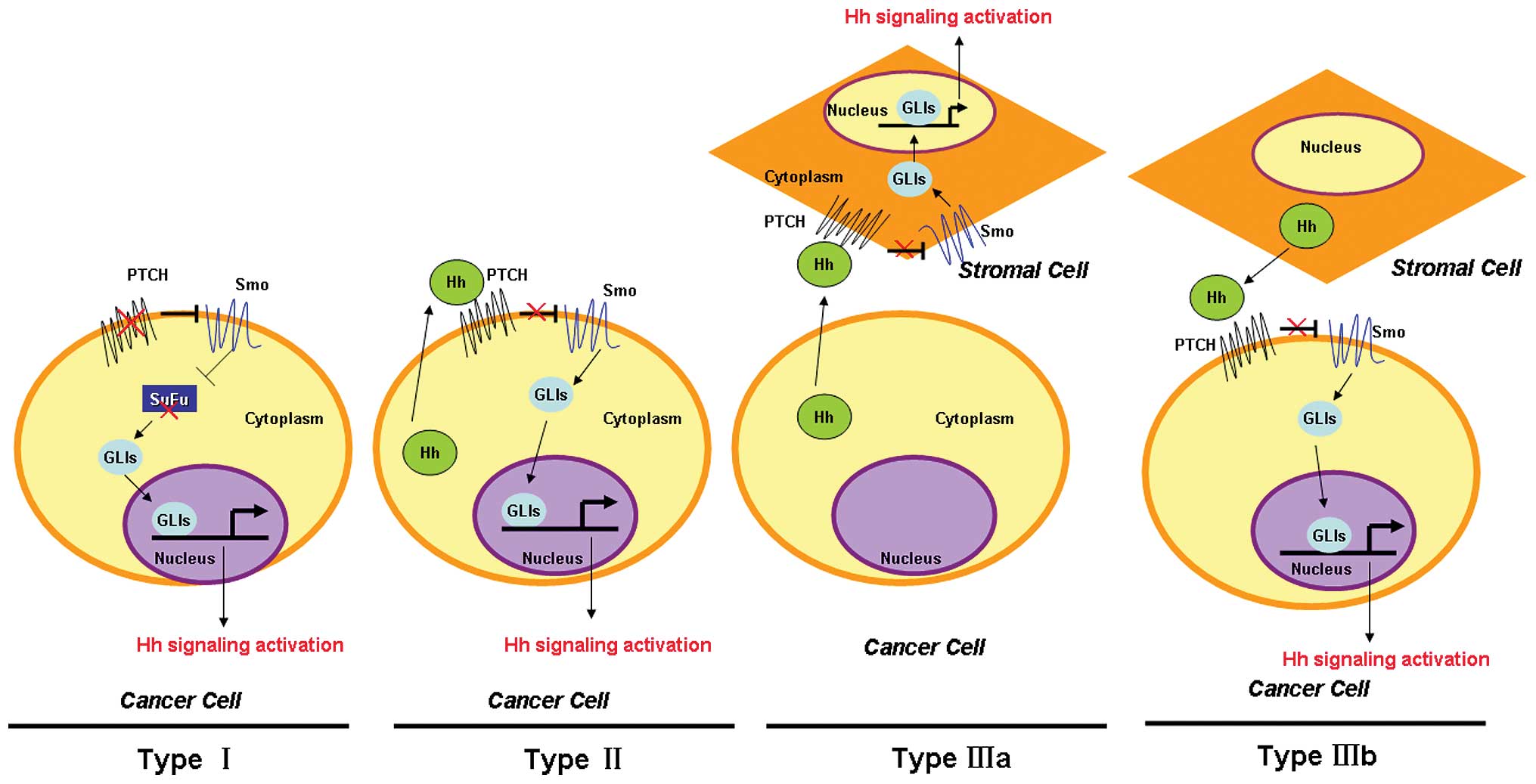
pathway is aberrantly activated in cancer (Fig. 2). Type I events underlie a
ligand-independent constitutive activation of Hh signaling due to
mutations that inactivate negative regulators including PTCH or
SuFu, and/or hyperactivation of Smo and GLI1, which are found in
basal cell carcinoma (BCC) (26)
and medulloblastoma (27). Type II
events lead to ligand-dependent autocrine signaling in which cancer
cells produce and secrete Hh ligands, which have been found in lung
cancer (28), gastrointestinal
cancer (29) and prostate cancer
(30). Type IIIa events cause
ligand-dependent paracrine affects by activating Hh signaling in
stromal cells as a result of Hh ligands excreted by cancer cells,
mostly in pancreatic cancer (31).
Type IIIb events arise as a ligand-dependent reverse paracrine
response in which the Hh pathway of cancer cells is activated by Hh
ligands excreted by stromal cells, which are found in malignant
lymphoma and plasmacytoma (32).
Hh signaling contributes to the pathogenesis of
various cancers. For example, GLI1 was found to be highly amplified
in human glioblastoma tissues and derived cell lines and is
attributed to gliomagenesis (33).
Aberration of Hh signaling was also found in medulloblastoma which
is the leading malignant pediatric brain tumor worldwide.
Experiments demonstrate that aberrant activation of
Hh signaling promotes brain tumor growth via upregulation of cell
proliferation (34). It has become
clear that a majority of basal cell carcinoma (BCC) patients have
aberrant activation of the Hh pathway (35). Consistently, there is often
constitutive activation of downstream targets of the Hh pathway in
BCC. For example, Watkins et al(36) found that Shh factors are
overexpressed in small-cell lung cancer (SCLC). Furthermore, the Hh
pathway in lung cancer can be activated in a Type IIIa manner by an
Hh ligand-dependent paracrine mechanism.
Lauth et al(37) report that Hh signaling of stromal
cells in lung cancer can be activated by Shh secreted by cancer
cells, and that activated stromal cells secrete numerous cytokines
to promote the malignant phenotype of lung cancer cells. In
addition, constitutive Hh signaling has also been identified as a
critical mechanism leading to breast cancer (38). Various mutations of Hh signaling,
including inactivating mutations of PTCH, activating missense
mutations of Smo, and loss of function mutations of SuFu are found
in breast cancer and are implicated in mammary carcinoma
development (39,40).
In addition to mutations, epigenetic regulatory
mechanisms can lead to activation of Hh signaling. These include
hypermethylation of the promoters of hedgehog interacting protein
(Hip) and PTCH, and hypomethylation of Shh promoters (41). Aberrant Hh signaling has been found
in colon cancer, and appears to promote the progression of colon
cancer from local adenoma in colon epithelium to advanced adenoma
with distant metastasis (42).
Multiple mechanisms mediated by ligand-dependent and
ligand-independent signaling cause hyperactivation of Hh signaling
in colon cancer. For example, overexpression of Shh has been found
in colon cancer tissues (42),
while both loss-of-function PTCH mutations and gain-of-function Smo
mutations are frequently found in colon cancers (43). Indeed, aberrant activation of Hh
signaling has been shown to promote proliferation, migration, and
invasion of colon cancer cells (44,45).
4. The role of Hh signaling in HCC
As mentioned above, the Hh pathway is altered and
contributes to the development and progression of many types of
cancer. These include liver cancer, prostate cancer, neuroblastoma
and ovarian cancer. The role of Hh in HCC is particularly
compelling.
Many groups have found aberrant activation of Hh
signaling in HCC (4,6,46).
However, the underlying mechanisms of Hh activation in HCC are
complex. Lu et al(46)
reported that Shh treatment at a concentration of 0.5 μg/ml
increased GLI1 expression and promoted HCC cell invasion and
migration. These results indicate that Hh signaling in HCC can be
activated by a ligand-dependent manner. Others have also found that
Shh treatment can stimulate Hh signaling in HCC cells (47).
Interestingly, Sicklick et al(48) found overexpression of Smo and an
increase in the stoichiometric ratio of Smo to PTCH mRNA levels in
HCC, and that this effect is related with tumor size and may be a
prognostic marker of liver cancer. Sicklick et al(48) also found mutant Smo expression in
Hep3B cells and that cyclopamine (an inhibitor against wild-type
Smo) did not affect Hh signaling activation and growth of these
cells. However, after treatment with KAAD-cyclopamine, which is a
blocker of mutant Smo, Hh signaling activity in Hep3B was repressed
by ~50% and the growth rate was decreased by 94%.
Tada et al(49) found hypermethylation of Hip
promoters and loss of heterozygosity (LOH) at the Hip locus, which
was attributed to downregulation of Hip in HCC tissues. Recent
experiments also found that HCC cells secrete Shh to induce
glycolysis of neighboring MFs, which consequently leads to the
production of myofibroblast-derived lactate that HCC cells use as
an energy source (50). These
results indicate that Hh ligand-dependent paracrine manner (Type
IIIa) may play an important role in the pathogenesis of HCC.
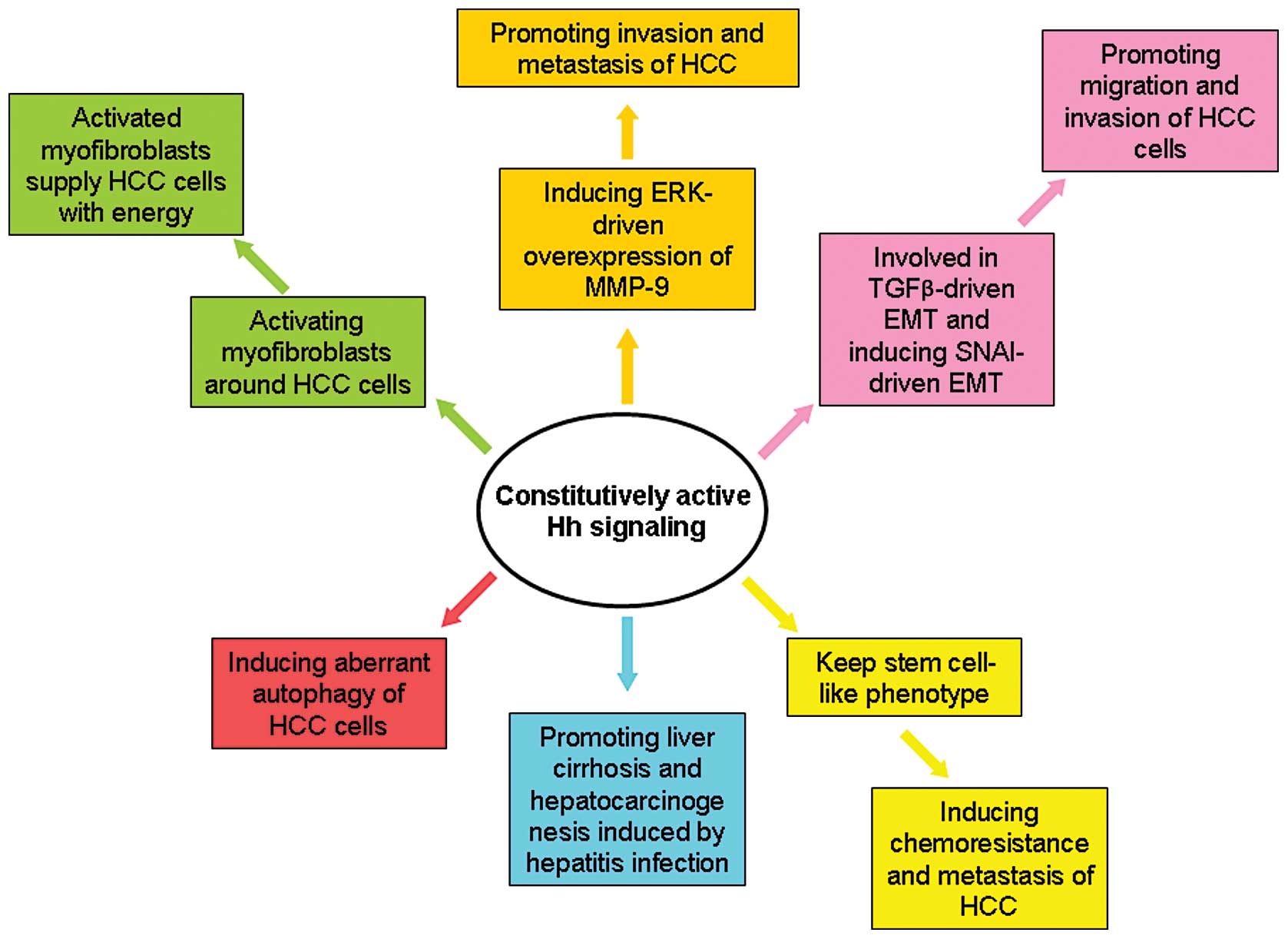
As shown in Fig. 3,
several studies revealed the key effects of Hh signaling on HCC.
Previous studies have found that GLI1 expression is positively
correlated with the EMT phenotype and with intrahepatic metastasis
and portal venous invasion of human HCCs (6). In addition, gene expression microarray
analysis found that GLI1 mRNA overexpression in HCC tissues was
associated with rapid recurrence of HCC tumors after surgery
(4). Other studies used
immunohistochemistry to find that GLI1 expression in HCC tissues is
associated with disease-free survival and overall survival. In
contrast, in vitro experiments indicated that forced
expression of GLI1 promotes proliferation, viability, colony
formation, migration and invasion of Huh7 cells, while silencing
GLI1 expression in SNU398 cells produced opposite results.
To elucidate the mechanisms that may underlie the
effects of GLI1 on HCC growth and motility, we searched the
sequence of the SNAI1 promoter to find potential GLI1 protein
binding sites. Through chromatin immunoprecipitation (ChIP) assays,
GLI1 was found to bind to a region in the SNAI1 promoter located
−1,417/−1,214 bp upstream of the transcriptional start site.
Additionally, overexpression of GLI1 induced upregulation of SNAI1
in Huh7 cells and knockdown of GLI1 in SNU398 cells decreased the
expression of SNAI1. Consistently, the EMT phenotype of Huh7 cells
was induced by upregulation of GLI1, and knockdown of GLI1 reversed
EMT to MET in SNU398 cells. These results strongly indicate that
GLI1 overexpression is essential for HCC cells to obtain and
maintain their EMT phenotype. On the other hand, we also found that
TGFβ1 treatment leads to upregulation of GLI1, which is necessary
for TGFβ1-driven EMT in Huh7 cells. During investigation of the
anti-HCC function of sulfatase 2 inhibitor OKN-007, we found that
activity of Hh signaling was modulated by sulfatase 2 in HCC cells
(3).
Hepatitis B virus encoded X protein (HBx) can also
contribute to the pathogenesis of HCC. Arzumanyan et
al(51) found a positive
relationship between HBx and Hh signaling components in HCC cell
lines, samples of HCC secondary to HBV infection and relevant mouse
specimens. Inhibiting Hh signaling eliminated the ability of HBx to
promote cell migration, anchorage-independent growth, and tumor
development of HCC. Hence, it seems that activation of Hh signaling
is necessary for HBx to accelerate hepatocarcinogenesis.
Pereira et al(52) found increased hepatic expression of
Hh ligands in all patients with chronic hepatitis during liver
cirrhosis and HCC. In addition, inhibiting Hh signaling in these
Hh-responsive cells blocked fibrosis. Based on these findings, it
seems that hepatitis infection can increase the production of Hh
ligands in hepatocytes and liver accumulation of Hh-responsive
cells, which aggravates liver cirrhosis and
hepatocarcinogenesis.
Autophagy plays a central role in controlling
apoptosis of HCC cells. Wang et al(47) found that activating the Hh pathway
through treatment of Shh and its agonists (SAG and purmorphamine)
blocked the induction of autophagy in several HCC cell lines. In
addition, inhibition of Hh signaling by GANT61, which is a
small-molecule inhibitor of GLI1, induced autophagy. These results
demonstrate that Hh signaling is involved in aberrant autophagy of
HCC cells.
Activation of Hh signaling was also found to promote
the invasion and metastasis of HCC via mediating the ERK-driven
overexpression of MMP-9 (46). EMT
is believed to be the critical event to promote migration and
invasion of cancer cells. Our preliminary clinical data indicate
that activation of Hh signaling is correlated positively with the
mesenchymal marker S100a4 and clinicopathological characteristics
indicative of an enhanced metastatic potential of HCC (6), and negatively correlated with
expression of epithelial marker E-cadherin. In addition, forced
expression of GLI1 was found to trigger HCC EMT in vitro.
Experiments utilizing Mdr2 knockout mice provide convincing
evidence of an important role of Hh signaling in the development of
HCC from liver cirrhosis (53).
These experiments found that Hh signaling was activated aberrantly
in Mdr2 knockout mice compared with wild-type mice. Chen et
al(54) reported that
well-differentiated CD133(+)/ALDH(high) or CD133(+)/EpCAM(+) HCC
cells (Huh7 and Hep3B cells) displayed features similar to HCC stem
cells. These cancer stem-like cells were found to have enhanced Hh
signaling activity which was responsible for their chemoresistance
and tumor invasion.
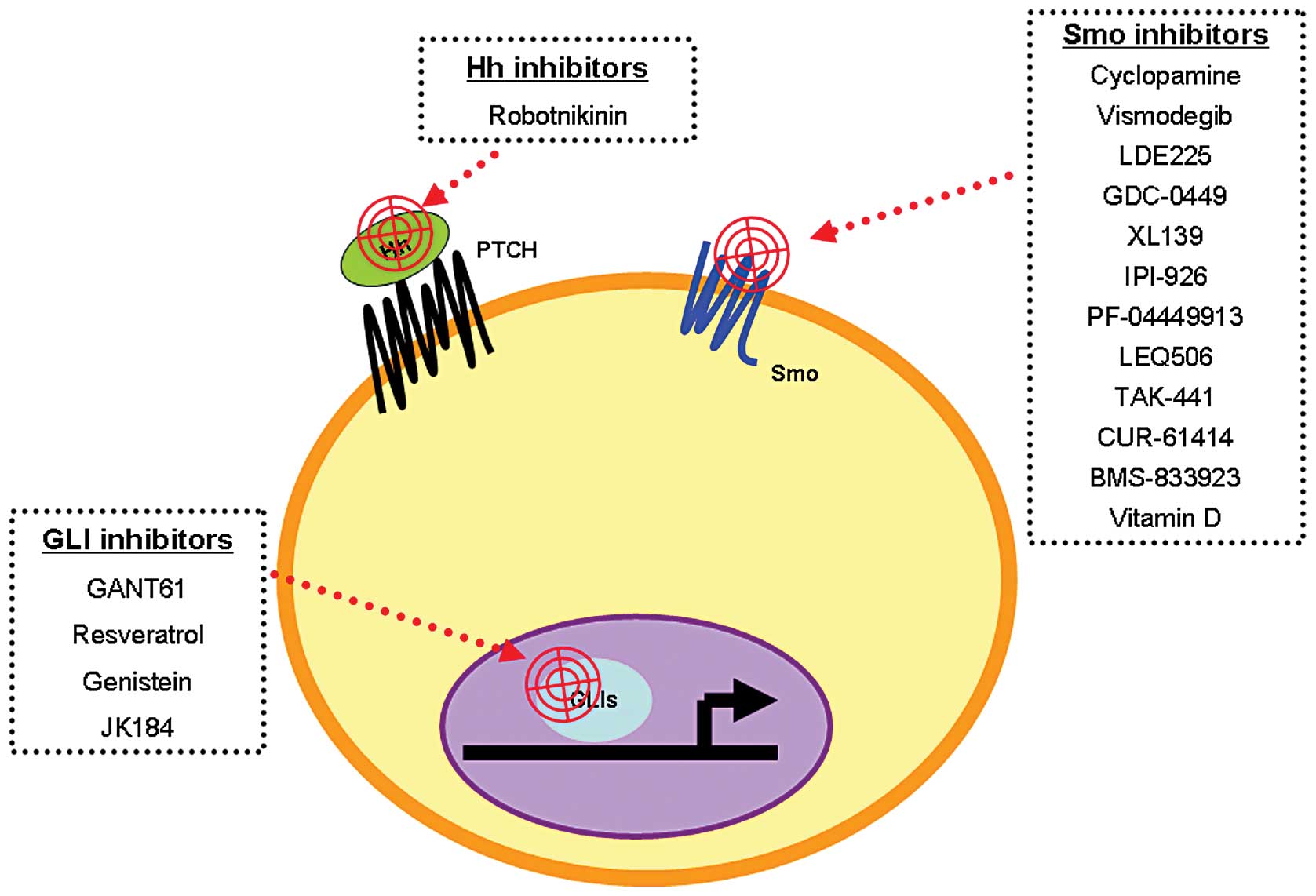
5. Hh target therapies for cancer
The first Hh signaling inhibitor, cyclopamine, was
found by Cooper et al(55)
in 1998. Since then, other small-molecule Hh signaling inhibitors
have been designed for in vitro and in vivo studies
of cancer treatment (briefly summarized in Fig. 4). These inhibitors target different
components of Hh signaling including Smo, Shh and GLI1 (56). Some of these compounds have been
tested in clinical trials; for example, vismodegib/GDC-0449, LED225
and GANT61.
Vismodegib, also named GDC-0449, is a small molecule
designed by Curis Genentech to target Smo. The antitumor effect of
vismodegib was first identified in a medulloblastoma allograft
mouse model, in which vismodegib treatment at doses of 12.5 mg/kg
BID repressed the growth of medulloblastoma completely (57). Preclinical studies also found
similar antitumor effects of vismodegib on colon cancer, pancreatic
cancer, lung cancer, esophageal cancer and gastric cancer (58–60).
These experiments found that GDC-0449 represses cell growth and
promotes the apoptosis of pancreatic cancer cells by activating
TRAIL-R/DR signaling, caspase-3 signaling, and inducing PARP
cleavage (61).
Most clinical trials of vismodegib have been carried
out in BCCs and ovarian cancers. In a phase I clinical trial of
BCCs, vismodegib showed a 55% response rate in 33 advanced BCC
patients and was well tolerated (62). The most common side-effects included
muscle spasms, altered taste, weight loss and hyponatremia.
Phase II clinical trials of vismodegib were
performed in advanced ovarian cancer and BCCs. These clinical
trials on advanced ovarian cancer conducted by Genentech found that
the average time for the disease to progress was 7.5 months in the
vismodegib group compared with 5.8 months in the placebo group
(63). Phase II clinical trials of
vismodegib on BCCs found an objective response rate in metastatic
basal cell carcinoma that reached up to 30.3%, and an objective
response rate in locally advanced basal cell carcinoma of 42.9%
(64). On the basis of these
clinical trials (65), the US Food
and Drug Administration approved the use of Erivedge™ (vismodegib)
capsules for the treatment of patients with recurrent, locally
advanced or metastatic BCC in January 30, 2012.
LDE225 is another inhibitor of Smo designed by
Novartis. Preclinical experiments found that it suppresses the
development of BCC. Novartis conducted 2 clinical trials of LDE225
for cancer therapy using different methods of administration. An
LDE225 cream was used to treat nevoid basal cell carcinoma syndrome
(NBCCS) in one phase I clinical trial which found a 92.3% (12/13)
clinical response rate and a mean volume reduction of 49.8% in the
LDE225 group compared to 9.1% in the placebo group (66). Interestingly, Hh signaling in NBCCS
tissues was also inhibited by this treatment.
Another phase I clinical trial was carried out to
test an oral dosing regimen of LDE225 in patients with advanced
solid tumors. As expected, GLI1 mRNA was reduced when the dose of
LDE225 was increased. Side-effects included fatigue, nausea,
vomiting, anorexia, muscle cramps and dysgeusia (67). Unfortunately, apparent
drug-resistance was observed in these clinical trials, which
thwarted its clinical development. This drug-resistance was
considered to be associated with overexpression of Gli2 and point
mutations in Smo, which maintained constitutive activation of Hh
signaling in solid tumors.
GANT61 is a hexahydropyrimidine derivative designed
as a small molecular inhibitor that targets GLI1. Lauth et
al(68) first identified its
ability to reduce GLI1-mediated transcription and named it GANT61
(for Gli-ANTagonist). Although it has not yet been tested in
clinical trials, GANT61 has displayed promising antitumor effects
in several preclinical studies. GANT-61 was found to inhibit cell
viability, spheroid formation, and GLI-DNA binding and
transcriptional activities in pancreatic cancer stem cells
(69). Moreover, GANT61 was found
to induce autophagy, apoptosis and cytotoxicity in HCC cells in
vitro and can inhibit tumor growth in SCID mice (47). Because of its favorable antitumor
effect in a variety of cancers, this compound may serve as a
potential targeting therapy against Hh signaling.
6. Conclusions
In normal adult liver tissues, Hh signaling is
inactivated. However, constitutive activation of Hh signaling is
found in lesions from chronic liver injuries and cirrhosis to HCC.
Evidence demonstrates that alterations of Hh signaling promote HCC
development and progression via different mechanisms. There are
several small-molecule drugs specifically designed to target Hh
signaling that are undergoing clinical trials. Some of these have
been approved to treat BCC in the clinic. It is likely that
targeting Hh signaling will develop into an important part of a
comprehensive strategy to combat advanced HCCs and bring a brighter
future to liver cancer patients.
Acknowledgements
This study was supported by grants from the National
Natural Scientific Foundation of China (nos. 81272645 and 81072052
to Q.L), the Research Fund for the Doctoral Program of High
Education of China from the Ministry of Education (no.
20120201120090 to X.Z.), and the Fundamental Research Funds for the
Basic Research Operating Expenses Program of Central College
sponsored by Xi’an Jiaotong University to X.Z. The authors thank
Professor Gary S. Goldberg (Rowan University, NJ, USA) for the
technical assistance.
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
Hh
|
Hedgehog
|
|
TACE
|
transcatheter arterial
chemoembolization
|
|
RFA
|
radiofrequency ablation
|
|
EMT
|
epithelial-mesenchymal transition
|
|
Shh
|
Sonic hedgehog
|
|
Ihh
|
Indian hedgehog
|
|
Dhh
|
Desert hedgehog
|
|
PTCH
|
Patched
|
|
Smo
|
Smoothened
|
|
Cos 2
|
kinesin-like protein Costal 2
|
|
Fu
|
fused
|
|
SuFu
|
suppressor of fused
|
|
BCC
|
basal cell carcinoma
|
|
Hip
|
hedgehog interacting protein
|
|
MF
|
myofibroblast
|
|
LOH
|
loss of heterozygosity
|
|
HBx
|
hepatitis B virus encoded X
protein
|
|
NBCCS
|
Nevoid basal cell carcinoma
syndrome
|
References
|
1
|
Yang JD and Roberts LR: Hepatocellular
carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 7:448–458.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sandhu DS, Tharayil VS, Lai JP and Roberts
LR: Treatment options for hepatocellular carcinoma. Expert Rev
Gastroenterol Hepatol. 2:81–92. 2008. View Article : Google Scholar
|
|
3
|
Zheng X, Gai X, Han S, et al: The human
sulfatase 2 inhibitor 2,4-disulfonylphenyl-tert-butylnitrone
(OKN-007) has an antitumor effect in hepatocellular carcinoma
mediated via suppression of TGFB1/SMAD2 and Hedgehog/GLI1
signaling. Genes Chromosomes Cancer. 52:225–236. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zheng X, Vittar NB, Gai X, et al: The
transcription factor GLI1 mediates TGFβ1 driven EMT in
hepatocellular carcinoma via a SNAI1-dependent mechanism. PLoS One.
7:e495812012.
|
|
5
|
Xu QR, Zheng X, Zan XF, Yao YM, Yang W and
Liu QG: Gli1 expression and its relationship with the expression of
Shh, Vimentin and E-cadherin in human hepatocellular carcinoma. Xi
Bao Yu Fen Zi Mian Yi Xue Za Zhi. 28:536–539. 2012.(In
Chinese).
|
|
6
|
Zheng X, Yao Y, Xu Q, Tu K and Liu Q:
Evaluation of glioma-associated oncogene 1 expression and its
correlation with the expression of sonic hedgehog, E-cadherin and
S100a4 in human hepatocellular carcinoma. Mol Med Rep. 3:965–970.
2010.PubMed/NCBI
|
|
7
|
Nüsslein-Volhard C and Wieschaus E:
Mutations affecting segment number and polarity in
Drosophila. Nature. 287:795–801. 1980.
|
|
8
|
Heretsch P, Tzagkaroulaki L and Giannis A:
Modulators of the hedgehog signaling pathway. Bioorg Med Chem.
18:6613–6624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pepinsky RB, Zeng C, Wen D, et al:
Identification of a palmitic acid-modified form of human Sonic
hedgehog. J Biol Chem. 273:14037–14045. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Porter JA, Young KE and Beachy PA:
Cholesterol modification of hedgehog signaling proteins in animal
development. Science. 274:255–259. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kinzler KW, Bigner SH, Bigner DD, et al:
Identification of an amplified, highly expressed gene in a human
glioma. Science. 236:70–73. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Corbit KC, Aanstad P, Singla V, Norman AR,
Stainier DY and Reiter JF: Vertebrate Smoothened functions at the
primary cilium. Nature. 437:1018–1021. 2005. View Article : Google Scholar
|
|
13
|
Huangfu D and Anderson KV: Cilia and
Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA.
102:11325–11330. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ruiz i Altaba A, Mas C and Stecca B: The
Gli code: an information nexus regulating cell fate, stemness and
cancer. Trends Cell Biol. 17:438–447. 2007.PubMed/NCBI
|
|
15
|
Kenney AM, Cole MD and Rowitch DH: Nmyc
upregulation by sonic hedgehog signaling promotes proliferation in
developing cerebellar granule neuron precursors. Development.
130:15–28. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kasper M, Schnidar H, Neill GW, et al:
Selective modulation of Hedgehog/GLI target gene expression by
epidermal growth factor signaling in human keratinocytes. Mol Cell
Biol. 26:6283–6298. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoon JW, Kita Y, Frank DJ, et al: Gene
expression profiling leads to identification of GLI1-binding
elements in target genes and a role for multiple downstream
pathways in GLI1-induced cell transformation. J Biol Chem.
277:5548–5555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Teh MT, Blaydon D, Chaplin T, et al:
Genomewide single nucleotide polymorphism microarray mapping in
basal cell carcinomas unveils uniparental disomy as a key somatic
event. Cancer Res. 65:8597–8603. 2005. View Article : Google Scholar
|
|
19
|
Eichberger T, Kaser A, Pixner C, et al:
GLI2-specific transcriptional activation of the bone morphogenetic
protein/activin antagonist follistatin in human epidermal cells. J
Biol Chem. 283:12426–12437. 2008. View Article : Google Scholar
|
|
20
|
Kump E, Ji J, Wernli M, Häusermann P and
Erb P: Gli2 upregulates cFlip and renders basal cell carcinoma
cells resistant to death ligand-mediated apoptosis. Oncogene.
27:3856–3864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hallikas O, Palin K, Sinjushina N, et al:
Genome-wide prediction of mammalian enhancers based on analysis of
transcription-factor binding affinity. Cell. 124:47–59. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Deng W, Lobo-Ruppert SM and Ruppert
JM: Gli1 acts through Snail and E-cadherin to promote nuclear
signaling by β-catenin. Oncogene. 26:4489–4498. 2007.PubMed/NCBI
|
|
23
|
Kasper M, Regl G, Frischauf AM and Aberger
F: GLI transcription factors: mediators of oncogenic Hedgehog
signalling. Eur J Cancer. 42:437–445. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fernandez-Zapico ME: Primers on molecular
pathways GLI: more than just Hedgehog? Pancreatology. 8:227–229.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stecca B, Mas C, Clement V, et al:
Melanomas require HEDGEHOG-GLI signaling regulated by interactions
between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci USA.
104:5895–5900. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kudchadkar R, Lewis K and Gonzalez R:
Advances in the treatment of Basal cell carcinoma: Hedgehog
inhibitors. Semin Oncol. 39:139–144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang X, Venugopal C, Manoranjan B, et al:
Sonic hedgehog regulates Bmi1 in human medulloblastoma brain
tumor-initiating cells. Oncogene. 31:187–199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Singh S, Wang Z, Liang Fei D, et al:
Hedgehog-producing cancer cells respond to and require autocrine
Hedgehog activity. Cancer Res. 71:4454–4463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Saqui-Salces M and Merchant JL: Hedgehog
signaling and gastrointestinal cancer. Biochim Biophys Acta.
1803:786–795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chung MK, Kim HJ, Lee YS, et al: Hedgehog
signaling regulates proliferation of prostate cancer cells via
stathmin1. Clin Exp Med. 10:51–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tian H, Callahan CA, DuPree KJ, et al:
Hedgehog signaling is restricted to the stromal compartment during
pancreatic carcinogenesis. Proc Natl Acad Sci USA. 106:4254–4259.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dierks C, Grbic J, Zirlik K, et al:
Essential role of stromally induced hedgehog signaling in B-cell
malignancies. Nat Med. 13:944–951. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Becher OJ, Hambardzumyan D, Fomchenko EI,
et al: Gli activity correlates with tumor grade in platelet-derived
growth factor-induced gliomas. Cancer Res. 68:2241–2249. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Romer JT, Kimura H, Magdaleno S, et al:
Suppression of the Shh pathway using a small molecule inhibitor
eliminates medulloblastoma in
Ptc1+/−p53−/− mice. Cancer
Cell. 6:229–240. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gailani MR, Ståhle-Bäckdahl M, Leffell DJ,
et al: The role of the human homologue of Drosophila patched
in sporadic basal cell carcinomas. Nat Genet. 14:78–81.
1996.PubMed/NCBI
|
|
36
|
Watkins DN, Berman DM, Burkholder SG, Wang
B, Beachy PA and Baylin SB: Hedgehog signalling within airway
epithelial progenitors and in small-cell lung cancer. Nature.
422:313–317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lauth M, Bergström A, Shimokawa T, et al:
DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling
by mutant RAS. Nat Struct Mol Biol. 17:718–725. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Katano M: Hedgehog signaling pathway as a
therapeutic target in breast cancer. Cancer Lett. 227:99–104. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hatsell S and Frost AR: Hedgehog signaling
in mammary gland development and breast cancer. J Mammary Gland
Biol Neoplasia. 12:163–173. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kasper M, Jaks V, Fiaschi M and Toftgård
R: Hedgehog signalling in breast cancer. Carcinogenesis.
30:903–911. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang LH, Choi YL, Hua XY, et al: Increased
expression of sonic hedgehog and altered methylation of its
promoter region in gastric cancer and its related lesions. Mod
Pathol. 19:675–683. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Varnat F, Duquet A, Malerba M, et al:
Human colon cancer epithelial cells harbour active HEDGEHOG-GLI
signalling that is essential for tumour growth, recurrence,
metastasis and stem cell survival and expansion. EMBO Mol Med.
1:338–351. 2009. View Article : Google Scholar
|
|
43
|
Mazumdar T, DeVecchio J, Agyeman A, Shi T
and Houghton JA: The GLI genes as the molecular switch in
disrupting Hedgehog signaling in colon cancer. Oncotarget.
2:638–645. 2011.PubMed/NCBI
|
|
44
|
Wang WS, Chen PM and Su Y: Colorectal
carcinoma: from tumorigenesis to treatment. Cell Mol Life Sci.
63:663–671. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Theunissen JW and de Sauvage FJ: Paracrine
Hedgehog signaling in cancer. Cancer Res. 69:6007–6010. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lu JT, Zhao WD, He W and Wei W: Hedgehog
signaling pathway mediates invasion and metastasis of
hepatocellular carcinoma via ERK pathway. Acta Pharmacol Sin.
33:691–700. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang Y, Han C, Lu L, Magliato S and Wu T:
Hedgehog signaling pathway regulates autophagy in human
hepatocellular carcinoma cells. Hepatology. Mar 16–2013.(Epub ahead
of print).
|
|
48
|
Sicklick JK, Li YX, Jayaraman A, et al:
Dysregulation of the Hedgehog pathway in human
hepatocarcinogenesis. Carcinogenesis. 27:748–757. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tada M, Kanai F, Tanaka Y, et al:
Down-regulation of hedgehog-interacting protein through genetic and
epigenetic alterations in human hepatocellular carcinoma. Clin
Cancer Res. 14:3768–3776. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chan IS, Guy CD, Chen Y, et al: Paracrine
Hedgehog signaling drives metabolic changes in hepatocellular
carcinoma. Cancer Res. 72:6344–6350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Arzumanyan A, Sambandam V, Clayton MM, et
al: Hedgehog signaling blockade delays hepatocarcinogenesis induced
by hepatitis B virus X protein. Cancer Res. 72:5912–5920. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
de Pereira TA, Witek RP, Syn WK, et al:
Viral factors induce Hedgehog pathway activation in humans with
viral hepatitis, cirrhosis, and hepatocellular carcinoma. Lab
Invest. 90:1690–1703. 2010.PubMed/NCBI
|
|
53
|
Philips GM, Chan IS, Swiderska M, et al:
Hedgehog signaling antagonist promotes regression of both liver
fibrosis and hepatocellular carcinoma in a murine model of primary
liver cancer. PLoS One. 6:e239432011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen X, Lingala S, Khoobyari S, Nolta J,
Zern MA and Wu J: Epithelial mesenchymal transition and hedgehog
signaling activation are associated with chemoresistance and
invasion of hepatoma subpopulations. J Hepatol. 55:838–845. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cooper MK, Porter JA, Young KE and Beachy
PA: Teratogen-mediated inhibition of target tissue response to Shh
signaling. Science. 280:1603–1607. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rubin LL and de Sauvage FJ: Targeting the
Hedgehog pathway in cancer. Nat Rev Drug Discov. 5:1026–1033. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Robarge KD, Brunton SA, Castanedo GM, et
al: GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med
Chem Lett. 19:5576–5581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yauch RL, Gould SE, Scales SJ, et al: A
paracrine requirement for hedgehog signalling in cancer. Nature.
455:406–410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tian F, Mysliwietz J, Ellwart J, Gamarra
F, Huber RM and Bergner A: Effects of the Hedgehog pathway
inhibitor GDC-0449 on lung cancer cell lines are mediated by side
populations. Clin Exp Med. 12:25–30. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ailles L and Siu LL: Targeting the
Hedgehog pathway in cancer: can the spines be smoothened? Clin
Cancer Res. 17:2071–2073. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Singh BN, Fu J, Srivastava RK and Shankar
S: Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits
pancreatic cancer stem cell characteristics: molecular mechanisms.
PLoS One. 6:e273062011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Von Hoff DD, LoRusso PM, Rudin CM, et al:
Inhibition of the hedgehog pathway in advanced basal-cell
carcinoma. N Engl J Med. 361:1164–1172. 2009.PubMed/NCBI
|
|
63
|
De Smaele E, Ferretti E and Gulino A:
Vismodegib, a small-molecule inhibitor of the hedgehog pathway for
the treatment of advanced cancers. Curr Opin Investig Drugs.
11:707–718. 2010.PubMed/NCBI
|
|
64
|
Rudin CM: Vismodegib. Clin Cancer Res.
18:3218–3222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Axelson M, Liu K, Jiang X, et al: U.S.
Food and Drug Administration approval: vismodegib for recurrent,
locally advanced, or metastatic basal cell carcinoma. Clin Cancer
Res. 19:2289–2293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Skvara H, Kalthoff F, Meingassner JG, et
al: Topical treatment of Basal cell carcinomas in nevoid Basal cell
carcinoma syndrome with a smoothened inhibitor. J Invest Dermatol.
131:1735–1744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li Y, Maitah MY, Ahmad A, Kong D, Bao B
and Sarkar FH: Targeting the Hedgehog signaling pathway for cancer
therapy. Expert Opin Ther Targets. 16:49–66. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lauth M, Bergström A, Shimokawa T and
Toftgård R: Inhibition of GLI-mediated transcription and tumor cell
growth by small-molecule antagonists. Proc Natl Acad Sci USA.
104:8455–8460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fu J, Rodova M, Roy SK, et al: GANT-61
inhibits pancreatic cancer stem cell growth in vitro and in
NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 330:22–32.
2013. View Article : Google Scholar : PubMed/NCBI
|