Introduction
Cell cycle progression is regulated by a series of
cyclin-dependent kinases (CDKs) and cyclin-dependent kinase
inhibitors (CDKIs). p16INK4a, a member of the INK4
family of CDKIs and a tumor suppressor, inhibits CDK4, maintaining
the hyperphosphorylation of retinoblastoma (Rb) that suppresses
cell cycle progression (1). The
p16-cyclin D1-CDK4-Rb pathway regulates cell cycle transition from
the G1 to the S phase; point mutations and/or epigenetic
modifications in this pathway are observed in almost all human
cancers (2,3). In addition to regulating the CDK4-Rb
pathway, p16INK4a decreases eukaryotic elongation factor
(eEF) 1A2 expression, reducing cancer cell proliferation (4). It also induces cellular senescence in
a cooperative manner with p21Waf1/Cip1, which was
confirmed in double-knockout mice that were more susceptible to
skin tumor formation (5). Moreover,
enforced expression of p16INK4a in cisplatin-resistant
non-small cell lung cancer cells increased their sensitivity to low
cisplatin concentrations (6). Thus,
treatments that increase p16INK4a expression may be
beneficial in reducing cancer cell growth.
Epigenetic modifications, such as the methylation of
CpG islands within gene promoters and histone deacetylation, can
inactivate tumor-suppressor genes, including
p16INK4a. Specifically, epigenetic
regulation of p16INK4a expression through
DNA methylation and/or histone deacetylation has been observed in
many types of human cancers (2).
Methylation of p16INK4a may be an early
event in carcinogenesis (7).
Furthermore, the detection of p16INK4a hypermethylation
at higher concentrations in the plasma of breast cancer patients
than in healthy control patients suggests that it may be a suitable
tool for cancer screening, and its prognostic value has been
suggested for breast and prostate cancer patients (7,8).
Epigallocatechin-3-gallate (EGCG), a green
tea-derived polyphenol, was found to reduce DNA methylation of the
p16INK4a promoter, reactivating its
expression in the human pancreatic carcinoma cell line, PANC-1 and
in human skin cancer cells (9,10). In
addition, trichostatin A (TSA), a histone deacetylase (HDAC)
inhibitor, elevated histone deacetylation of the HBP1 region,
thereby inducing p16INK4a expression and subsequent cell
apoptosis and differentiation (11). Thus, DNA methyltransferases (DNMTs)
and HDACs may represent important therapeutic targets for the
design of antitumor drugs (12,13).
The present study sought to prove the hypothesis
that EGCG and TSA may synergistically enhance p16INK4a
expression via epigenetic modification. After treatment with EGCG
and TSA alone or in combination, cell proliferation, cell cycle
progression, promoter methylation, and p16INK4a
expression were examined in human Burkitt lymphoma CA46 cells.
Materials and methods
Reagent preparation and cell culture
EGCG (Sigma, St. Louis, MO, USA) was diluted to 10
mg/ml with phosphate-buffered saline (PBS) (pH 7.4). TSA (Sigma)
was diluted to 2 mg/ml with dimethyl sulfoxide (DMSO). Both were
stored at −20°C until further use. The CA46 human Burkitt lymphoma
cell line (CRL-1648™, ATCC, Atlanta, GA, USA) was maintained in
RPMI-1640 complete medium (Gibco, Carlsbad, CA, USA) containing 10%
fetal bovine serum (FBS) (Sijiqing, Zhejiang, China) at 37°C with
5% CO2.
Cell proliferation analysis
Cell proliferation was assessed with the MTT assay
(Sigma) following the manufacturer’s instructions. Cells
(4×103/well) were seeded onto 96-well plates in 90 μl of
growth medium containing the indicated treatment. On days 1, 2, 3
and 4, 10 μl of MTT solution was added. After 2–3 h at 37°C,
optical density (OD) was measured at 450 nm, and the inhibition
rate was determined as follows: [1 − (ODexperiment −
ODblank)/(ODnegative − ODblank)] ×
100%. This experiment was performed three times.
Cell cycle analysis
CA46 cells (1×106/well) were cultured
with the indicated treatment for 48 h after which the cells were
collected and washed twice with PBS. After fixing with 70% ethanol
at 4°C overnight, the cells were stained with 40 μg/ml propidium
iodide (PI; Sigma). Flow cytometry was performed with a FACSCalibur
flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA). WinList
v6.0 software (Topsham, ME, USA) was used to set the gate, and data
were analyzed with ModFit LT V3.0 (Invitrogen). The proliferative
index was determined as follows, where the G1, S, G2, and M values
represent the proportion of cells in the corresponding phase: (S +
G2/M)/(GO/G1 + S +
G2/M) ×100%. This experiment was performed three
times.
Nested methylation-sensitive PCR
(n-MSP)
Genomic DNA was isolated from 2×106 CA46
cells using the traditional chloroform extraction method followed
by bisulfite modification using the EpiTect Bisulfite kit (Qiagen,
Gaithersburg, MD, USA) following the manufacturer’s instructions
and was stored at −20°C. n-MSP was performed with a two-stage
nested approach as previously described by Palmisano et
al(14) using the 2720 Thermal
Cycler (Applied Biosystems, Foster City, CA, USA). Briefly, PCR
amplification of a sequence of the p16 gene that was 280 bp
in length and included a promoter region rich in CpG was undertaken
using primers specific for bisulfite modification (primers for the
first stage; Table I) and the
following PCR conditions: one cycle at 95°C for 10 min; 40 cycles
at 95°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec; and one cycle
at 72°C for 10 min. All primers were synthesized by Invitrogen.
After the products were diluted 30-fold, 2 μl was used for the
second stage PCR using primers specific for methylated and
unmethylated templates (Table I).
The PCR conditions were as follows: 40 cycles of 95°C for 30 sec,
60°C for 30 sec and 72°C for 30 sec and a final extension at 72°C
for 10 min. The final products were subjected to gel
electrophoresis and sequencing. Sensitivity for detecting
methylated alleles was determined using DNA from peripheral
lymphocytes of healthy subjects which served as a control.
 | Table IPrimer sequences for n-MSP and
real-time PCR. |
Table I
Primer sequences for n-MSP and
real-time PCR.
| Primer sequence
(5′-3′) |
|---|
| n-MSP |
| p16 for first
stage | F:
GAAGAAAGAGGAGGGGTTGG
R: CTACAAACCCTCTACCCACC |
| p16 for
methylated | F:
TTATTAGAGGGTGGGGCGGATCGC
R: GACCCCGAACCGCGACCGTAA |
| p16 for
unmethylated | F:
TTATTAGAGGGTGGGGTGGATTGT
R: CAACCCCAAACCACAACCATAA |
| q-PCR |
| p16 | F:
GAATTGGAATCAGGTAGC
R: GAGGAGGTCTGTGATTAC |
| GAPDH | F:
GAAGGTGAAGGTCGGAGTCAAC
R: CAGAGTTAAAAGCAGCCCTGGT |
Real-time PCR analysis
Total RNA was extracted from cells
(2×106) with TRIzol (Invitrogen) according to the
manufacturer’s instructions. The Reverse Transcription system
(Promega, Madison, WI, USA) was used for the reverse transcription
into cDNA, and real-time PCR was undertaken using the SYBR Green
Master (ROX, Roche, Germany), primers (Table I) and an ABI 7500 thermal cycler.
The conditions were as follows: 40 cycles of 95°C for 1 min, 95°C
for 15 sec and 60°C for 30 sec. The melting curve was subsequently
performed to ensure primer specificity. Relative gene expression
was determined using the 2−ΔΔCt method. Untreated cells
served as the control. This experiment was carried out seven times
in triplicate.
Western blot analysis
Cells (2×106) were collected after the
indicated treatments for 72 h and lysed in RIPA buffer (Thermo
Fisher Scientific, Waltham, MA, USA). The protein concentration was
determined with a BCA kit (Pierce, Rockford, IL, USA) after which
60 μg was separated by SDS-PAGE and transferred onto nitrocellulose
membranes (Bio-Rad Laboratories, Hercules, CA, USA). After the
membrane was blocked in non-fat milk at room temperature for 2 h,
it was incubated with primary antibodies specific for p16 or
β-actin (Abcam®, Hong Kong) at 4°C overnight. After
washing, the membrane was incubated with a horseradish
peroxidase-conjugated secondary antibody (Pierce) at room
temperature for 2 h. Visualization was carried out using the ECL
Plus kit (Pierce).
Statistical analysis
The continuous variables are presented as the mean
and standard deviation. For comparisons of the experimental groups,
one-way ANOVA was used. When a significant difference between
groups was apparent, multiple comparisons were performed using the
Bonferroni procedure with type-I error adjustment. SAS software
package, version 9.2 (SAS Institute, Inc., Cary, NC, USA) was used
for the statistical analysis. All statistic assessments were
evaluated at a two-sided α level of 0.05.
Results
Effects of EGCC and TSA on CA46 cell
growth
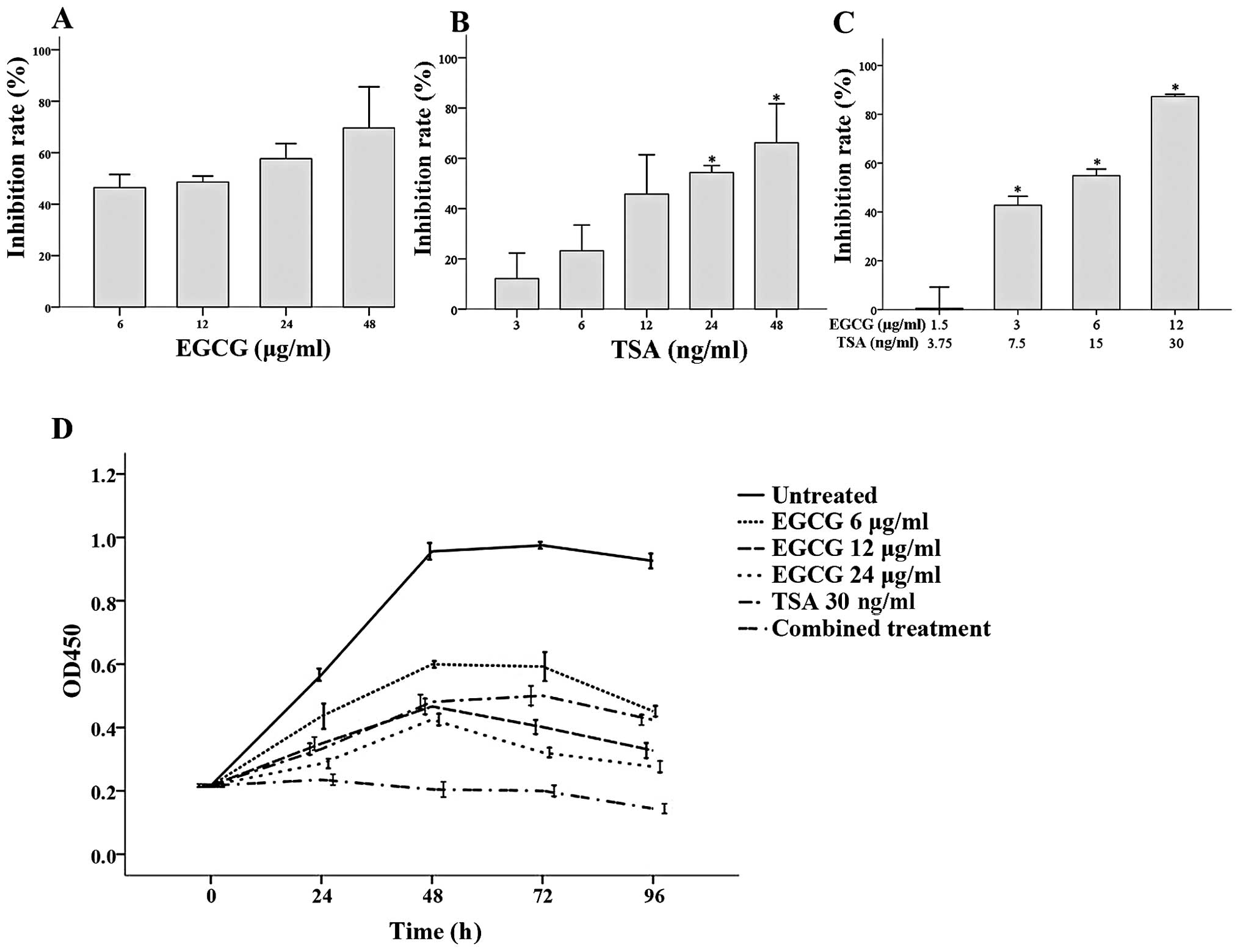
As shown in Fig. 1,
both EGCG and TSA inhibited CA46 cell proliferation after 48 h.
Increasing EGCG concentrations did not further inhibit CA46 cell
growth when compared to the growth of cells treated with 6 μg/ml
EGCG (Fig. 1A), while significant
inhibition was observed with 24 and 48 ng/ml TSA at 48 h when
compared to the cells treated with 3 ng/ml TSA (both P≤0.001;
Fig. 1B). The effects of EGCG and
TSA co-treatment on CA46 cell proliferation was assessed using
various concentrations of each. Significantly greater growth
inhibition was observed with 12 μg/ml EGCG + 30 ng/ml TSA, 6 μg/ml
EGCG + 15 ng/ml TSA and 3 μg/ml EGCG + 7.5 ng/ml TSA when compared
to cells treated with 1.5 μg/ml EGCG + 3.75 ng/ml TSA (all
P<0.001; Fig. 1C).
Analysis of the effects of EGCG and TSA alone or in
combination on CA46 proliferation over time was also assessed
(Fig. 1D). From 24 to 96 h, CA46
cell survival was significantly higher in the untreated group when
compared to cell survival in all of the treatment groups (all
P<0.001). Furthermore, significantly reduced CA46 cell growth
was observed in the combined treatment group (6 μg/ml EGCG and 15
ng/ml TSA) at 24 h and the effect continued until the end of the
study (all P<0.001; Fig.
1D).
Effects of EGCG and TSA on CA46 cell
cycle progression
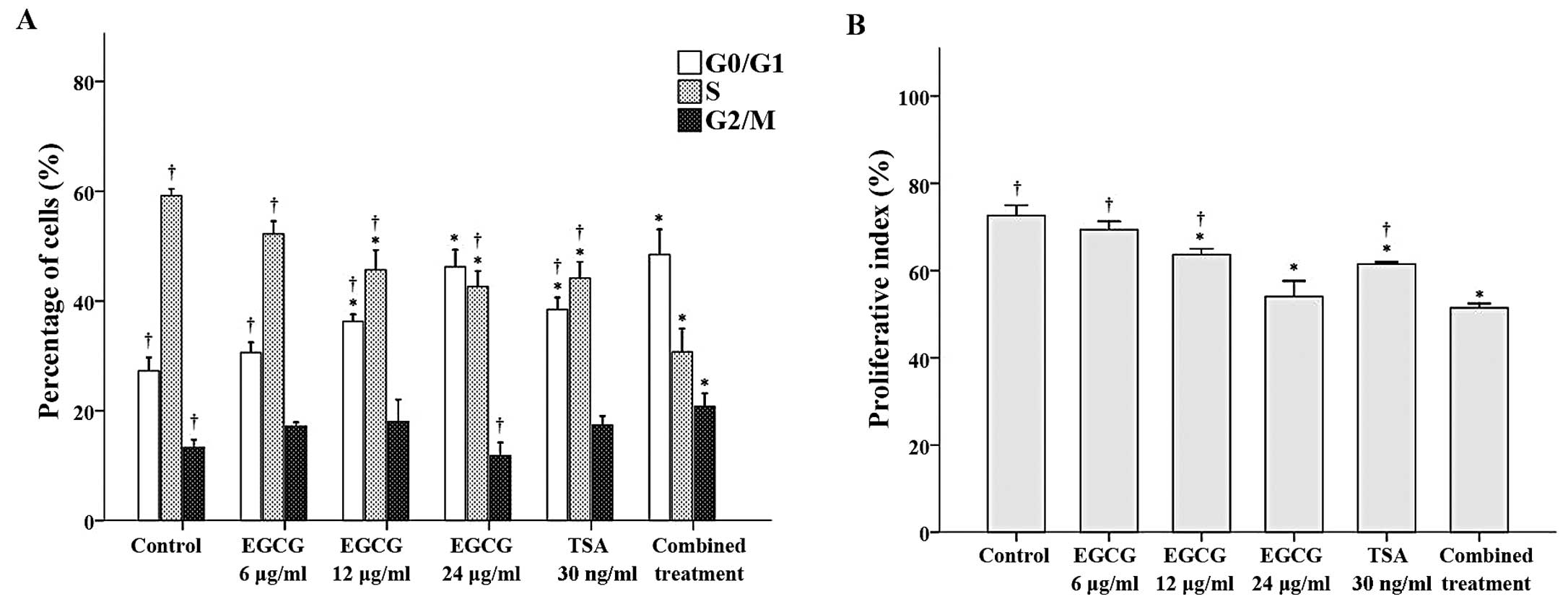
Flow cytometry was performed to observe the effects
of EGCG and TSA alone and in combination on cell cycle progression.
With the exception of CA46 cells treated with 6 μg/ml EGCG, the
proportion of cells in the G0/G1 phase was significantly higher in
the treatment groups when compared to the proportion of cells in
the control group (all P≤0.002; Fig.
2A). Among the groups tested, the cells treated with 24 μg/ml
EGCG and the combined treatment had the greatest proportion of
cells in the G0/G1 phase. In contrast, with the exception of CA46
cells treated with 6 μg/ml EGCG, the proportion of cells in the S
phase was significantly lower in the treatment groups when compared
to the proportion of cells in the control group (all P<0.001;
Fig. 2A). Finally, the proportion
of cells in the G2/M phase was significantly higher in the combined
treatment group when compared to this proportion in the control
group (P≤0.002; Fig. 2A).
Subsequent analysis of the proliferative index revealed that the
cells treated with 12 μg/ml EGCG, 24 μg/ml EGCG, 30 ng/ml TSA or
the combination treatment had lower proliferative indices than the
control group (Fig. 2B).
Effects of EGCG and TSA on the
methylation of the p16INK4a gene in CA46 cells
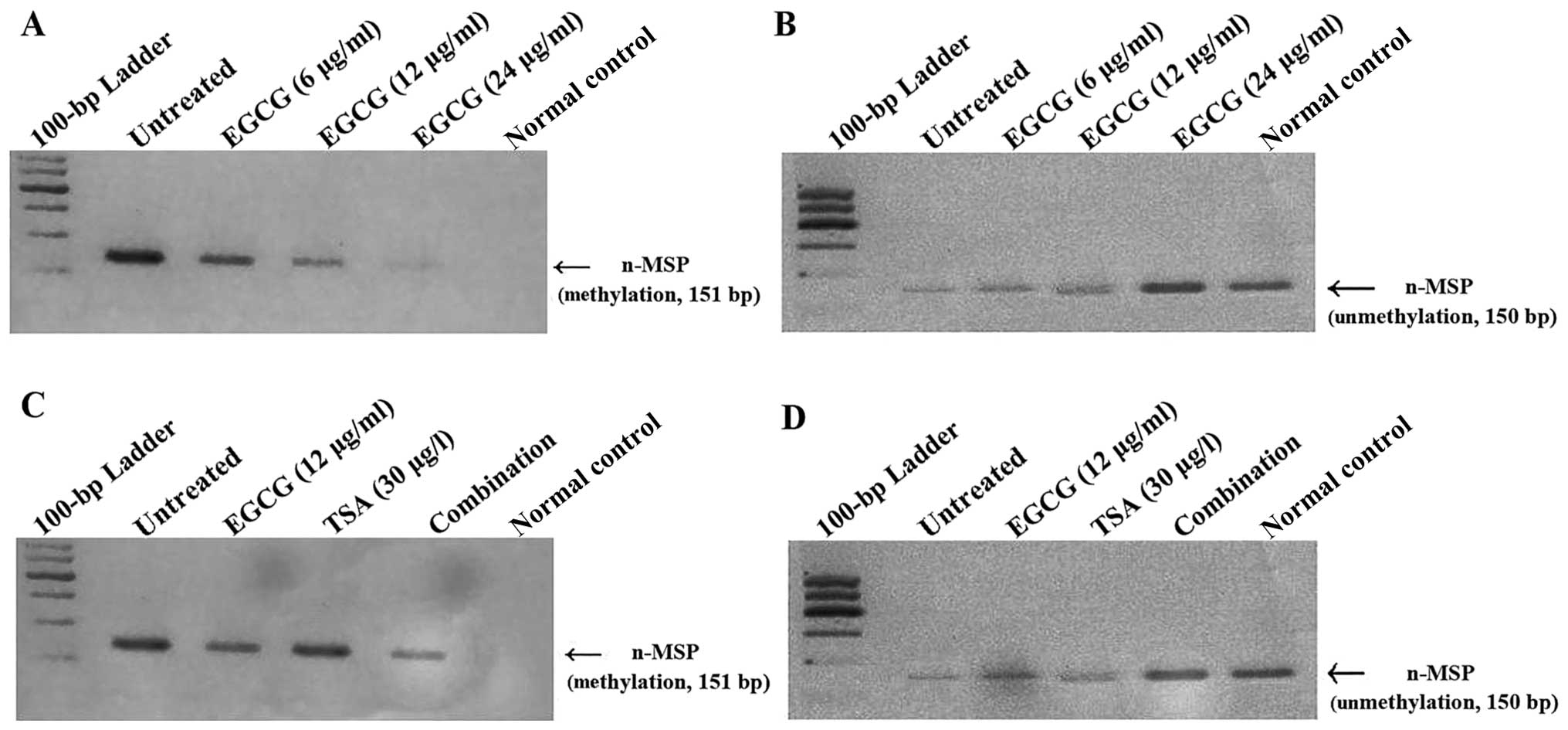
As shown in Fig. 3A,
n-MSP analysis revealed a 151-bp band, corresponding to a
methylated product, in the untreated controls, which decreased with
increasing concentrations of EGCG. A corresponding increase in the
150-bp unmethylated product was observed with EGCG treatment at
increasing concentrations (Fig.
3B). Whereas TSA did not dramatically alter the methylation
status of the p16INK4a gene, the
combination treatment (6 μg/ml EGCG and 15 ng/ml TSA) decreased
p16INK4a methylation and concurrently
increased the unmethylated status of the
p16INK4a gene to a greater degree than the
untreated control (Fig. 3C and
D).
Effects of EGCG and TSA on
p16INK4a expression
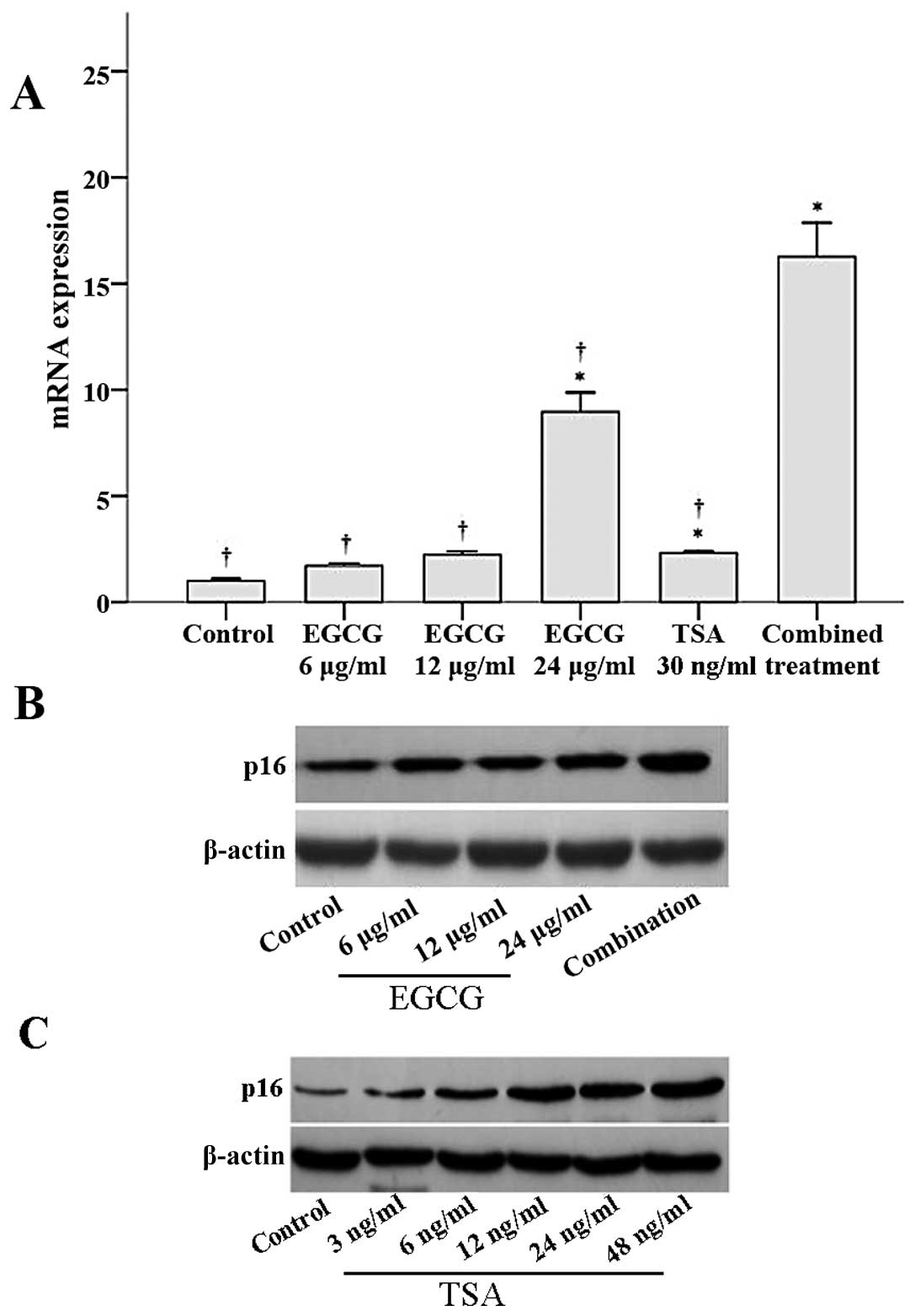
The effects of EGCG and TSA on p16INK4a
mRNA and protein expression were determined by RT-PCR and western
blot analyses, respectively. As shown in Fig. 4A, p16INK4a mRNA
expression significantly increased with 24 μg/ml EGCG, 30 ng/ml
TSA, and the combined treatment when compared to
p16INK4a mRNA expression in the control group (all
P≤0.003). Notably, p16INK4a mRNA expression in the
combined treatment group was significantly higher than that of the
other groups (all P<0.001; Fig.
4A). Similar results were observed for p16 protein expression.
Both EGCG and TSA increased p16 protein expression in a
dose-dependent manner (Fig. 4B and
C). Furthermore, a combination of EGCG and TSA increased p16
expression to a greater extent than that observed with EGCG
treatment alone (Fig. 4B).
Discussion
Methylation of the p16INK4a
promoter and histone deacetylation have been associated with the
occurrence and development of many types of cancers (2,15–18).
Abnormal CpG methylation of the tumor-suppressor gene,
p16INK4a, may significantly inhibit its
expression. In the present study, co-treatment of CA46 cells with
EGCG and TSA reduced p16INK4a gene
methylation, reactivated p16INK4a
expression and reduced cell proliferation to a greater extent than
either agent alone. EGCG and TSA may modulate
p16INK4a expression through epigenetic
modification, reducing DNA methylation and histone acetylation.
In the present study, co-treatment with EGCG and TSA
inhibited the proliferation of CA46 cells, by arresting these cells
in the G0–G1 phase, to a greater extent than either agent alone.
These results are similar to the additive effects of EGCG and TSA
on NF-κB activity and cell invasion as previously reported
(19). These effects may be
mediated at least in part through increased p16INK4a
expression, which inhibits cancer cell proliferation and induces
cellular senescence (4,5). Reduced p16INK4a expression
may result in sustained binding of CDK4/CDK6 to cyclin D and
subsequent phosphorylation of Rb protein, resulting in uncontrolled
cell proliferation (2,3). p16INK4a expression may also
mediate cell senescence (5,20); it may also inhibit cell growth by
directly interacting with the eukaryotic elongation factor 1A2
(eEF1A2), reducing its expression (4). In addition, EGCG-induced apoptosis of
Jurkat cells through hydrogen peroxide production has been
demonstrated (21). Further
analyses may determine whether the anti-proliferative effects of
EGCG and TSA combination therapy are mediated by altered
p16INK4a expression.
DNMT inhibitors not only interfere with the binding
of methylation-sensitive transcription factors to target genes and
directly inhibit RNA polymerase activity, but also indirectly alter
histone acetylation (10). The
methyl-CpG-binding protein (MeCP) can specifically bind to
methylated DNA and recruit HDACs, thereby inducing focal histone
deacetylation and transcription inhibition (22–24).
Furthermore, TSA can reactivate the expression of genes which were
reduced as a result of DNA methylation (25,26).
In AML1/ETO-positive leukemia cells, the methyltransferase
inhibitor, 5-azacytidine, increased the activity of FR901228, a
histone deacetylase inhibitor, to elevate histone acetylation
(27). In the present study,
co-treatment with EGCG and TSA increased DNA demethylation of the
p16INK4a gene to a greater extent than
EGCG alone, which upregulated its expression. These results are
similar to a study with leukemia cell lines (28), in which TSA and
5-aza-2′-deoxycytidine co-treatment reactivated the expression of
genes with high methylation and static transcription; however,
similar changes were not observed after treatment with TSA alone.
Similarly, in the present study, TSA treatment alone did not
dramatically alter the methylation status of the
p16INK4 gene, suggesting that chromatin is
maintained in a closed formation through DNA methylation, thus
impairing the effect of TSA. In human skin cancer cells, EGCG not
only inhibited DNA methylation but also increased histone
acetylation (10). Therefore,
further studies are necessary to analyze the levels of histone
acetylation following EGCG treatment with and without TSA.
The mechanism underlying the EGCG-induced DNA
demethylation is largely unclear. For example, it may inhibit
dihydrofolate reductase and subsequent folate metabolism, thereby
inhibiting DNA and RNA synthesis and reversing
p16INK4a methylation (29,30).
Conversely, EGCG may directly inhibit DNMT activity, inducing
reactivation of methylation-silenced genes as previously reported
(10,31–34).
Direct interaction of EGCG to the catalytic site of DNMT 1 has been
reported (32), in addition to
oxidation of DNMT by EGCG (20).
Furthermore, inhibition of HDACs by EGCG has been reported
(21). Although the mechanism by
which EGCG altered p16INK4a methylation in the present
study was not determined, we speculate that it may directly inhibit
DNMT activity or indirectly reduce their expression via interfering
with folate metabolism. Further studies will be undertaken to test
these postulations.
Reactivation of p16INK4a will likely be
of clinical benefit to cancer patients as its expression has been
associated with longer disease-free survival and better prognosis
in human breast cancer (35). In
addition, the prognostic value of determining p16INK4a
promoter methylation has been reported for breast (7) and prostate cancers (8). Further in vivo studies are
needed to evaluate the possible benefits of combining EGCG with a
clinically useful HDAC inhibitor, such as suberoylanilide
hydroxamic acid.
The present study is limited in that the mechanisms
by which EGCG and TSA co-treatment reduced CA46 cell proliferation
and reversed p16INK4a promoter methylation were not
assessed. Further studies will be conducted to analyze HDAC and
DNMT activity and histone acetylation after treatment with TSA and
EGCG. In addition, the IC50 values of EGCG and TSA were
not determined in the present study and, therefore, will be
undertaken in further analyses.
Taken together, the additive effect of EGCG and TSA
co-treatment on the increase in the expression of
p16INK4a tumor suppressor and the inhibition of CA46
cell proliferation suggests that combination treatment containing
HDAC and DNMT inhibitors may be useful in cancer treatment. Further
in vivo study will be undertaken to fully elucidate the
benefits of epigenetic modifications using EGCG with an HDAC
inhibitor, such as suberoylanilide hydroxamic acid.
Acknowledgements
The present study was supported by grants from the
Fujian Medical University (09-ZD021), the National Natural Science
Foundation of China (81370629 and 81300428), the Natural Science
Foundation of Fujian Province (2011J01179), the Fujian Provincial
Health Bureau Youth Research Projects (2010-1-12), and National
Clinical Key Specialty Construction Project.
References
|
1
|
Li G, Ji Y, Liu C, Li J and Zhou Y:
Reduced levels of p15INK4b, p16INK4a, p21cip1 and p27kip1 in
pancreatic carcinoma. Mol Med Rep. 5:1106–1110. 2012.PubMed/NCBI
|
|
2
|
Herman JG, Merlo A, Mao L, et al:
Inactivation of the CDKN2/p16/MTS1 gene is frequently
associated with aberrant DNA methylation in all common human
cancers. Cancer Res. 55:4525–4530. 1995.
|
|
3
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee MH, Choi BY, Cho YY, et al: Tumor
suppressor p16INK4a inhibits cancer cell growth by
down-regulating eEF1A2 through a direct interaction. J Cell Sci.
126:1744–1752. 2013.PubMed/NCBI
|
|
5
|
Takeuchi S, Takahashi A, Motoi N, et al:
Intrinsic cooperation between p16INK4a and
p21Waf1/Cip1 in the onset of cellular senescence and
tumor suppression in vivo. Cancer Res. 70:9381–9390.
2010.
|
|
6
|
Fang K, Chiu CC, Li CH, Chang YT and Hwang
HT: Cisplatin-induced senescence and growth inhibition in human
non-small cell lung cancer cells with ectopic transfer of p16INK4a.
Oncol Res. 16:479–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee JJ, Ko E, Cho J, et al: Methylation
and immunoexpression of p16INK4a tumor
suppressor gene in primary breast cancer tissue and their
quantitative p16INK4a hypermethylation in
plasma by real-time PCR. Korean J Pathol. 46:554–561.
2012.PubMed/NCBI
|
|
8
|
Ameri A, Alidoosti A, Hosseini SY, et al:
Prognostic value of promoter hypermethylation of retinoic acid
receptor beta (RARB) and CDKN2 (p16/MTS1) in prostate
cancer. Chin J Cancer Res. 23:306–311. 2011.PubMed/NCBI
|
|
9
|
Zhu Z, Wang Y, Liu Z, Wang F and Zhao Q:
Inhibitory effects of epigallocatechin-3-gallate on cell
proliferation and the expression of HIF-1α and P-gp in the human
pancreatic carcinoma cell line PANC-1. Oncol Rep. 27:1567–1572.
2012.PubMed/NCBI
|
|
10
|
Nandakumar V, Vaid M and Katiyar SK:
(-)-Epigallocatechin-3-gallate reactivates silenced tumor
suppressor genes, Cip1/p21 and
p16INK4a, by reducing DNA methylation and
increasing histones acetylation in human skin cancer cells.
Carcinogenesis. 32:537–544. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang W, Pan K, Chen Y, Huang C and Zhang
X: The acetylation of transcription factor HBP1 by p300/CBP
enhances p16INK4A expression. Nucleic Acids Res.
40:981–995. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zanders ED: Overview of chemical genomics
and proteomics. Methods Mol Biol. 800:3–10. 2012. View Article : Google Scholar
|
|
13
|
Sandoval J and Esteller M: Cancer
epigenomics: beyond genomics. Curr Opin Genet Dev. 22:50–55. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Palmisano WA, Divine KK, Saccomanno G, et
al: Predicting lung cancer by detecting aberrant promoter
methylation in sputum. Cancer Res. 60:5954–5958. 2000.PubMed/NCBI
|
|
15
|
Choung HK, Kim YA, Lee MJ, Kim N and
Khwarg SI: Multigene methylation analysis of ocular adnexal MALT
lymphoma and their relationship to Chlamydophila psittaci
infection and clinical characteristics in South Korea. Invest
Ophthalmol Vis Sci. 53:1928–1935. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zainuddin N, Kanduri M, Berglund M, et al:
Quantitative evaluation of p16INK4a
promoter methylation using pyrosequencing in de novo diffuse large
B-cell lymphoma. Leukemia Res. 35:438–443. 2011.
|
|
17
|
Sato H, Oka T, Shinnou Y, et al:
Multi-step aberrant CpG island hyper-methylation is associated with
the progression of adult T-cell leukemia/lymphoma. Am J Pathol.
176:402–415. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kondo T, Oka T, Sato H, et al:
Accumulation of aberrant CpG hypermethylation by Helicobacter
pylori infection promotes development and progression of
gastric MALT lymphoma. Int J Oncol. 35:547–557. 2009.PubMed/NCBI
|
|
19
|
Kim SO and Kim MR: (-)-Epigallocatechin
3-gallate inhibits invasion by inducing the expression of Raf
kinase inhibitor protein in AsPC 1 human pancreatic adenocarcinoma
cells through the modulation of histone deacetylase activity. Int J
Oncol. 42:349–358. 2013.
|
|
20
|
Rajarajacholan UK, Thalappilly S and
Riabowol K: The ING1a tumor suppressor regulates endocytosis to
induce cellular senescence via the Rb-E2F pathway. PLoS Biol.
11:e10015022013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakagawa H, Hasumi K, Woo JT, Nagai K and
Wachi M: Generation of hydrogen peroxide primarily contributes to
the induction of Fe(II)-dependent apoptosis in Jurkat cells by
(-)-epigallocatechin gallate. Carcinogenesis. 25:1567–1574. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Satoh A, Toyota M, Itoh F, et al: DNA
methylation and histone deacetylation associated with silencing DAP
kinase gene expression in colorectal and gastric cancers. Br J
Cancer. 86:1817–1823. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Steele N, Finn P, Brown R and Plumb JA:
Combined inhibition of DNA methylation and histone acetylation
enhances gene re-expression and drug sensitivity in vivo. Br J
Cancer. 100:758–763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dannenberg LO and Edenberg HJ: Epigenetics
of gene expression in human hepatoma cells: expression profiling
the response to inhibition of DNA methylation and histone
deacetylation. BMC Genomics. 7:1812006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oh HJ, Lee TH, Lee JH and Lee BC:
Trichostatin A improves preimplantation development of bovine
cloned embryos and alters expression of epigenetic and pluripotency
genes in cloned blastocysts. J Vet Med Sci. 74:1409–1415. 2012.
View Article : Google Scholar
|
|
26
|
Wu X, Li Y, Li GP, et al: Trichostatin A
improved epigenetic modifications of transfected cells but did not
improve subsequent cloned embryo development. Anim Biotechnol.
19:211–224. 2008. View Article : Google Scholar
|
|
27
|
Klisovic MI, Maghraby EA, Parthun MR, et
al: Depsipeptide (FR 901228) promotes histone acetylation, gene
transcription, apoptosis and its activity is enhanced by DNA
methyltransferase inhibitors in AML1/ETO-positive leukemic cells.
Leukemia. 17:350–358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shaker S, Bernstein M, Momparler LF and
Momparler RL: Preclinical evaluation of antineoplastic activity of
inhibitors of DNA methylation (5-aza-2′-deoxycytidine) and histone
deacetylation (trichostatin A, depsipeptide) in combination against
myeloid leukemic cells. Leuk Res. 27:437–444. 2003.
|
|
29
|
Beltz LA, Bayer DK, Moss AL and Simet IM:
Mechanisms of cancer prevention by green and black tea polyphenols.
Anticancer Agents Med Chem. 6:389–406. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Navarro-Martínez MD, García-Cánovas F and
Rodríguez-López JN: Tea polyphenol epigallocatechin-3-gallate
inhibits ergosterol synthesis by disturbing folic acid metabolism
in Candida albicans. J Antimicrob Chemother. 57:1083–1092.
2006.PubMed/NCBI
|
|
31
|
Fang M, Chen D and Yang CS: Dietary
polyphenols may affect DNA methylation. J Nutr. 137:223S–228S.
2007.PubMed/NCBI
|
|
32
|
Fang MZ, Wang Y, Ai N, et al: Tea
polyphenol (-)-epigallo-catechin-3-gallate inhibits DNA
methyltransferase and reactivates methylation-silenced genes in
cancer cell lines. Cancer Res. 63:7563–7570. 2003.PubMed/NCBI
|
|
33
|
Li Y and Tollefsbol TO: Impact on DNA
methylation in cancer prevention and treatment by bioactive dietary
components. Curr Med Chem. 17:2141–2151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee WJ, Shim JY and Zhu BT: Mechanisms for
the inhibition of DNA methyltransferases by tea catechins and
bioflavonoids. Mol Pharmacol. 68:1018–1030. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peurala E, Koivunen P, Haapasaari KM,
Bloigu R and Jukkola-Vuorinen A: The prognostic significance and
value of cyclin D1, CDK4 and p16 in human breast cancer. Breast
Cancer Res. 15:R52013. View Article : Google Scholar : PubMed/NCBI
|