Introduction
Although the incidence of gastric cancer has been on
the decrease in most industrialized nations over the past two
decades, it remains the second leading cause of cancer-related
mortalities worldwide. The incidence of this cancer type is highest
in Japan, Korea, China, Latin America and Eastern Europe (1). In Western countries such as the USA
the incidence is lower, with 21,000 new cases diagnosed each year.
Despite recent advances in surgery and chemotherapy, the 5-year
survival rate for gastric cancer patients remains extremely low
(2). Therefore, identifying
alternative factors that may reduce the initiation and promotion of
gastric cancer is important to minimize the incidence and severity
of this disease.
Diallyl disulfide (DADS), a main organosulfur
component responsible for the diverse biological effects of garlic,
exhibits a wide variety of internal biological activities and has
shown potential as a therapeutic agent in various types of cancer
(3–7). The antiproliferative property of DADS
in cultured human colon tumor cells (HCT-116 and SW480), leukemic
and prostate (PC-3) cancer cell lines is connected with its ability
to arrest cells in the G2/M phase (8–11).
Changes in activity may also result from a combination of the
quantity and activity of specific cellular proteins. For example,
the hyperphosphorylation of p34(cdc2) kinase was increased by 15%
following the exposure of colon cells to DADS, and DADS also
reduced CDC25C protein expression (12). Ashra and Rao (13) demonstrated that the elevated
phosphorylation of checkpoint kinase 1 (Chk1), decreased
phosphorylation of Chk2, and decreased levels of CDC25C, 14-3-3 and
cyclin B1 were the critical changes associated with the abrogation
of the G2/M checkpoint control.
Previously, we reported that DADS inhibits the
growth of gastric cancer in vitro and in vivo
(6,14) and that it induces cell cycle
G2/M arrest in human MGC803 gastric cancer cells
(15). Decreased CDC25C expression
is crucial in G2/M arrest following treatment with DADS
(15). The aim of the present study
was to investigate the underlying mechanism involved in the
induction of G2/M phase cell cycle arrest by DADS in the
human MGC803 gastric cancer cell line, with special emphasis on its
role in the key G2/M checkpoint kinase-Chk1 and -Chk2.
We showed that DADS inhibited the growth of BGC823 cells, which was
associated with the phosphorylation of ATR (ATM-RAD3-related gene)
and Chk1 while suppressing the expression of CDC25C and cyclin B1
(16). In the present study, DADS
was shown to induce phosphorylation of ATR and Chk1 while
suppressing the expression of CDC25C and cyclin B1 in the human
MGC803 gastric cancer cell line. The results demonstrated that DADS
selectively causes Chk1-mediated G2/M arrest in this
cell line. These observations contribute to understanding the
mechanisms of the antitumor effect of DADS in gastric cancer cells,
and indicate the potential of DADS for clinical development as a
therapeutic drug to combat gastric cancer.
Materials and methods
Chemicals and reagents
DADS was purchased from Fluka Chemika (Ronkonkoma,
NY, USA). Phospho-Chk1 (Ser345) antibody, phospho-ATR (Ser428) and
ECL LumiGLO reagent were obtained from Cell Signaling Technology,
Inc. (CST, USA). The phospho-Chk2 (Thr68) antibody, ATR antibody
and anti-rabbit IgG (HRP-linked) Chk1 and Chk2 antibodies were
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
The CDC25C, cyclin B1 and β-actin antibodies were from Boster
Company (Boster, China).
Cell line and cell culture
The human MGC803 gastric cancer cell line was
obtained from the Cell Research Institute of the Chinese Academy of
Science (China). The cells were maintained in RPMI-1640 medium
(Sigma, St. Louis, MO USA), supplemented with 10% heat-inactivated
fetal bovine serum (FBS; Life Technologies, Grand Island, NY, USA),
100 μg/ml streptomycin and 100 U/ml penicillin G (Life
Technologies) in a humidified atmosphere of 5% CO2 and
95% air at 37°C. The cells were suspended at a final concentration
of 1×104 cells/ml in RPMI-1640 medium containing 10% FBS
in culture flasks. Reagents were added to each flask in various
combinations and incubated at 37°C in 5% CO2.
Northern blot analysis
RNA from the cells obtained at each time point was
extracted with TRIzol (Invitrogen). Reverse transcription (RT) was
performed with an RT kit (Promega, Madison, WI, USA). Total RNA (1
mg) was used as a template, and RT-generated cDNA encoding Chk1,
Chk2 and β-actin (internal control) was amplified by PCR. Primer
sequences were: forward 5′-CTG AAG AAG CAG TCG CAGTG-3′ and reverse
5′-TTC CAC AGG ACC AAA CATCA-3′ for Chk1; forward 5′-TCC GCT TGC
TGA TGA TCT TTA TGG-3′ and reverse 5′-GAC CTA CTC CTT GGG CTC GGC
TAT-3′ for Chk2; and forward 5′-CGT CAT ACT CCT GCTT-3′ and reverse
5′-ATC TGG CAC CAC ACCT-3′ for β-actin. The primers were designed
using Primer Premier 5.0 software (Premier Company, Canada) and
were synthesized by the Shanghai Sangon Biological Technology and
Services Co., Ltd. (Shanghai, China). Amplification parameters were
95°C for 5 min, and 30 cycles of 95°C (30 sec), 60°C (15 sec), 72°C
(60 sec) and 72°C (60 sec) using 3 μl cDNA.
Northern blots were performed according to standard
procedures (17). Total RNA (20–30
μg) was separated on 1.2% formaldehyde agarose gel and transferred
to a polyamide membrane (Millipore, Billerica, MA, USA). cDNA
oligonucleotide probes were labeled with α32P-ATP using
terminal deoxynucleotidyl transferase recombinant enzyme (Promega)
as recommended by the manufacturer’s instructions, followed by
pre-hybridization (2 h) at 68°C, heat denaturation (5 min) at 100°C
and hybridization (16 h) at 68°C. The membrane was air-dried on
blotting paper and then exposed to X-ray film by autoradiography at
−70°C for 24–48 h in a cassette containing an intensifying
screen.
Western blot analysis
Human gastric cancer cells were cultured with or
without DADS at the indicated concentrations for various periods of
time, washed once with ice-cold phosphate-buffered saline (PBS),
and lysed in a buffer consisting of 20 mM Tris/HCl, pH 8.0, 137 mM
NaCl, 1.5 mM MgCl2, 1 mM ethylene glycol tetraacetic
acid (EGTA), 10% glycerol, 100 mM NaF and 1% Triton X-100. Protein
concentrations in the lysates were measured with the Protein BCA
sssay kit (Bio-Rad, Hercules, CA, USA). Protein lysate (30 μg) was
subjected to 10% SDS-polyacrylamide gel electrophoresis and the
proteins separated were transferred to polyvinylidene difluoride
membranes (Millipore). To block non-specific binding, the membranes
were incubated at room temperature for 1 h with 5% skim milk
powder, followed by a 12 h incubation at 4°C with an anti-serum
containing antibodies against Chk1, Chk2, ATR, phospho-Chk1
(Ser345), phospho-Chk2 (Thr68), phospho-ATR (Ser428), CDC25C and
cyclin B1. A peroxidase-conjugated secondary antibody (1:5,000
dilution) and ECL western blotting detection reagents were used to
visualize the target proteins (ECL New England BioLabs, Ipswich,
MA, USA), which were quantified with a BioImage Intelligent
Quantifier 1-D (version 2.2.1; Nihon-BioImage Ltd., Japan).
Immunoprecipitation
Human gastric cancer cells were cultured with or
without DADS at the indicated concentrations for various periods of
time. The cells were characterized using a Seize (R) Classic
Mammalian Immunoprecipitation kit (Piece Technology). Then cells
were removed from the culture medium, washed once with PBS (0.1 M
phosphate, 0.15 M NaCl, pH 7.2), harvested and lysed using lysis
buffer of the M-PER reagent. The lysates were collected,
transferred to a microcentrifuge tube and centrifuged at 13.000 × g
for 5–10 min to separate the cell debris. The supernatants were
transferred to another tube for subsequent analysis. Purified Chk1
or Chk2 antibody was added to the sample, followed by overnight
incubation at 4°C. The immune complex was added to the spin cup
containing equilibrated protein G beads. Elution buffer (190 μl)
was added to the spin cup and the samples underwent elution.
Immunoprecipitated protein levels were determined by western blot
analysis with the anti-CDC25C antibody and the ECL detection
system.
Establishment of a stable MGC803 cell
line expressing Chk1 or Chk2
PCR primers were designed and synthesized according
to human Chk1 or Chk2 cDNA sequences (GenBank), including
BamHI and HindIII restriction sites using these
sequences: forward, 5′-CGG AAG CTT ATG GCA GTG CCC TTTG-3′ and
reverse, 5′-CGG CGA ATT CTC ATG TGG CAGGA-3′ for Chk1; forward,
5′-CGC CAA GCT TAT GTC TCG GGA GTC-3′ and reverse, 5′-CGG AAT TCT
CAC AAC ACA GCA GCA CAC-3′ for Chk2. The Chk1 or Chk2 gene was
amplified from the plasmid expression vector pcDNA3.1(+)-Chk1 and
pcDNA3.1(+)-Chk2 by PCR. The amplified Chk1 or Chk2 gene products
and the plasmid vector pcDNA3.1(+) were digested with restriction
endonucleases BamHI and HindIII (Jingmei Biotech Co.,
Ltd., China), and linked with T4 DNA ligase (Shanghai Sangon
Biological Technology and Services Co., Ltd.). The new recombinant
plasmid vectors were transferred into competent DH5α bacteria to
obtain a recombinant pcDNA3.1(+)-Chk1 or pcDNA3.1(+)-Chk2 vector,
and sub-cloned PCR fragments were confirmed by DNA sequencing
analysis (Shanghai Sangon Biological Technology and Services Co.,
Ltd.).
MGC803 cells were seeded in a 24-well plate and
infected with a viral medium of 500 μl. Plasmid DNA (0.8 μg) in 50
μl non-serum Opti-MEM medium containing 2 μl Lipofectamine 2000
(Invitrogen Life Technologies) was subsequently added to each well.
The mixtures were incubated for 5 h, and 3 ml fresh medium was
added. On the day following infection, the cells were passaged at a
1:10 ratio into selective medium containing G418 (400 μg/ml). When
cloned MGC803 cells became distinct, the drug concentration used
for screening was reduced and the MGC803 cells were transferred
into a new culture flask for cell growth. Chk1 or Chk2 protein
expression was detected by western blot analysis.
RT-PCR
Chk1 or Chk2 mRNA was detected by RT-PCR according
to the manufacturer’s instructions. The primers for Chk1, Chk2 and
β-actin were the same as those for northern blotting. Amplification
parameters were 95°C for 5 min, and 30 cycles of 95°C (30 sec),
60°C (15 sec), 72°C (60 sec) and 72°C (60 sec) using 3 μl cDNA.
Standards were run in duplicate and samples in triplicate. The
expression levels for all the genes analyzed were normalized to
β-actin.
Chk1 or Chk2 siRNA transfection
Cells were seeded at 3×105 cells/well in
6-well plates and allowed to grow overnight. The following day, the
cells were left untreated, Lipofectamine-transfected, or
transfected with Chk1 siRNA or Chk2 siRNA (both from Santa Cruz
Biotechnology, Inc.). Transfections were performed using
Lipofectamine 2000 and Opti-MEM-reduced serum medium (both from
Santa Cruz Biotechnology, Inc.). The cells were transiently
transfected with siRNA specifically targeting Chk1 or Chk2 (both
from Santa Cruz Biotechnology, Inc.) at a final concentration of
100 nM using a transfection reagent for 24 h. Following
transfection, the cells were left untreated or exposed to DADS for
the indicated time. Cellular protein was extracted and subjected to
western blot analysis of the targeted proteins (Chk1 and Chk2).
Real-time PCR and western blot
analysis
Chk1 or Chk2 mRNA were detected according to the
manufacturer’s instruction. The used were: Chk1, forward 5′-CGG TAT
AAT AAT CGT GAGCG-3′ and reverse 5′-TTC CAA GGG TTG AGG TATGT-3′;
Chk2, forward 5′-CTC GGG AGT CGG ATG TTGAG-3′ and reverse 5′-GAG
TTT GGC ATC GTG CTGGT-3′; and β-actin forward 5′-AAG AAG GTG GTG
AAG CAGGC-3′ and reverse 5′-TCC ACC ACC CTG TTG CTGTA-3′. Chk1 or
Chk2 protein expression was detected by western blot analysis.
Cell cycle analysis
Cells were incubated in culture medium alone or in
culture medium containing 30 mg/l DADS, at 37°C for 12, 24, 36 and
48 h. The cells were harvested in cold PBS, fixed in 700 ml/l
ethanol and stored at 4°C for subsequent cell cycle analysis. Fixed
cells were washed with PBS once and suspended in 1 ml propidium
iodide-staining reagents (20 mg/l ribonuclease and 50 mg/l
propidium iodide). Samples were incubated in the dark for 30 min
prior to cell cycle analysis, and cell distribution in the various
phases of the cell cycle was measured with a flow cytometer
(Coulter EPICS XL; Beckman, Miami, FL, USA). The percentage of
cells in the G1, S and G2/M phases was then
calculated by CellQuest software on the flow cytometer (Coulter
EPICS XL).
Statistical analysis
Results were analyzed by the SPSS 11.5 statistical
software package. Data were expressed as means ± SD (standard
deviation). Comparisons between different groups were made by
one-way ANOVA or the Student’s t-test. P<0.05 was considered to
indicate a statistically significant result.
Results
Expression and activation of Chk1 and
Chk2 induced by DADS
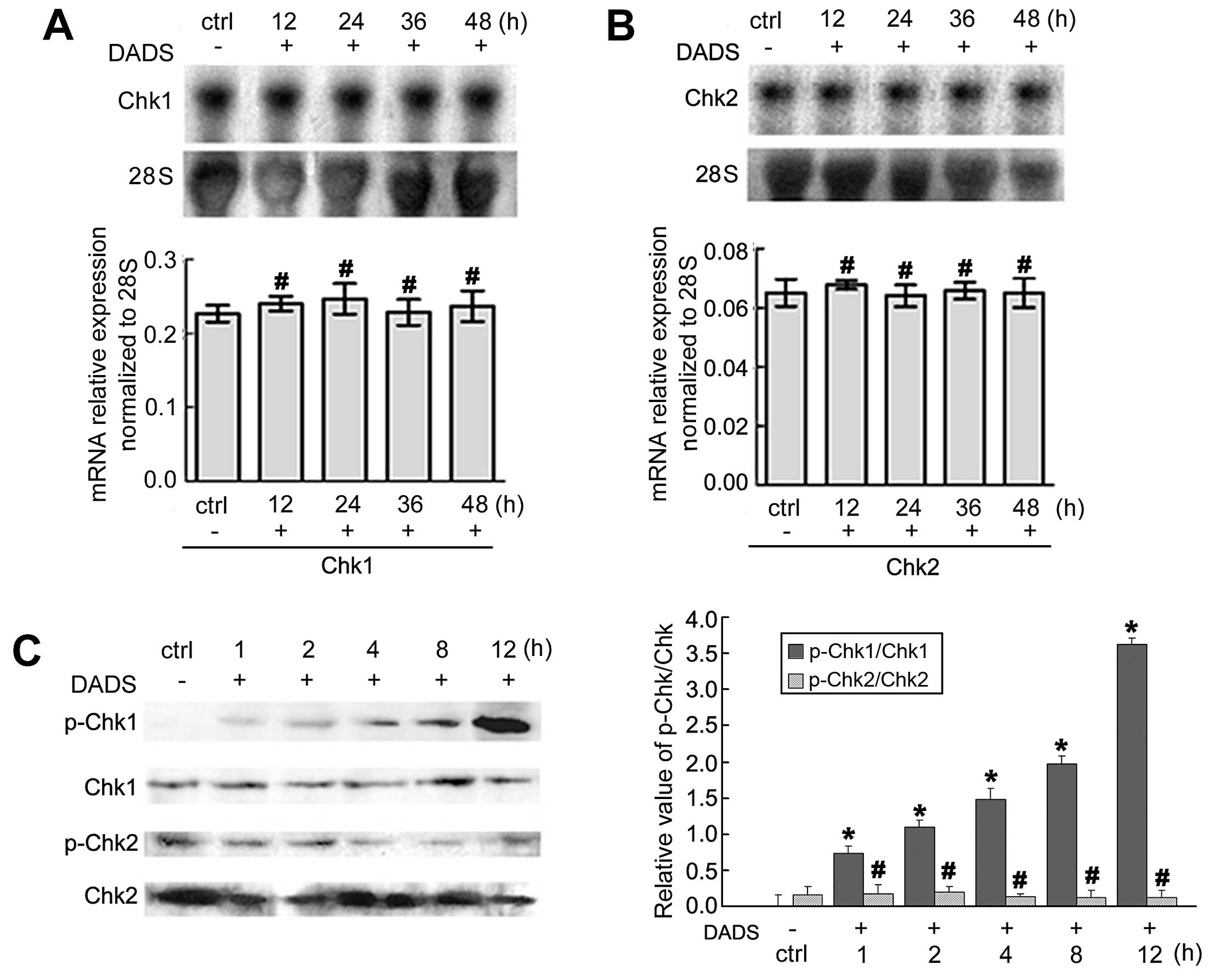
The expression of Chk1 and Chk2 mRNA associated with
the cell cycle arrest of MGC803 cells was revealed by northern
blotting. After treatment with 30 mg/l DADS for 12, 24, 36 and 48
h, the expression of Chk1 and Chk2 mRNA showed no significant
changes when compared to the untreated cells (p>0.05).
Immunoblot analyses of phospho-Chk1 and Chk2 protein
in MGC803 cells revealed that 30 mg/l DADS enhanced the levels of
phospho-Chk1 protein in a time-dependent manner as compared with
the untreated cells from 1 to 12 h (p<0.05). However, the
phospho-Chk2 protein was not activated by DADS (p>0.05)
(Fig. 1).
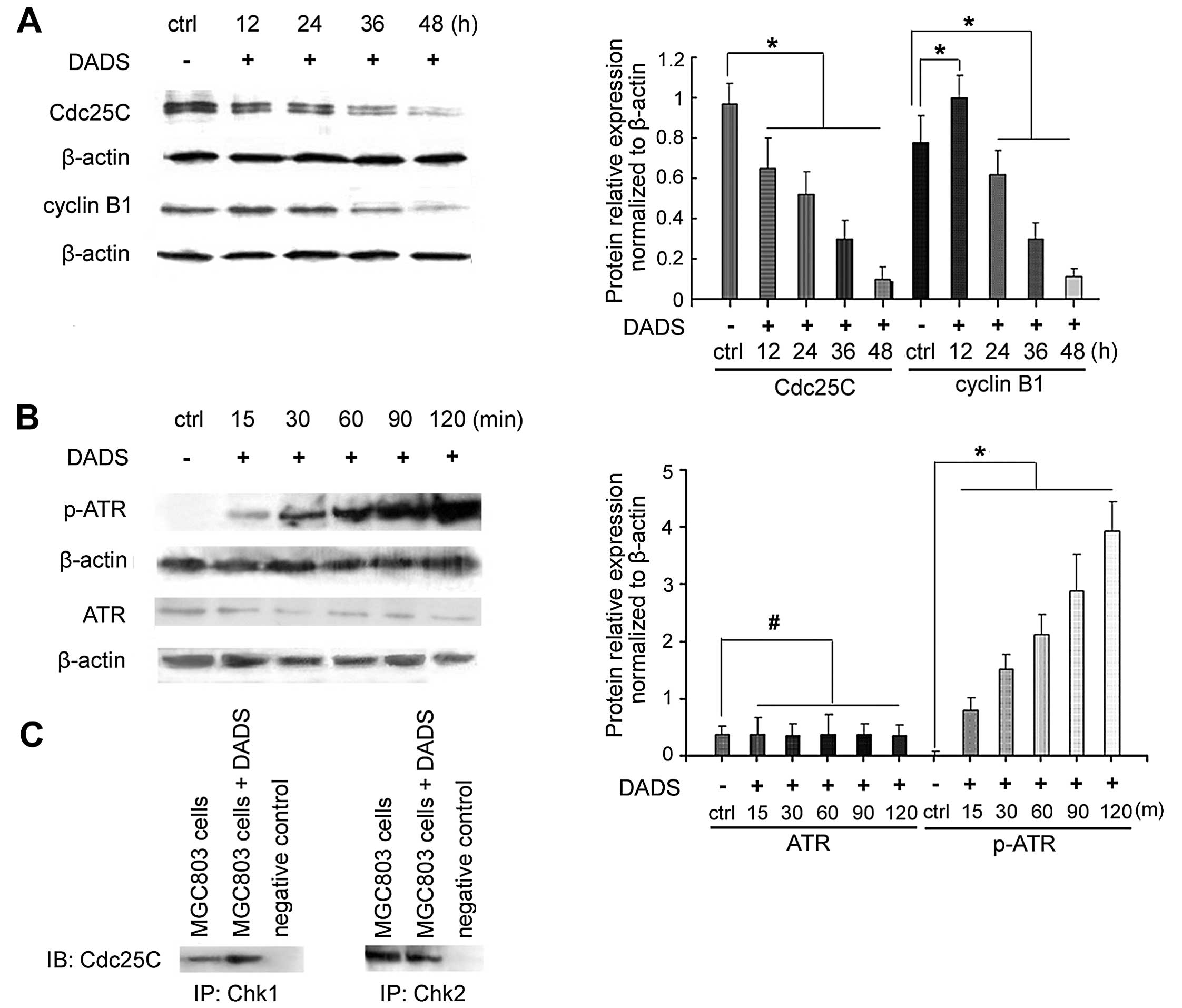
DADS regulates the protein expression
levels in the G2/M cell cycle
DADS (30 mg/l) inhibited the expression of the cell
cycle-associated phosphatase CDC25C in MGC803 cells after 12 h,
which was decreased by 81% after 48 h (p<0.05) (Fig. 2). As shown in Fig. 2A, the expression of cyclin B1
increased after 12 h of DADS treatment, and then decreased after 36
h, being reduced by 87.5% after 48 h (p<0.05).
ATR mediates checkpoint signaling through its
downstream effect or Chk1 (14). As
shown in Fig. 2B, 30 mg/l DADS
treatment of MCC803 cells for 15 min to 2 h resulted in an increase
in phospho-ATR expression, whereas no change was found in ATR
expression.
The interaction between endogenous Chk1 and CDC25C
was determined by co-immunoprecipitation using anti-Chk1 or
anti-CDC25C. Western blot analysis showed that CDC25C was present
in the immunoprecipitate of anti-Chk1 (Fig. 2C). Chk1 was also
co-immunoprecipitated with anti-CDC25C (Fig. 2C). Chk1 and CDC25C were not detected
in the immunoprecipitate of rabbit IgG, which was used as a
negative control. This result indicated that Chk1 may interact with
CDC25C.
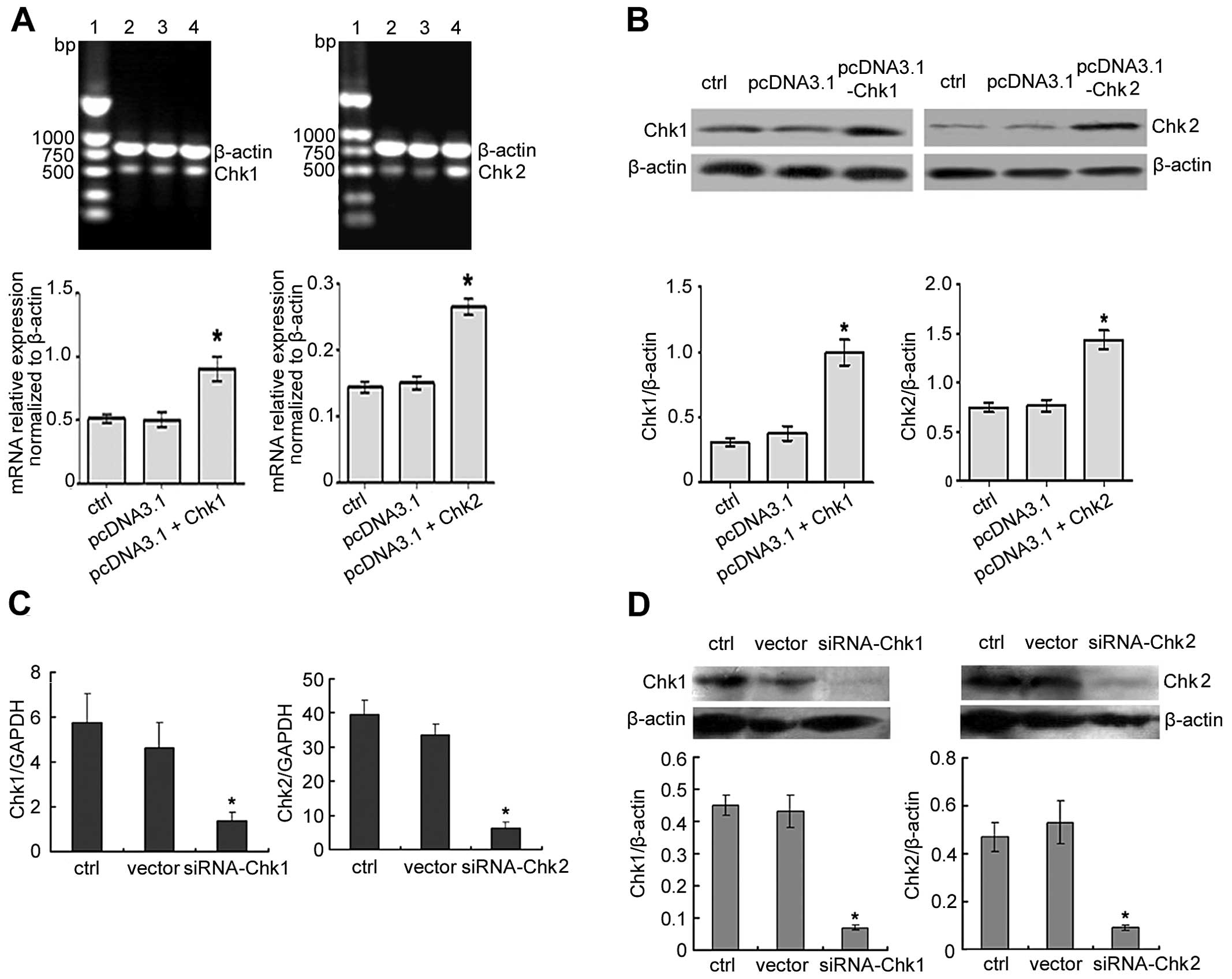
Generation of Chk1 or Chk2
overexpressing/knockdown MGC803 human gastric cancer cell line
To investigate the functional role of Chk1 or Chk2
in the cell cycle arrest of DADS-induced human MGC803 gastric
cancer cell line, cell lines were produced with overexpressed or
reduced levels of Chk1 or Chk2. Overexpression and knockdown were
confirmed by RT-PCR, RT-qPCR and western blotting. Antibody
labeling showed that levels of Chk1 or Chk2 protein were increased
in overexpressed cells compared to the controls (Fig. 3A and B). By contrast, the knockdown
cells showed reduced levels of Chk1 or Chk2 expression compared to
controls in the MGC803 cell line (Fig.
3C and D).
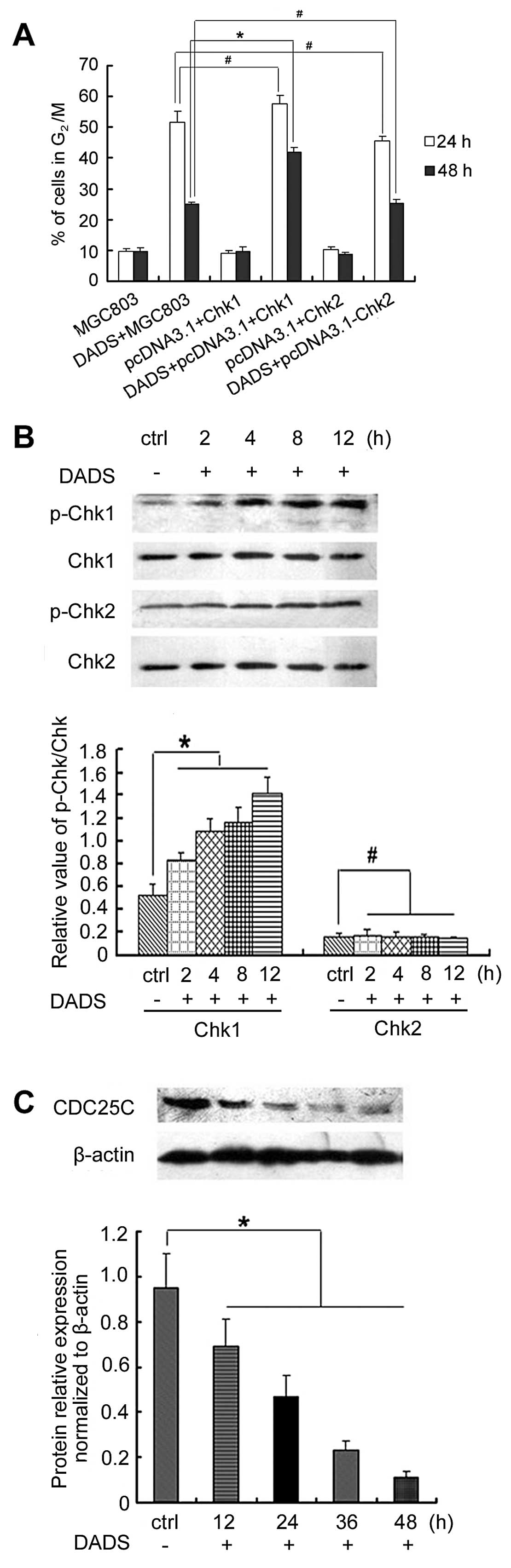
Overexpression of Chk1 but not Chk2
promotes DADS-induced G2/M arrest by activating
phospho-Chk1 and decreasing CDC25C expression
We evaluated whether Chk1 and Chk2 expression
enhanced DADS-induced cell cycle arrest in MGC803 cells. Flow
cytometry revealed the proportion of cells in the G2/M
phase. FACS analysis indicated there were no difference in the
G2/M phase between pcDNA3.1(+)/Chk1-transfected cells or
pcDNA3.1(+)/Chk2-transfected cells and untransfected control cells
(p>0.05) (Fig. 4). However, 30
mg/l DADS induced a higher percentage of G2/M phase
cells in pcDNA3.1(+)/Chk1-transfected cells (41.9%, 48 h) than in
untransfected control cells (25%, 48 h) (p<0.05) (Fig. 4). By contrast, DADS did not have
this effect on pcDNA3.1(+)/Chk2-transfected cells. Thus, the
results suggested that the overexpression of Chk1, but not Chk2,
enhanced DADS-induced G2/M arrest in MGC803 cells.
The effect of DADS on the expression and
phosphorylation of Chk1 or Chk2 was also examined. Western blot
analysis showed that Chk1 phosphorylation was increased in
Chk1-transfected MGC803 cells in a time-dependent manner following
treatment with DADS (30 mg/l) at 2, 4, 8 and 12 h (p<0.05). By
contrast, Chk2 phosphorylation remain unchanged in Chk2-transfected
MGC803 cells by DADS (p>0.05). DADS reduced the expression of
CDC25C in pcDNA3.1(+)/Chk1-transfected cells (Fig. 4). After stimulation with 30 mg/l
DADS for 12 h the CDC25C expression showed a significant decrease,
and continued to decrease gradually in a time-dependent manner
(p<0.05). A high Chk1 expression was able to increase
G2/M arrest induced by DADS in MGC803 cells, and the
Chk1/CDC25C pathway was involved in DADS-induced G2/M
arrest.
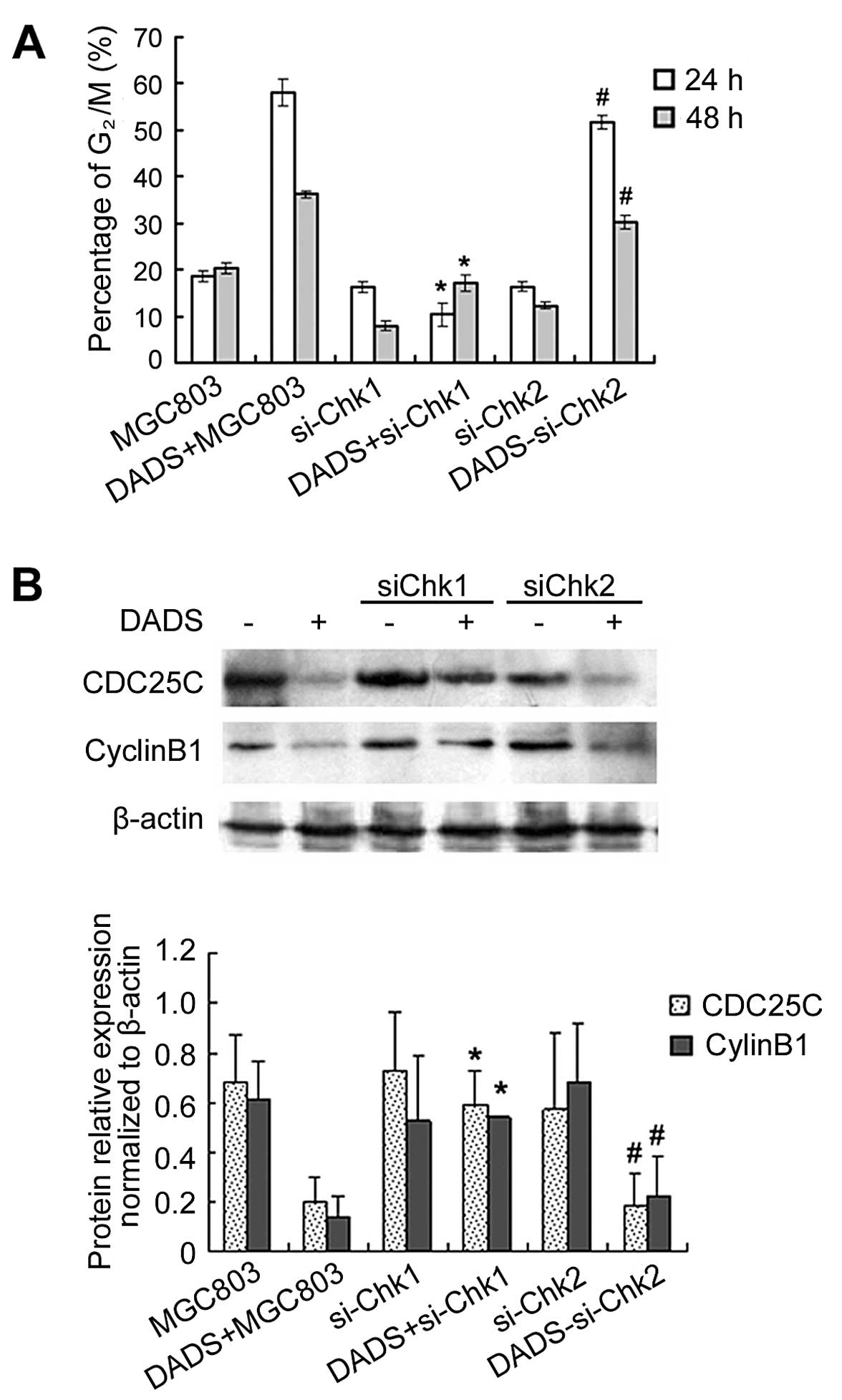
Knockdown of Chk1 but not Chk2 inhibits
DADS-induced G2/M arrest by inhibiting expression of
CDC25C and cyclin B1
After MGC803 cells were treated with 30 mg/l DADS
for 24 h, G2/M cells increased to 51.5% (24 h) and 25%
(48 h), which was significantly higher than those of the untreated
control group. However, siChk1 transfection markedly reduced the
proportion of DADS-induced G2/M cells, which showed that
the G2/M phase ratio of siChk1-transfected cells was
significantly reduced after treatment with 30 mg/l DADS compared to
the untransfected cells, particularly in the 24 h treatment group,
which was reduced from 58.1 to 10.4%. These results demonstrated
that DADS-induced G2/M arrest was significantly
inhibited by siChk1, but that siChk2 transfection had no effect on
DADS-induced G2/M ratio in MGC803 cells (Fig. 5).
Western blotting showed that although DADS reduced
the expression of CDC25C and cyclin B1 in untransfected cells,
inhibition of CDC25C and cyclin B1 expression treated by DADS was
blocked by Chk1 gene silencing (p<0.05). By contrast, Chk2 gene
silencing did not have this effect (Fig. 5). These results suggested that Chk1
gene silencing abrogated G2/M arrest induced by DADS in
the MGC803 cell line, and that the Chk1/CDC25C/cyclin B1 pathway
was involved in the G2/M arrest induced by DADS.
Discussion
The focus of the present study was to understand the
mechanisms responsible for activation of the G2/M
checkpoint in response to DADS treatment of human gastric cancer
cells. Previous studies have shown that DADS, the major component
of cooked garlic, suppresses the proliferation of cancer cells in,
for example, gastric and colon cancer, and leukemic cells (8–11,16).
However, the mechanisms by which DADS inhibits the proliferation of
cancer cells remain to be determined, since little is known about
DADS-induced G2/M arrest.
Cell cycle arrest and apoptosis are two important
mechanisms involved in anticancer drug treatment (18,19).
In a previous study, we reported that DADS-induced G2/M
arrest and differentiation of MGC803 cells involved activation of
the p38 MAP kinase and ERK1/2 signaling pathway (6,15).
Although these phenomena occurred in a time-dependent manner, the
G2/M arrest-inducing mechanisms were not investigated in
depth.
Many compounds have been developed to inhibit
specific checkpoint components, particularly checkpoint kinase 1
(Chk1), an active transducer kinase, at the S and G2 checkpoints,
rendering it a target for rational anticancer drug development
(20,21). The hyperactive cell cycle Chk1 and
Chk2 play a pivotal role in the DNA damage response including
radiation and chemotherapy, and the therapy targeting Chk1 gene may
be a novel method of treating glioblastoma (22). Chk1 is required for the intra-S
phase and G2/M checkpoints in the cell cycle, and plays
a critical role in maintaining genomic stability and transducing
the DNA damage response (23). The
duration of Chk1-activated G2/M cell cycle arrest
determines the level of autophagy following teh DNA mismatch repair
(MMR) processing of these nucleoside analogs (24).
2-(3-Methoxyphenyl)-6,7-methylenedioxoquinolin-4-one (MMEQ) induced
G2/M arrest through the promotion of Chk1, Chk2 and
CDC25C in TSGH8301 cells (25).
Pabla et al reported that Chk1-S, an N-terminally truncated
alternative splice variant of Chk1, is an endogenous repressor and
regulator of Chk1. During DNA damage Chk1 is phosphorylated, which
disrupts Chk1-Chk1-S interaction and results in free active Chk1 to
arrest the cell cycle (26).
Deletion of a single Chk1 allele compromises G2/M
checkpoint function, which is not further affected by Chk2
depletion (27). Loratadine
directly damages DNA and activates Chk1, thereby promoting
G2/M arrest, making cells more susceptible to
radiation-induced DNA damage. Additionally, loratadine
downregulates total Chk1 and cyclin B, abrogating the
radiation-induced-G2/M checkpoint and allowing cells to
re-enter the cell cycle despite the persistence of damaged DNA
(28). In the present study, no
change was observed in the the mRNA and protein expression of Chkl
and Chk2 following treatment with DADS. However, DADS induced the
accumulation of phosphorylated Chk1 and ATR, while downregulating
the expression of CDC25C and cyclin B1 expression, without Chk2
phosphorylation. Furthermore, CDC25C was found to be
immunoprecipitated by anti-Chk1 but not anti-Chk2. In order to
clarify the function of Chk1 and Chk2 in DADS-induced
G2/M arrest in the MGC803 cell line, this cell line was
generated with increased and decreased levels of Chk1 or Chk2
protein. We assessed the cell cycle G2/M arrest in these
cells. In the MGC803 cell lines tested, increasing the levels of
Chk1 increased the percentage of G2/M phase cells,
demonstrating that Chk1 is required for DADS-induced
G2/M arrest. Conversely, decreasing the levels of Chk1
decreased DADS-induced G2/M phase cells, suggesting that
Chk1 is involved in DADS-induced G2/M arrest in the
MGC803 cell line. By contrast, Chk2 overexpression or knockdown had
no significant effect on DADS-induced G2/M arrest in the
MGC803 cell line. These results show that Chk1 is crucial in
DADS-induced G2/M arrest in MGC803 cells.
Pectenotoxin-2 induces G2/M phase cell
cycle arrest in human breast cancer cells, while ATM- and
Chk1/2-mediated phosphorylation of CDC25C plays an important role
in G2/M arrest (29).
Activation of the G2/M checkpoint involves a series of
signaling events, including activation of ATR and Chk1 kinases and
inhibition of Cdc2/cyclin B activity (31). Serine (Ser)/threonine (Thr) protein
phosphatase 2A (PP2A) has an important role in G2/M
checkpoint activation in response to γ-irradiation (IR). Specific
PP2A inhibition abrogates the IR-induced activation of ATR and Chk1
kinases, as well as the phosphorylation of Cdc2-Tyr15, and
attenuates IR-induced G2/M arrest (30), an important role of the DNA damage
response mediated by ATR-Chk1 in p53/p21(Waf/CIP1) activation and
downstream G2/M arrest during treatment with gambogic
acid (GA) (31).
A crucial step in G2/M phase arrest was
reported as Cdc2 and CDC25C activated by Chk1/2 in response to DNA
damage (32–34). The arresting effect of gossypin on
the cell cycle in G2/M phase was involved in the
phosphorylation of CDC25C tyrosine phosphatase via the activation
of Chk1 (35). Human CDC25C is one
of the central targets and regulators of the G2/M
checkpoint mechanisms activated in response to DNA injury (36). CDC25C is thought to be the major
effector of the G2/M DNA damage checkpoint kinase, Chk1
and/or Chk2, which triggers the cyclin B1/CDK1 complex (37,38).
The present study has demonstrated that DADS reduced the expression
of CDC25C in pcDNA3.1(+)/Chk1-transfected cells. After stimulation
with 30 mg/l DADS for 12 h CDC25C expression showed a significant
decrease, and this expression continued to decrease gradually with
time. Thus, a high expression of Chk1 was able to increase
G2/M arrest induced by DADS in MGC803 cells, and the
Chk1/CDC25C pathway was involved in DADS-induced G2/M
arrest. Furthermore, inhibition of the expression of CDC25C and
cyclin B1 treated with DADS was blocked by Chk1 gene silencing. By
contrast, Chk2 gene silencing did not have this effect. These
results suggest that Chk1 gene silencing abrogated G2/M
arrest induced by DADS in MGC803 cells, and that the
Chk1/CDC25C/cyclin B1 pathway was involved in the G2/M
arrest induced by DADS. These results are important in determining
the mechanism of action of DADS and its future clinical use.
The subsequent degradation of CDC25C in response to
DADS was mediated by Chk1. Chk1 and Chk2 were not directly
responsible for the inhibition of DNA synthesis induced by DADS.
However, Chk1 negatively regulated the entry of DADS-treated cells
into mitosis. These findings suggest that DADS stimulates Chk1 to
initiate a G2/M cell cycle checkpoint. Furthermore, Chk1
acts to coordinate the cell cycle with DNA synthesis, thus
preventing premature mitotic entry in DADS-treated cells.
Co-immunoprecipitated Chk1 or Chk2 was detected by anti-Chk1 or
anti-Chk2 immunoprecipitation followed by anti-CDC25C
immunoblotting. DADS treatment enhanced the binding activity of
Chk1 with CDC25C in MGC803 cells, however, it did not influence the
binding activity of Chk2 with CDC25C. This finding confirms that
DADS induces G2/M arrest through the interaction of Chk1
and CDC25C in MGC803 cells.
There are two possible explanations for how DNA
synthesis may be arrested: either directly mediated by checkpoint
activation, or by other mechanisms that activate other checkpoints,
for example chain termination.
Compounds induced the activation of ATR as well as
Chk1 protein, suggesting that the compound caused effective DNA
damage. During DNA damage, Chk1 is activated by ataxia
telangiectasia and Rad3-related (ATR)-mediated phosphorylation.
Chk1 is a key regulator of checkpoint signaling in the unperturbed
cell cycle and the DNA damage response. Ataxia telangiectasia
[(ATR) and Rad-3-related)] protein kinases are important in the
cell response to DNA damage (39).
Once the DNA damage has occurred, checkpoint effector proteins such
as Chk1 and Chk2 are phosphorylated and activated by ATR, leading
to cell cycle arrest in G1, S, G2 and M phases. As radio- and
chemotherapy activate the checkpoints, it can be expected that
abrogating the DNA damage checkpoints, particularly
G2/M, by certain agents would sensitize DNA damage,
leading to the death of cancer cells (40). ATR is capable of specifically
phosphorylating Chk1 (37). Another
important finding of the present study was that ATR is required for
the maximal phosphorylation of Chk1 checkpoint proteins following
DADS treatment. However, ATR only phosphorylates Chk1 (Ser345), and
not Chk2. This substrate specificity may reflect the initial
oxidative DNA strand breaks. Furusawa et al demonstrated
that Chk1 plays a crucial role in the apoptosis and regulation of
cell cycle progression in human leukemia Jurkat cells under
hyperthermia induced by heat stress (HS) (41). The ATR-Chk1 pathway was
preferentially activated under HS. Inhibition of ATR and Chk1 also
abrogated G2/M checkpoint activation by HS. In addition,
the efficiency of Chk1 inhibition on G2/M checkpoint
abrogation and apoptosis induction was confirmed in the adherent
HeLa, HSC3 and PC3 cancer cell lines, suggesting that the targeting
of Chk1 can be effective in solid tumor cells (41).
The results of the present study have shown that
DADS induces G2/M arrest in MGC803 gastric
adenocarcinoma cell lines. These effects may be related to the
activation of phospho-checkpoint kinase-1. The DADS-induced
G2/M checkpoint response is mediated by Chk1 signaling
through ATR/Chk1/CDC25C/cyclin B1 and is independent of Chk2.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 30600285 and 81071966), the
Key Project of Hunan Province Science and Technology Plan (no.
2012SK2012), the Platform Open Innovation Fund Project for Hunan
Province Universities (grant no. 12K093), the Foundation of the
Construct Program of the Key Discipline in Hunan Province of China
[no. (2011)76], and the Aid Program for Science and Technology
Innovative Research Team in University of South China.
Abbreviations:
|
DADS
|
diallyl disulfide
|
|
ATR
|
ATM-RAD3-related gene
|
|
Chk1
|
checkpoint kinase 1
|
|
CDC25C
|
cell division cycle 25C
|
|
PBS
|
phosphate-buffered saline
|
References
|
1
|
Steigman SA, Kunisaki SM, Wilkins-Haug L,
Takoudes TC and Fauza DO: Optical properties of human amniotic
fluid: implications for videofetoscopic surgery. Fetal Diagn Ther.
27:87–90. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kunisaki C, Makino H, Kimura J, et al:
Impact of lymphovascular invasion in patients with stage I gastric
cancer. Surgery. 147:204–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arunkumar R, Sharmila G, Elumalai P, et
al: Effect of diallyl disulfide on insulin-like growth factor
signaling molecules involved in cell survival and proliferation of
human prostate cancer cells in vitro and in silico approach through
docking analysis. Phytomedicine. 19:912–923. 2012. View Article : Google Scholar
|
|
4
|
Lai KC, Kuo CL, Ho HC, et al: Diallyl
sulfide, diallyl disulfide and diallyl trisulfide affect drug
resistant gene expression in colo 205 human colon cancer cells in
vitro and in vivo. Phytomedicine. 19:625–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yi L, Ji XX, Tan H, et al: Involvement of
Mcl1 in diallyl disulfide-induced G2/M cell cycle arrest in HL-60
cells. Oncol Rep. 27:1911–1917. 2012.PubMed/NCBI
|
|
6
|
Ling H, Zhang LY, Su Q, et al: Erk is
involved in the differentiation induced by diallyl disulfide in the
human gastric cancer cell line MGC803. Cell Mol Biol Lett.
11:408–423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yi L and Su Q: Molecular mechanisms for
the anti-cancer effects of diallyl disulfide. Food Chem Toxicol.
57:362–370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song JD, Lee SK, Kim KM, et al: Molecular
mechanism of diallyl disulfide in cell cycle arrest and apoptosis
in HCT-116 colon cancer cells. J Biochem Mol Toxicol. 23:71–79.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liao QJ, Su J, He J, Song Y, Tang HL and
Su Q: Effect of diallyl disulfide on cell cycle arrest of human
colon cancer SW480 cells. Ai Zheng. 28:138–141. 2009.PubMed/NCBI
|
|
10
|
Dasgupta P and Bandyopadhyay SS: Role of
di-allyl disulfide, a garlic component in NF-κB mediated transient
G2-M phase arrest and apoptosis in human leukemic cell-lines. Nutr
Cancer. 65:611–622. 2013.PubMed/NCBI
|
|
11
|
Arunkumar A, Vijayababu MR, Srinivasan N,
Aruldhas MM and Arunakaran J: Garlic compound, diallyl disulfide
induces cell cycle arrest in prostate cancer cell line PC-3. Mol
Cell Biochem. 288:107–113. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Knowles LM and Milner JA: Diallyl
disulfide inhibits p34cdc2 kinase activity through
changes in complex formation and phosphorylation. Carcinogenesis.
21:1129–1134. 2000.PubMed/NCBI
|
|
13
|
Ashra H and Rao KV: Elevated
phosphorylation of Chk1 and decreased phosphorylation of Chk2 are
associated with abrogation of G2/M checkpoint control during
transformation of Syrian hamster embryo (SHE) cells by Malachite
green. Cancer Lett. 237:188–198. 2006. View Article : Google Scholar
|
|
14
|
Xiang SL, Xiao XL, Ling H, et al:
Antitumor effect of diallyl disulfide on human gastric cancer
MGC803 cells xenograft in nude mice. Ai Zheng. 24:940–944. 2005.(In
Chinese).
|
|
15
|
Yuan JP, Wang GH, Ling H, et al: Diallyl
disulfide-induced G2/M arrest of human gastric cancer MGC803 cells
involves activation of p38 MAP kinase pathways. World J
Gastroenterol. 10:2731–2734. 2004.
|
|
16
|
Ling H, Wen L, Ji XX, et al: Growth
inhibitory effect and Chk1-dependent signaling involved in
G2/M arrest on human gastric cancer cells induced by
diallyl disulfide. Braz J Med Biol Res. 43:271–278. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shioya K, Michaux C, Kuenne C, Hain T, et
al: Genome-wide identification of small RNAs in the opportunistic
pathogen Enterococcus faecalis V583. PLoS One. 6:e239482011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dickson MA and Schwartz GK: Development of
cell-cycle inhibitors for cancer therapy. Curr Oncol. 16:36–43.
2009.PubMed/NCBI
|
|
19
|
Call JA, Eckhardt SG and Camidge DR:
Targeted manipulation of apoptosis in cancer treatment. Lancet
Oncol. 9:1002–1011. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bucher N and Britten CD: G2 checkpoint
abrogation and checkpoint kinase-1 targeting in the treatment of
cancer. Br J Cancer. 98:523–528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gong QF, Liu EH, Xin R, Huang X and Gao N:
2ME and 2OHE2 exhibit growth inhibitory effects and cell cycle
arrest at G2/M in RL95-2 human endometrial cancer cells through
activation of p53 and Chk1. Mol Cell Biochem. 352:221–230. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu J, Lai G, Wan F, et al: Knockdown of
checkpoint kinase 1 is associated with the increased
radiosensitivity of glioblastoma stem-like cells. Tohoku J Exp Med.
226:267–274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu W, Zong Q, John-Baptiste A and Jessen
B: Transient knock down of checkpoint kinase 1 in hematopoietic
progenitors is linked to bone marrow toxicity. Toxicol Lett.
204:141–147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeng X and Kinsella TJ: BNIP3 is essential
for mediating 6-thioguanine- and 5-fluorouracil-induced autophagy
following DNA mismatch repair processing. Cell Res. 20:665–675.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hsu SC, Yu CC, Yang JS, et al: A novel
synthetic 2-(3-methoxyphenyl)- 6,7-methylenedioxoquinolin-4-one
arrests the G2/M phase arrest via Cdc25c and induces apoptosis
through caspase- and mitochondria-dependent pathways in TSGH8301
human bladder cancer cells. Int J Oncol. 40:731–738. 2012.
|
|
26
|
Pabla N, Bhatt K and Dong Z: Checkpoint
kinase 1 (Chk1)-short is a splice variant and endogenous inhibitor
of Chk1 that regulates cell cycle and DNA damage checkpoints. Proc
Natl Acad Sci USA. 109:197–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Niida H, Murata K, Shimada M, et al:
Cooperative functions of Chk1 and Chk2 reduce tumour susceptibility
in vivo. EMBO J. 29:3558–3570. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Soule BP, Simone NL, DeGraff WG, Choudhuri
R, Cook JA and Mitchell JB: Loratadine dysregulates cell cycle
progression and enhances the effect of radiation in human tumor
cell lines. Radiat Oncol. 5:82010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Moon DO, Kim MO, Nam TJ, Kim SK, Choi YH
and Kim GY: Pectenotoxin-2 induces G2/M phase cell cycle
arrest in human breast cancer cells via ATM and Chk1/2-mediated
phosphorylation of cdc25C. Oncol Rep. 24:271–276. 2010.PubMed/NCBI
|
|
30
|
Yan Y, Cao PT, Greer PM, et al: Protein
phosphatase 2A has an essential role in the activation of
γ-irradiation-induced G2/M checkpoint response. Oncogene.
29:4317–4329. 2010.
|
|
31
|
Rong JJ, Hu R, Song XM, et al: Gambogic
acid triggers DNA damage signaling that induces
p53/p21Waf1/CIP1 activation through the ATR-Chk1
pathway. Cancer Lett. 296:55–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bonnet J, Mayonove P and Morris MC:
Differential phosphorylation of Cdc25C phosphatase in mitosis.
Biochem Biophys Res Commun. 370:483–488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reinhardt HC and Yaffe MB: Kinases that
control the cell cycle in response to DNA damage: Chk1, Chk2, and
MK2. Curr Opin Cell Biol. 21:245–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zegerman P and Diffley JF: DNA replication
as a target of the DNA damage checkpoint. DNA Repair. 8:1077–1088.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi L, Chen J, Wang YY, et al: Gossypin
induces G2/M arrest in human malignant glioma U251 cells by the
activation of Chk1/Cdc25C pathway. Cell Mol Neurobiol. 32:289–296.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aressy B and Ducommun B: Cell cycle
control by the CDC25 phosphatases. Anticancer Agents Med Chem.
8:818–824. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chaudhary P, Sharma R, Sahu M, Vishwanatha
JK, Awasthi S and Awasthi YC: 4-Hydroxynonenal induces
G2/M phase cell cycle arrest by activation of the ataxia
telangiectasia mutated and Rad3-related protein (ATR)/checkpoint
kinase 1 (Chk1) signaling pathway. J Biol Chem. 288:20532–20546.
2013.PubMed/NCBI
|
|
38
|
Duan J, Yu Y, Li Y, et al: Toxic effect of
silica nanoparticles on endothelial cells through DNA damage
response via Chk1-dependent G2/M checkpoint. PLoS One.
8:e620872013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Matsuoka S, Ballif BA, Smogorzewska A, et
al: ATM and ATR substrate analysis reveals extensive protein
networks responsive to DNA damage. Science. 316:1160–1166. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nishida H, Tatewaki N, Nakajima Y, et al:
Inhibition of ATR protein kinase activity by schisandrin B in DNA
damage response. Nucleic Acids Res. 37:5678–5689. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Furusawa Y, Iizumi T, Fujiwara Y, et al:
Inhibition of checkpoint kinase 1 abrogates G2/M checkpoint
activation and promotes apoptosis under heat stress. Apoptosis.
17:102–112. 2012. View Article : Google Scholar : PubMed/NCBI
|