Introduction
In our previous studies, a functionally unknown gene
FAM172A, the family with sequence similarity 172, member A, was
identified. Bioinformatics analysis demonstrated that FAM172A
(C5orf21, NM_032042.5) contains an open reading frame composed of
1251 nucleotides encoding a protein comprised of 416 amino acids
and an Arb2 conserved domain located in gene sequence. Online
software CELLO 2.5 (http://cello.life.nctu.edu.tw/) to forcast the
subcellular location of the human protein (1), was indicative of localization in the
nucleus and/or cytoplasm of human FAM172A protein.
FAM172A was first identified in human aortic
endothelial cells, THP-1-derived macrophages, and human aortic
smooth muscle cells at the translation level through western
blotting (2,3). Feng et al found that FAM172A
was notably downregulated among hepatocellular carcinoma or
cirrhotic patients. It indicated that FAM172A might be a novel
anticancer gene, which enphasizes a crucial role in the control of
cell cycle and proliferation of tumor cells (4,5).
STAT1, signaling transducer and transcription
activator 1, belongs to the STAT protein family. This protein,
activated by ligands including interferon-α, PDGF, and IL6,
mediates the expression of various genes, which is considered to be
crucial for cell activity in response to pathogens and cell stimuli
(6). STAT-proteins are activated by
tyrosine phosphorylation, usually by JAK kinases. They dimerize,
translocate to the nucleus and activate their specific target genes
(7–9). STAT1 is required for apoptosis induced
by ischemia in cardiac myocytes and by tumor necrosis factor,
oxysterols, and DNA damage (10,11).
As an important transcription factor, STAT1 may exert an essential
role in the expression of FAM172A.
We have found that FAM172A protein expressed
moderately in normal tissue, but decreased significantly in
colorectal cancer tissue (4).
However, the functions of FAM172A on colon cancer cells are
unknown, and the regulatory mechanism of its expression remains
unclear. In the current study, our results demonstrated that
FAM172A inhibited proliferation and promoted apoptosis or
differentiation of colon cancer cells. In addition, we cloned the
functional promoter variants in FAM172A and presented that the DNA
fragment from −112 and +48 of the FAM172A promoter is crucial for
its transcription in LoVo cells. Above all, we elaborated the
interaction between transcription factors STAT1 and FAM172A
promoter in regulating expression of FAM172A. These findings
contribute to clarification of the regulatory mechanisms of FAM172A
transcription and to comprehend the functions of FAM172A.
Materials and methods
Cell culture
Human LoVo and SW480 cells were obtained from The
General Hospital of People's Liberation Army (Beijing, China).
Cells were cultured in a 5% Co2-humidified atmosphere
using DMEM medium containing 10% fetal bovine serum (FBS) at
37°C.
Genomic DNA preparation and construction
of plasmids
Genomic DNA and cDNA were amplified and sequenced as
previously described (12). Genomic
DNA was prepared from LoVo cells using a genomic DNA Purification
kit (Promega, Madison, WI, USA). DNA fragments of FAM172A [P1 (−740
to +205), P2 (−740 to −260), P3 (−260 to +205), P4 (−112 to +205),
P5 (+48 to +205) and P6 (−112 to +48)] upstream of the
transcription initiation site were cloned into a pGL4.10 Basic
vector with the restriction sites of XholI and EcoRV
(Promega). The followings were PCR primers: P1 sense,
5′-CTCGAGTTGCAAAGTACAAACAGTGTG-3′; P2 antisense,
5′-GATATCCAGACTTTACCCTGTCCATTC-3′; P3 sense,
5′-CTCGAGACACACTCTGAGTAGCGGAG-3′; P4 sense,
5′-CTCGAGAGTGCATAAGAGAACTACACTTAATTC-3′; P5 sense,
5′-CTCGAGAGTGCAACTCGAACTTGGTC-3′; P6 antisense,
5′-GATATCTCCGGGGTCTTCAGGAG-3′; P1 antisense,
5′-GATATCCAAACGGCAGTCCTACCTG-3′. The STAT1 expression plasmid was
amplified from STAT1 mRNA (PubMed: NM_007315.3) using the upstream
primer (5′-GATATCATGTCTCAGTGGTACGAACTTCAGC-3′) and the downstream
primer (5′-GGATCCTACTGTGTTCATCATACTGTCGAAT-3′) inserted into
pcDNA3.1/myc-His (−) plasmid with restriction sites EcoRV
and BamHI.
Dual-luciferase reporter assay
The method of Luciferase reporter assay was
previously described (13). LoVo
cells were cultured in a 48-well plates in advance and transfected
with 0.23 μg of FAM172A promoter plasmids and 0.02 μg
of endogenous control phRL-TK plasmids using jetPRIME™ (Polyplus
Transfection, Illkirch, France) in accordance with the
manufacturer's protocol. The medium was changed 4 h after
transfection, then continued for approximately 24 h. Afterwards,
cells were collected and lysed in 70 μl of passive lysis
buffer. Luciferase reporter assay was implemented with a
dual-luciferase reporter assay system (Promega) and measured using
a Veritas Microplate Luminometer (Turner BioSystems, USA). The
transfections were conducted in triplicate, and the activities of
promoter was recorded as Firefly/Renilla ratio using mean ± SEM of
three independent experiments. For STAT1 responses, 0.11 μg
of FAM172A promoter plasmids, 0.03 μg of phRL-TK plasmids as
internal control and 0.11 μg of pcDNA3.1 (−)-STAT1 plasmids
were co-transfected into LoVo cells.
Cell counting and cell cycle assay
Colon cancer cells were added with diverse
concentrations of FAM172A recombinant proteins (0, 0.1, 1.0, 10 and
100 ng/ml). Then, cells were trypsinized and washed lightly in PBS
followed by fixation for 30 min in ice-cold 70% ethanol.
Subsequently, cells were harvested and then stained with 7-AAD (5
mg/ml) for 30 min. The group treated by PBS was applied to the
controls. Cell cycle was determined by flow cytometer (BD
Biosciences, San Diego, CA, USA). The results were analysed with
Modifit software.
Determination of apoptosis
Annexin V-FLUOS (Biolegend) staining was performed
on the cells and analyzed using a fluorescence-activated cell
sorting (FACS) following the described protocol (14). Cells (3×105) were
cultured onto dishes for 48 h, and subsequently washed at 4°C for
30 min with PBS and then incubated at 4°C for 3 h with Annexin
V-FLUOS staining solution (Roche Diagnostics, Indianapolis, IN,
USA). Afterwards, gels were washed 4 times in PBS at 4°C and fixed
with 1% paraformaldehyde (Sigma-Aldrich) at room temperature for 15
min. The total number of cells were identified separately by FACS
technique. Apoptosis was verified by detection of activated
caspases (12).
Chromatin immunoprecipitation (ChIP)
The process for ChIP was previously clarified
(15,16). LoVo cells (1×106) were
cultured for this experiment. Immunoprecipitation was performed
overnight at 4°C with 5 μg of anti-STAT1 (Cell Signaling
Technology inc., Danvers, MA, USA) or negative control, normal
anti-IgG antibody or positive control, anti-acetyl histone H3
antibody and 20 μl of protein A agarose beads. The
immunoprecipitated complex was washed multiple times, and then
DNA-antibody-protein complex was detached from the beads. Followed
by decross-link incubation, the RNA and protein were removed by
RNAse and proteinase K, and a quantitative real-time PCR was
carried out to amplify the eluted DNA with specific primers for the
STAT1 binding site. A 1% agarose gel was used to separate the PCR
products.
Electrophoretic mobility shift assay
(EMSA)
The EMSA annlysis was condocted as previously
described (17). Probes for EMSA
were synthesized by a biotin label at the 3′ end (Invitrogen,
Shanghai, China). To prepare Nuclear extracts from LoVo cells, a
Nuclear Extraction kit (Pierce, Rockford, IL, USA) was utilized.
Protein concentration was quantitated by BCA protein assay kit
(Pierce). EMSA assays were done using LightShift chemiluminescence
EMSA kit (Pierce). DNA-protein binding reactions were executed in a
20 μl volume using 3 μg of nuclear extract in a
binding buffer containing different volumes of 50% glycerol, 100 mM
MgCl2, and 0.02 mM EDTA. The complexes were separated
using 6.5% non-denaturing polyacrylamide gels. To analyze the
supershift, additional 5 μg anti-STAT1 antibody was added 30
min prior to the addition of complexes.
Western blot analysis and polyclonal
antibody
Preparation and generation of FAM172A recombinant
protein was previously described (2). Then protein was boiled with 5X SDS gel
loading buffer. Equal amounts of sample were subjected to 10%
SDS-PAGE electrophoresis, transferred to PVDF membrane (Millipore,
Billerica, MA, USA), and incubated with specific antibodies,
respectively, followed by secondary antibodies IgG. Expression of
GAPDH was considered as an internal control. The band intensities
of protein were quantitated using the GE Typhoon Trio (GE
Healthcare, Piscataway, NJ, USA).
RNA isolation and real-time PCR
We used Total RNA kit I (Promega) to extract total
RNA from cultured cells. Extracted RNA was quantified by
spectrophotometry. The 0.1 μg of RNA was reversely
transcribed by PrimeScript™ RT reagent kit (Takara, Otsu, Japan).
Power SYBR® Green PCR Master Mix was performed for
real-time PCR using an ABI Prism 7500 (USA). β-actin was considered
as an internal control. The specific primers of FAM172A were as
follows: sense, 5′-cgactggcgaactggaag-3′; antisense,
5′-gagctcaaggaaatagacatcaatc-3′.
Statistical analysis
The results are presented as means ± SEM. For
statistical analysis, the two-tailed Student's t-test or one-way
analysis of variance (ANOVA) was done to compare the differences of
means using GraphPad Prism 5 or SPSS 20 software. p-value <0.05
was set as statistically significant. All experiments were repeated
three separate times unless stated otherwise.
Results
FAM172A inhibits proliferation of colon
cancer cells
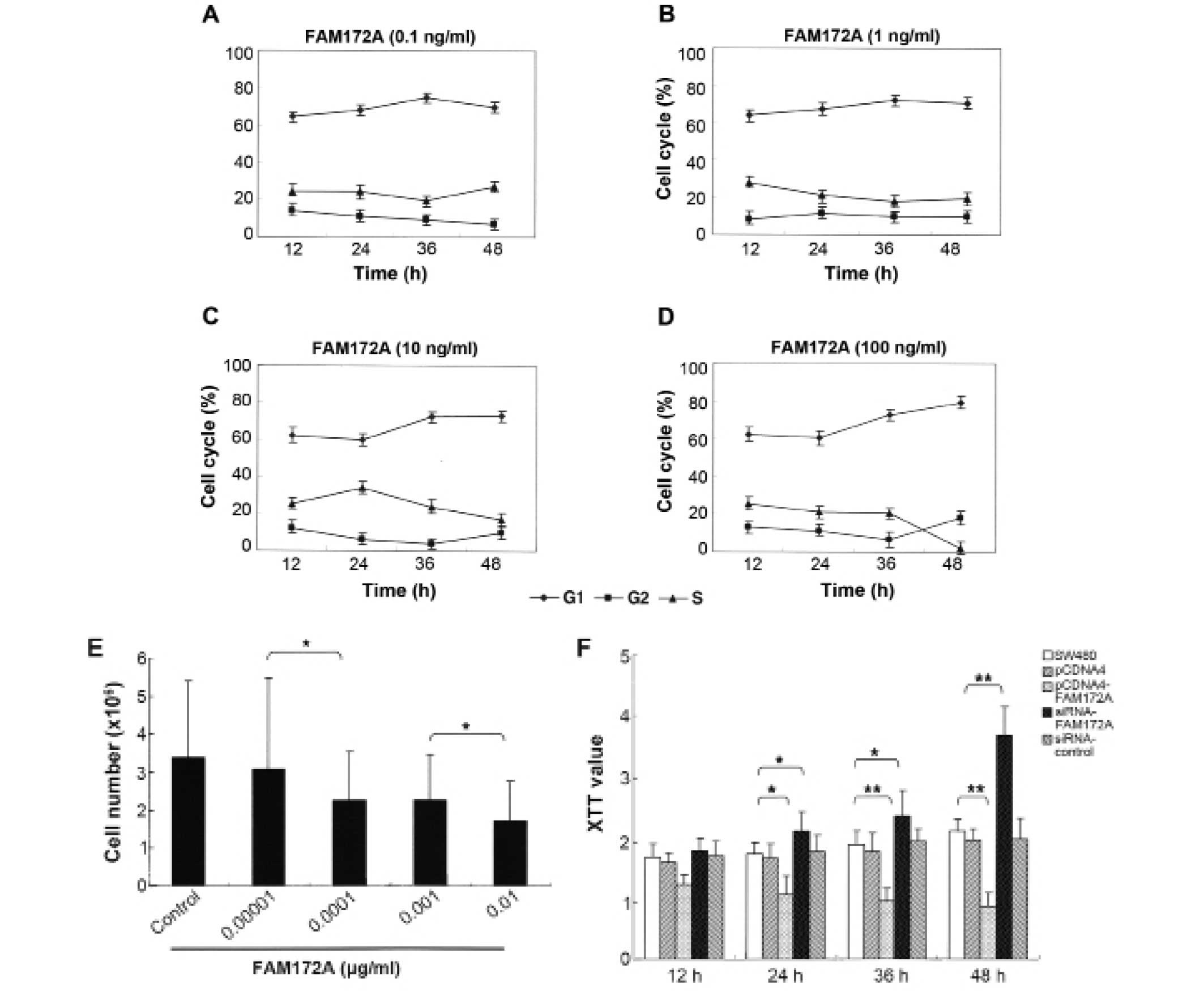
To determine the influence of FAM172A on the cell
cycle, SW480 colon cancer cells were co-cultured with different
concentrations of FAM172A recombinant protein. We found that the
percentage of cells in S phase and the total number of colon cancer
cells decreased in a dose-dependent manner (Fig. 1A–E). Morever, when at the highest
concentration (0.01 μg/ml), the percentage of colon cancer
cells in S phase reduced to 3.18%. These results demonstrated that
FAM172A could significantly suppress the proliferation of colon
cancer cells, especially at a concentration >0.01
μg/ml.
Using XTT proliferation assay we found that FAM172A
significantly inhibited cell growth of SW480 cells compared to
control group at each time point (p<0.01) (Fig. 1F). The cells transfected with
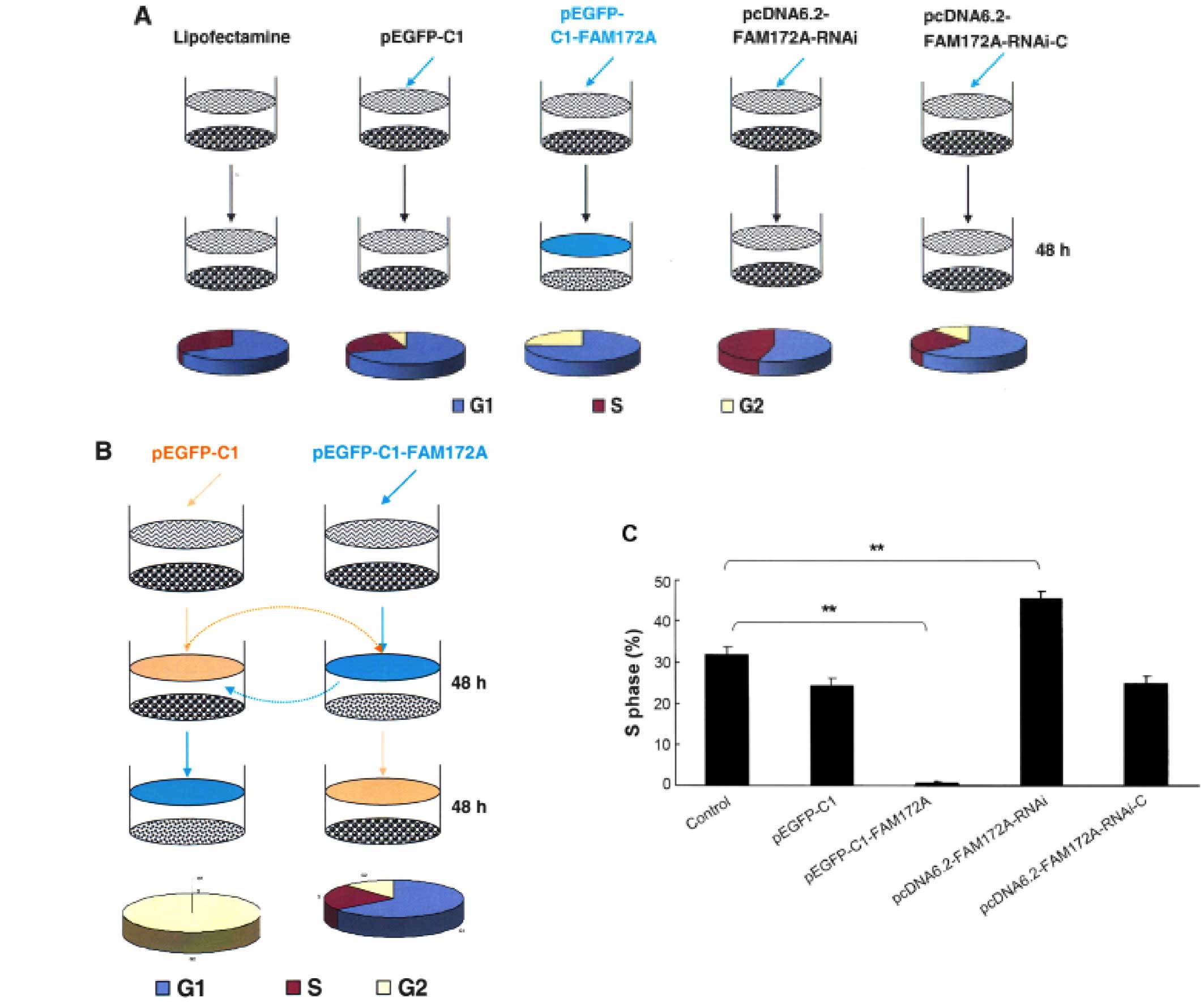
FAM172A were arrested at G1/S phase. Following transfection with
different vectors for 48 h, the S-phase of SW480 transfected with
pEGFP-C1, pEGFP-C1-FAM172A, pcDNA6.2-FAM172A shRNA and
pcDNA6.2-negative were 31.73, 24.32, 0.18, 45.42 and 24.85%,
respectively (Fig. 2A). After
changing the culture supernatant of cells, the influence of FAM172A
on the cell cycle was investigated (Fig. 2B). After incubation for 48 h, the
S-phase of SW480 cells was 40.1% (Fig.
2B). However, when SW480 cells were cultured for an additional
48 h with the culture supernatant of SW480 cells transfected with
pEGFP-C1- FAM172A, the S-phase was undetectable.
SW480 cells transfected with pEGFP-C1- FAM172A were
cultured for 48 h, and then further cultured for another 48 h with
interchanged culture supernatant from SW480 cells cultured for 48
h, but we found that the S-phase was unchanged. Compared with the
control cells, the cell cycles of the cells transfected with
pEGFP-C1-FAM172A were arrested at G1/S phase (p<0.05). The
results of pEGFP-C1-transfected group was the same as the
pcDNA6.2-RNAi-c-transfected group, and the cells of the two groups
were lower in number than the control cells (p>0.05).
Furthermore, the S-phase of pcDNA6.2-FAM172A-RNAi-3-transfected
group was higher than the other groups (p<0.05) (Fig. 2C).
FAM172A promotes apoptosis and
differentiation of colon cancer cells
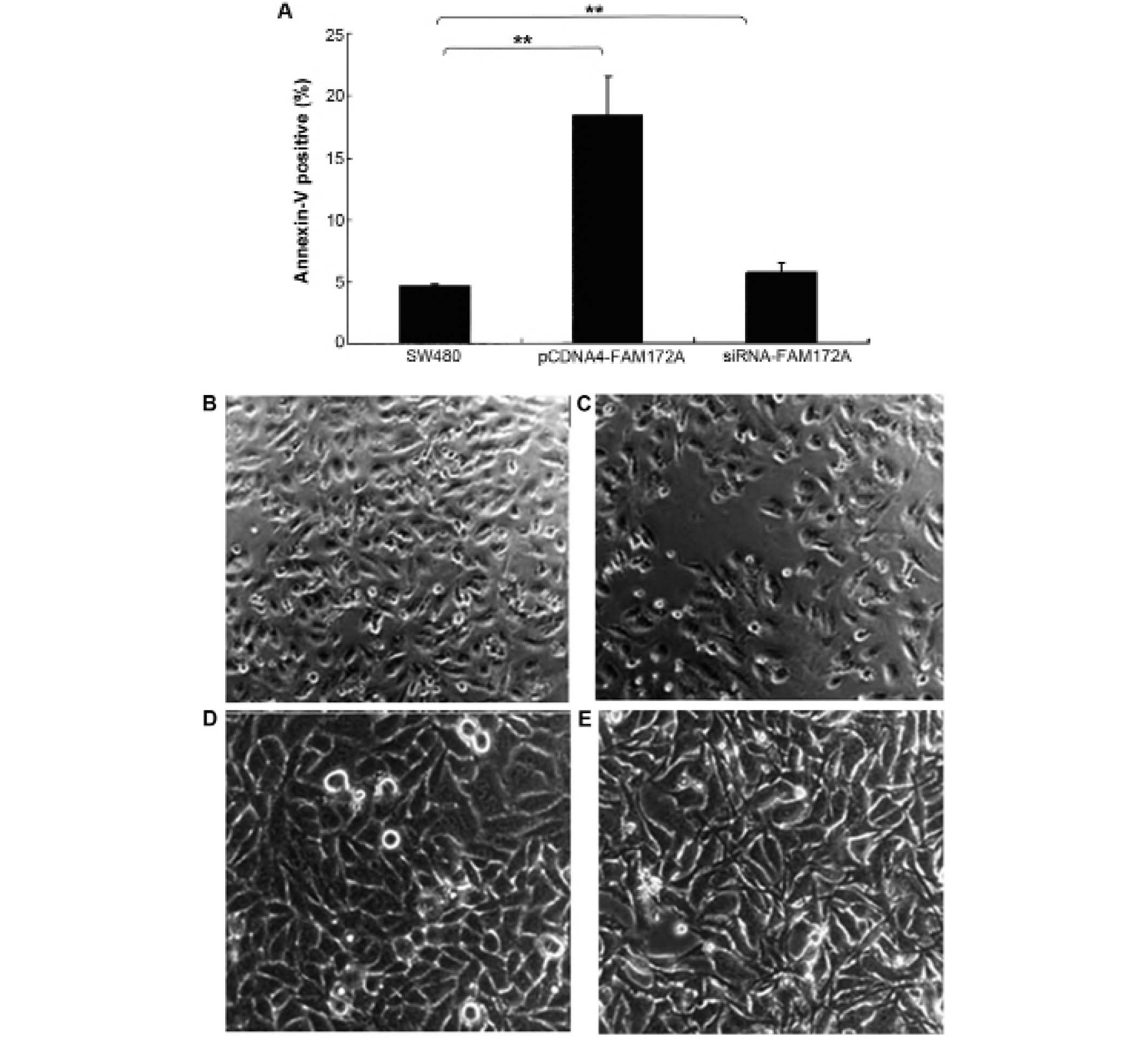
The effect of FAM172A on apoptosis was assessed in
colon cancer cells by flow cytometry. Upregulation of FAM172A in
SW480 cells induced increase of apoptosis (p<0.01). Moreover,
SW480 cells treated with shRNA of FAM172A suppressed apoptosis
significantly (p<0.01) (Fig.
3A). FAM172A recombinant protein caused differentiation
(Fig. 3C, magnification ×100; and
3E, magnification ×400) of SW480 cells compared with control
(Fig. 3B, magnification ×100; and
3D, magnification ×400).
Identification and analysis of FAM172A
promoter
In order to identify the FAM172A promoter, we
obtained the FAM172A gene mRNA and analyzed and predicted the
transcription start sites based on NCBI database and UCSC genome
Browser (http://genome.ucsc.edu/). Then the
genomic DNA fragment from −740 bp to +205 bp in the predicted
promoter of FAM172A was amplified from LoVo cells. The putative
FAM172A promoter region was inserted into a pGL4.10 Basic vector
with the restriction sites XholI and EcoRV.
pGL4.10-FAM172A promoter was named P1 and used for further
shortened promoter plasmids as the template. After transfection for
24 h, Luciferase reporter assay was carried out. The results were
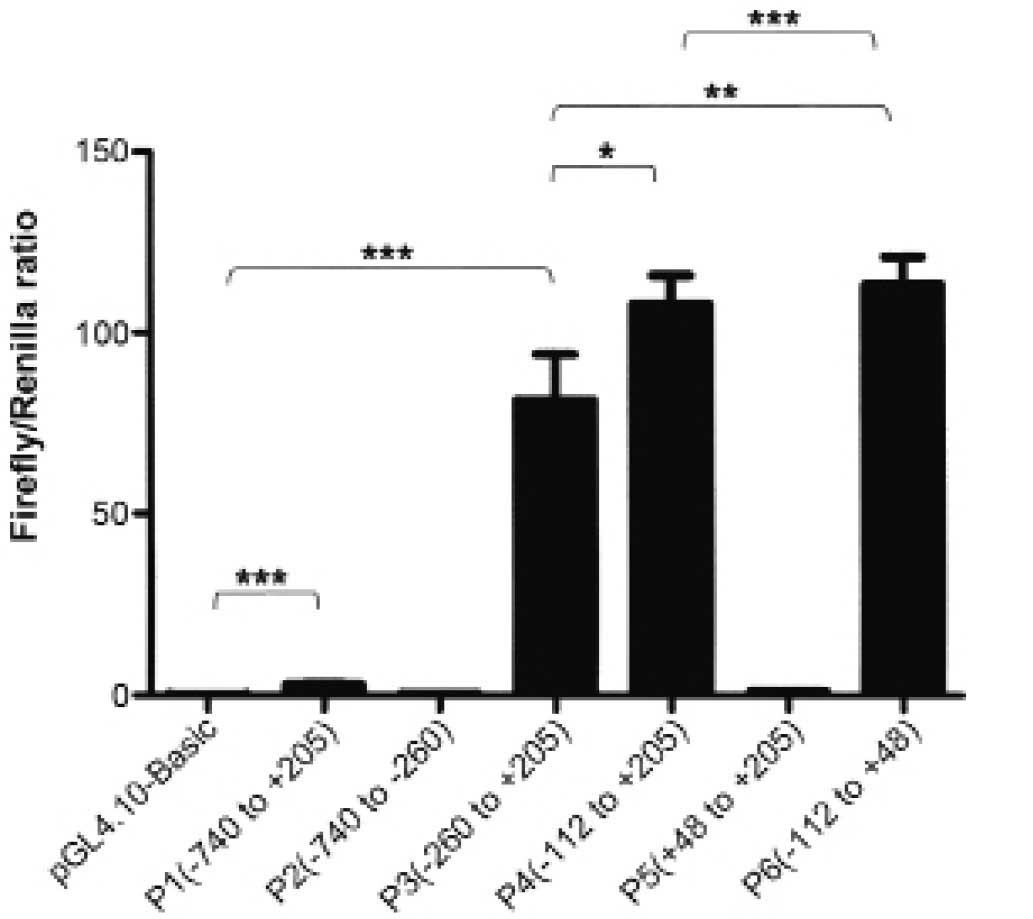
recorded as Firefly/Renilla ratio. Compared with pGL4.10 Basic, P1
had ~40-fold luciferase activity, which indicated that the putative
FAM172A promoter was active in the LoVo cells. Then, we studied the
minimal FAM172A promoter. Five recombinants plasmids P2, P3, P4, P5
and P6 were constructed, respectively by sequentially truncating P1
plasmid. Subsequently, luciferase assays were carried out as above.
The luciferase activity results are shown in Fig. 4. The serial truncation analysis
suggested that P6, −112 to +48, contained the essential sequences
for transcriptional activity.
Prediction of transcription factor and
its binding sites
We predicted possible transcription factors by
searching TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html) and
GeneCards (http://www.genecards.org/). Based on
our analysis, one major common transcription factor with the
highest score, STAT1, was within the minimum promoter. The STAT1
binding site was between −68 and −58 within the mimimal promoter
region (−112 to +48).
Specific binding of STAT1 transcription
factors to the functional promoter
In order to investigate whether STAT1 could bind to
FAM172A promoter in vivo, chromatin immunoprecipitation
(ChIP) was performed followed by the PCR assay to amplify the
region between −112 and +48. These results demonstrated that the
binding bite of STAT1 was presented on P6 compared with
IgG-precipitated controls. This revealed that STAT1 could
specifically bind to the FAM172A promoter (Fig. 5).
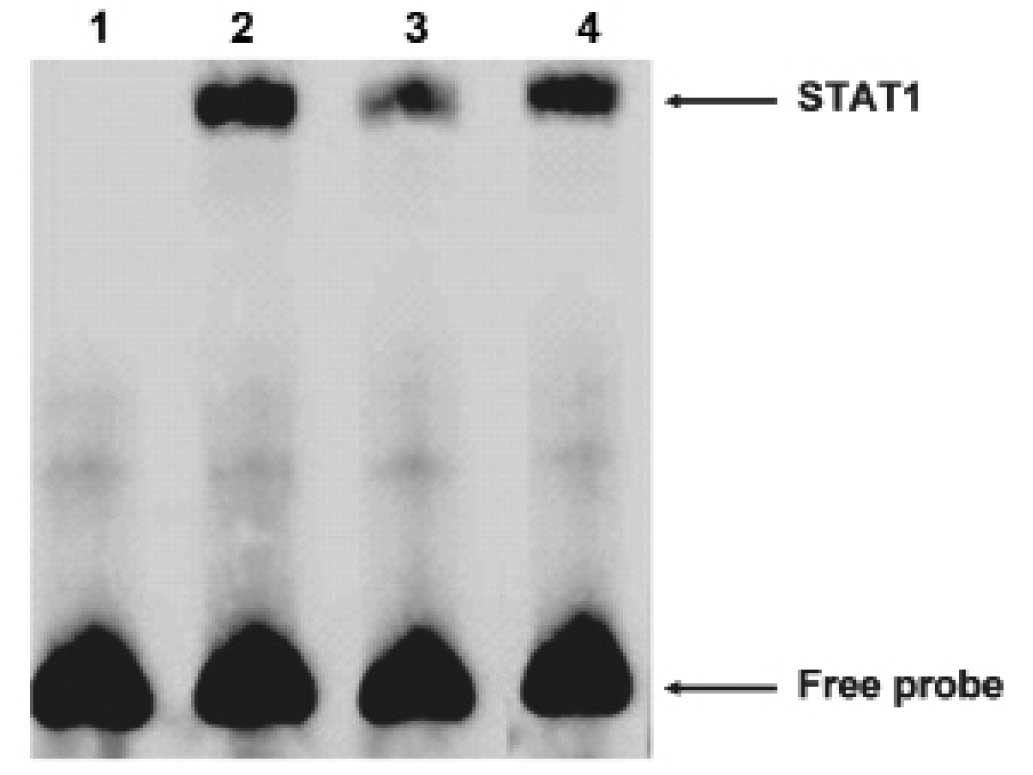
Then we carried out electrophoretic mobility shift
assays (EMSA) to explore whether STAT1 could bind the FAM172A
promoter. The oligonucleotide contained sequences which matched a
consensus binding site for STAT1 was chose for EMSA. The nuclear
extracts from the LoVo cells revealed strong binding to a labeled
probe embracing an STAT1 binding sites. We then conducted
competition experiments with unlabeled probes. DNA-protein
complexes were specific, as demonstrated by the fact that it could
be completely out-competed by a 100-fold excess of unlabeled probe
(Fig. 6). These results suggested
that STAT1 was able to bind to the FAM172A minimum promoter. As
shown above, these results revealed that STAT1 could specifically
bind to the FAM172A promoter.
Upregulation of the expression of FAM172A
by STAT1 transcription factor
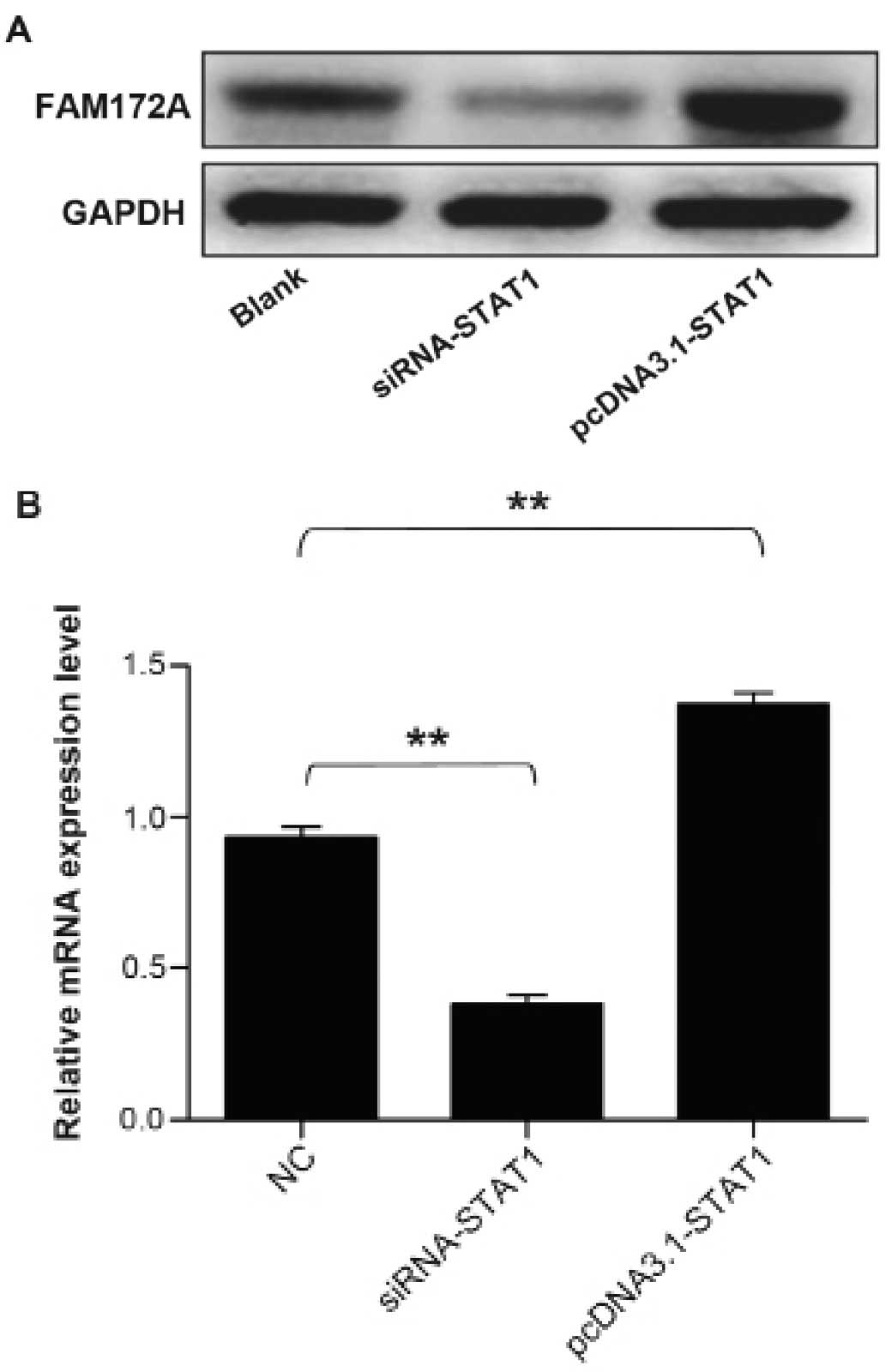
To address whether STAT1 transcription factor could
facilitate the expression of FAM172A, we performed western blot
analysis together with quantitative real-time PCR to validate the
upregulation of FAM172A. The protein samples and total RNA were
extracted from cultured LoVo cells transfected with STAT1 gene
overexpression plasmid and siRNA. The results demonstrated that the
mRNA and protein expression level of FAM172A increased prominently
induced by STAT1, which infered that STAT1 could promote the
expression of FAM172A. The results are presentrd in Fig. 7.
Discussion
Our previous study (4) found that FAM172A significantly
decreased in colorectal cancer tissues, but its functions were
unknown and few relevant studies on FAM172A in colorectal cancer
were reported. In this investigation, we explored the functions of
FAM172A gene and the mechanism of its transcriptional regulation.
Firstly, XTT assays showed that FAM172A was able to significantly
inhibit proliferation of colon cancer cells. Subsequently, we
evaluated the effect of FAM172A on the cell cycle and apoptosis.
Our results elaborated that FAM172A could promote apoptosis of
colon cancer cells and might be involved in G1/S arrest. In
summary, we considered that FAM172A might be an important target
gene for anti-colon cancer therapy in the future. Then the
regulation of expression of FAM172A was studied because
transcription process is one of the most important factors. A
putative promoter of FAM172A, a region ~945 bp upstream of
transcription start site, was cloned in a reporter vector. Then
truncated promoters were constructed as indicated above. These
promoters were analyzed using dual-luciferase reporter assay system
in LoVo cells. The fragment P6 had the highest promoter activity,
which indicated P6 possessed the essential sequences for
transcription activity and might be a minimum promoter region. This
minimum promoter fragment, from −112 bp to +48 bp, was verified to
drive the expression of FAM172A.
In addition, the major cis-acting element
STAT1 was present in this minimum promoter region. Further studies
showed that STAT1 could bind to the minimum FAM172A promoter. We
considered that STAT1 might affect the expression of FAM172A. After
co-transfection of STAT1 over expression plasmid or siRNA with
minimum promoter in vitro, the results revealed that STAT1
strongly increased the transcriptional activity of FAM172A minimum
promoter.
STAT proteins emphasize a vital role in
signal-transduction pathways activated by cytokines (18,19).
The JAK-STAT pathway is essential for converting cytokine signals
into gene expression programs and then mediates the cell
proliferation and differentiation (12,20,21).
As a transcription factor, we predicted that STAT1 could affect the
FAM172A expression. After transfected with overexpression plasmid
and siRNA of STAT1, the western blot analysis and real-time PCR
demonstrated that FAM172A was significantly upregulated by STAT1
compared to control groups. The result indicated that STAT1 could
enhance the activity of FAM172A promoter, prompting its
transcription, thus upregulating the expression of FAM172A.
In conclusion, this study identified the minimum
FAM172A promoter and demonstrated that FAM172A is upregulated by
STAT1 transcription factor. We also elucidated the regulatory
mechanisms of FAM172A transcription. This will add to better
understanding of the functions of FAM172A.
In conclusion, FAM172A modulated apoptosis and the
proliferation process of colon cancer cells. We will conduct
further studies to explore the functions of FAM172A for the
biological treatment of colorectal cancer.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (81172383), Guangdong Natural
Science Foundation (2014A030313324) and the Key Clinical Specialty
Discipline Construction Program.
References
|
1
|
Yu CS, Chen YC, Lu CH and Hwang JK:
Prediction of protein subcellular localization. Proteins.
64:643–651. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li L, Dong X, Leong MC, Zhou W, Yang Z,
Chen F, Bao Y, Jia W and Hu R: Identification of the novel protein
FAM172A, and its up-regulation by high glucose in human aortic
smooth muscle cells. Int J Mol Med. 26:483–490. 2010.PubMed/NCBI
|
|
3
|
Li LX, Tao Z, Dong XH, Liang WC, Yang Zh,
Mou B, Bao YQ, Wang C, Jia WP and Hu RM: Molecular cloning of a
novel gene, C5orf21 gene and its roles in diabetic macroangiopathy.
Zhonghua Yi Xue Za Zhi. 89:2574–2577. 2009.in Chinese.
|
|
4
|
Feng Z, Li H, Liu S, Cheng J, Xiang G and
Zhang J: FAM172A induces S phase arrest of HepG2 cells via Notch 3.
Oncol Rep. 29:1154–1160. 2013.PubMed/NCBI
|
|
5
|
Ehebauer M, Hayward P and Arias AM: Notch,
a universal arbiter of cell fate decisions. Science. 314:1414–1415.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DeVries TA, Kalkofen RL, Matassa AA and
Reyland ME: Protein kinase Cdelta regulates apoptosis via
activation of STAT1. J Biol Chem. 279:45603–45612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heim MH: The Jak-STAT pathway: Cytokine
signalling from the receptor to the nucleus. J Recept Signal
Transduct Res. 19:75–120. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rawlings JS, Rosler KM and Harrison DA:
The JAK/STAT signaling pathway. J Cell Sci. 117:1281–1283. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kisseleva T, Bhattacharya S, Braunstein J
and Schindler CW: Signaling through the JAK/STAT pathway, recent
advances and future challenges. Gene. 285:1–24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Agrawal S, Agarwal ML, Chatterjee-Kishore
M, Stark GR and Chisolm GM: Stat1-dependent, p53-independent
expression of p21(waf1) modulates oxysterol-induced apoptosis. Mol
Cell Biol. 22:1981–1992. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Townsend PA, Scarabelli TM, Davidson SM,
Knight RA, Latchman DS and Stephanou A: STAT-1 interacts with p53
to enhance DNA damage-induced apoptosis. J Biol Chem.
279:5811–5820. 2004. View Article : Google Scholar
|
|
12
|
Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi
C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C,
Eid P, et al: Impaired response to interferon-alpha/beta and lethal
viral disease in human STAT1 deficiency. Nat Genet. 33:388–391.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Datta M and Bhattacharyya NP: Regulation
of RE1 protein silencing transcription factor (REST) expression by
HIP1 protein interactor (HIPPI). J Biol Chem. 286:33759–33769.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haybaeck J, Obrist P, Schindler CU, Spizzo
G and Doppler W: STAT-1 expression in human glioblastoma and
peritumoral tissue. Anticancer Res. 27:3829–3835. 2007.
|
|
15
|
Das S and Bhattacharyya NP: Transcription
regulation of HYPK by Heat Shock Factor 1. PLoS One. 9:e855522014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao X, Wang Q, Li W, Yang B, Song H, Ju W,
Liu S and Cheng J: Identification of nucleolar and coiled-body
phosphoprotein 1 (NOLC1) minimal promoter regulated by NF-κB and
CREB. BMB Rep. 44:70–75. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pradhan SA, Rather MI, Tiwari A, Bhat VK
and Kumar A: Evidence that TSC2 acts as a transcription factor and
binds to and represses the promoter of Epiregulin. Nucleic Acids
Res. 42:6243–6255. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheon H, Yang J and Stark GR: The
functions of signal transducers and activators of transcriptions 1
and 3 as cytokine-inducible proteins. J Interferon Cytokine Res.
31:33–40. 2011. View Article : Google Scholar :
|
|
19
|
Chapgier A, Boisson-Dupuis S, Jouanguy E,
Vogt G, Feinberg J, Prochnicka-Chalufour A, Casrouge A, Yang K,
Soudais C, Fieschi C, et al: Novel STAT1 alleles in otherwise
healthy patients with mycobacterial disease. PLoS Genet.
2:e1312006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stark GR and Darnell JE Jr: The JAK-STAT
pathway at twenty. Immunity. 36:503–514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pfitzner E, Kliem S, Baus D and Litterst
CM: The role of STATs in inflammation and inflammatory diseases.
Curr Pharm Des. 10:2839–2850. 2004. View Article : Google Scholar : PubMed/NCBI
|