Introduction
The cancer microenvironment has been implicated in
playing a critical role(s) in the development of drug resistance
(1). Mesenchymal stem cells (MSCs)
comprise a cell type found in the cancer microenvironment (2). It has been reported that stromal cells
protect tumor cells from apoptosis induced by chemotherapy either
through cell-cell interactions with tumor cells or by the local
release of soluble factors, such as interleukin-6, which promote
survival and tumor growth (3,4). In
contrast to solid tumors that invade bone marrow, leukemia
originates in the marrow in close proximity to stromal cells that
provide growth and survival signals (5). These signals provide the primary drug
resistance mechanism in leukemia, also referred to as cell
adhesion-mediated drug resistance, and may account for the minimal
residual disease in tissue (6,7). We
recognized the importance of MSC in drug resistance, and we propose
that determining how leukemia cells communicate with the
surrounding tissue will pave the way for eliminating residual
leukemia cells that are 'hiding' in stromal niches.
Leukemia-stroma interactions and the consequences on
tumor survival involve CXCR4/CXCL12 signaling (3). C-X-C motif chemokine 12 (CXCL12),
previously called stromal cell-derived factor-1 (SDF-1), is a
chemokine that is constitutively secreted by MSC and facilitates
the interaction between leukemia cells and stromal cells via its
cognate receptor CXCR4 (8).
Intracellular CXCR4 levels are significantly elevated in B-CLL
(9,10), B-cell acute lymphoblastic leukemia
(11), multiple myeloma (12), some AML (13), and CML (3). The CXCR4 antagonist, AMD3100
(Plerixafor), has been in clinical trials to resensitize leukemia
cells to cytotoxic drugs in co-cultures with MSC (14). However, blocking the CXCR4 only
partially overcomes stromal cell-emediated drug resistance
(14–17). Therefore, other interactions between
leukemia cells and the tumor microenvironment may provide
alternative therapeutic targets.
Arsenic trioxide (As2O3) is a
clinically effective agent in the treatment of acute promyelocytic
leukemia (APL) in primary cases (18,19),
relapsed cases (20), and in
ATRA-resistant patients (21).
As2O3 has also been shown to induce apoptosis
and display antiproliferative effects in Hodgkin's lymphoma,
myeloid leukemias that express BCR-ABL tyrosine kinase as well as
HL-60 cell types that overexpress Bcl-2, Bcl-X (L), MPR, and MRP
proteins (22–25). Low-dose As2O3
induces partial differentiation, while high-dose
As2O3 triggers apoptosis (26). Degradation of PML-RARα and PML is
the most important role that As2O3 plays in
the differentiation of APL cells (27). Mitochondrial pathway is the
principle mechanism of As2O3-induced
apoptosis (28). Intracellular
reactive oxygen species (ROS) also play a role in apoptosis
(29). However, the molecular
mechanism by which As2O3 modulates apoptosis
remains unclear. Identification of the genes regulated by
As2O3 will aid in developing our
understanding of the mechanisms of As2O3
therapy for the treatment of leukemia.
A downstream target of the TGFβ tumor suppressor
pathway, RUNX3, functions as a candidate tumor suppressor (30,31),
RUNX3 modulates cellular proliferation and apoptosis through
transcriptional regulation of genes, such as the growth regulator
p21 and the pro-apoptotic gene Bim (32,33).
RUNX3 is an important molecule that links the oncongenic Wnt and
the tumor-suppressive TGFβ pathways in intestinal carcinogenesis
(34). RUNX3 exhibits dynamic
shuttling between the nuclear and cytoplasmic compartments
(35), and RUNX3 is found tightly
associated with components of the nuclear architecture. The
targeting of RUNX3 to the nuclear matrix may have significant
biological consequences and may regulate RUNX3 activity (35,36).
In our study, we report that RUNX3 plays a critical role in
As2O3-induced apoptosis and MSC-mediated drug
resistance in leukemia cells.
Materials and methods
Generation of mesenchymal stem cells
Three milliliters of bone marrow cells were obtained
from normal donors aged between 20 and 50 years old who had
provided written consent under the Ethics Committee of Sun Yat-sen
University, which approved the protocol according to the
Declaration of Helsinki. To isolate MSC, bone marrow mononuclear
cells were isolated via density gradient centrifugation over
Percoll (Amersham Pharmacia Biotech, Little Chalfont, UK).
Immediately after centrifugation, isolated mononuclear cells were
resuspended at 5×103 cells/cm2 in α-modified
eagle's medium (αMEM) with high glucose concentration (Gibco, UK),
10% fetal bovine serum (FBS; Lonza, UK), and 1%
penicillin-streptomycin (Gibco). Cells were cultured at 37°C in a
humidified atmosphere containing 5% carbon dioxide. After 72 h,
non-adherent cells were removed. When the cells were 70–80%
confluent, adherent cells were trypsinized and expanded for 3–5
weeks. Before use in experiments, the identity of MSC was verified
by checking for positivity of CD105, CD106, CD73,CD90,CD144,
HLA-class I, and the lack of expression of CD45, CD34, CD133.
Co-culture experiments
For co-culture experiments, mesenchymal stem cells
were seeded the day before the experiment onto 6-well plates
(Corning Life Sciences) at a concentrationof 1×105
cells/well and incubated at 37°C in 5% CO2. After
confirming the confluence of the stromal layer by phase contrast
microscopy, K562 or NB4 cells were added onto the MSC layers
separately at a ratio of 20:1. For assessment of MSC-derived drug
resistance, cells were treated with As2O3 at
the indicated time points. Next, K562 or NB4 cells were collected
from the top layer leaving the adherent stromal layer intact. Then,
K562 and NB4 cells were assayed for cell viability. MSC
contamination, assessed by FACS as the fraction of CD19-negative
cells, was always <1%. For experiments with AMD3100 and
As2O3 treatment, K562 cells were
pre-incubated for 4 h with AMD3100 and seeded onto confluent marrow
stromal cell layers. After 4 h, As2O3 was
added for a further 24 h. Then, the K562 cell layer was vigorously
washed off and collected as previously described.
Cell transfection
A short hairpin RNA (shRNAs) plasmid against human
RUNX3 and control shRNA were purchased from Joekai Biotechonology
LLC (Shanghai, China) and transduced into cells by electroporation.
Electroporation was performed with a Gene Pulser Xcell Unit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) under conditions of
250 V, 1200 µF, and exponential wave. Clones were selected
with G418 (500 µg/ml). The RUNX3 expression plasmid
PCDNA3.1/RUNX3 was constructed using full-length RUNX3 cDNA, as
described previously (37). K562
cells were transfected using Lipofectamine LTX from Invitrogen.
Empty vectors were transfected as controls.
Cell cycle analysis
K562 cells expressing scrambled or RUNX3-targeting
shRNA were incubated with As2O3. After
incubation, the cells were harvested and resuspended in 500
µl of cold PBS and cold ethanol (1.5 ml) for 2 h at 4°C,
followed by incubation with 0.1% sodium citrate containing
propidium iodide (PI) 0.05 mg and 50 µg RNase for 30 min.
Finally, the cell cycle analysis was detected by flow cytometry
(FC500; Beckman Coulter, Miami, FL, USA), and the proportion of
cells within the G0/G1, S, and G2/M phases of the cell cycle were
analyzed using the MultiCycle AV DNA Analysis software (Phoenix
Flow Systems).
Quantitative reverse
transcriptase-polymerase chain reaction
Total RNA was extracted from each sample using the
Total RNA Extraction kit (Invitrogen), following the manufacturer's
instructions. The concentration of RNA was measured by
spectrophotometry. Total RNA was reverse transcribed to cDNA with
reverse transcriptase reagents (Takara, Nanjing, China), according
to the manufacturer's protocol. Specific primers for RUNX3 and
GAPDH genes were designed based on sequence data from the GenBank
database. The primers were purchased from Invitrogen Biological
Engineering Technology & Services Co., Ltd. For human RUNX3
transcripts, 5′-AGGCATTGCGCAGCTCAGCGGAGTA-3′ was used as a sense
primer and 5′-TCTGCTCCGTGCTGCCCTCGCACTG-3′ as an antisense primer.
For the human GAPDH transcripts, 5′-TCACAGGGGTTCCCTTCTCTC-3′ was
used as a sense primer and 5′-CACTTCTTTGTGCCATCCATGG-3′ as an
antisense primer. Conditions for all PCRs were optimised on an
iCycler iQ (Bio-Rad Laboratories, Inc.), and the optimum annealing
temperature was 60°C.
Immunofluorescence microscopy
Cells were incubated on poly-L-lysine-coated slides
for 30 min, washed gently with PBS and fixed in 10% methanol at
room temperature. After washing with PBS, the cells were
permeabilized with 0.1% Triton in PBS for 60 min at room
temperature. The cells were then incubated with the rabbit
anti-RUNX3 antibodies (BD Biosciences) overnight at 4°C. After
washing with PBS, the slides were incubated with the secondary
antibody, Alexa 555-conjugated goat anti-rabbit immunoglobulin G
(IgG; Molecular Probes), for 2 h and then washed with PBS. Nuclei
were stained with 10 nM DAPI, and cells were then subjected to
fluorescence microscopy.
Morphological detection of apoptosis
Morphological evaluation of apoptotic cell death was
performed using the Hoechst 33258 kit (KeyGen Biotech), according
to the manufacturer's instructions. Cells were cytospun, stained
with Hoechst 33258, and observed with light microscopy.
CXCL12 enzyme-linked immunosorbent
assay
After culturing mesenchymal stem cells in 24-well
plates for 24 h, medium was collected and concentrated by
ultrafiltration. CXCL12 levels in the medium were assayed with a
Human CXCL12/SDF-1 α immunoassay kit (R&D Systems, Abingdon,
UK) according to the manufacturer's instructions. Samples were run
in triplicate.
Flow cytometry analysis of CXCR4
expression
Monoclonal antibodies against human CXCR4-PE (BD
Pharmingen, DakoCytomation) were used for flow cytometry analysis.
PE-conjugated IgG1 and IgG2a control monoclonal antibodies were
from Cell Signaling Technology, Inc.
Transmigration assays
For quantitative transmigration assays, K562 cells
(5×104 cells) were placed in the upper chamber of a
Transwell system (24-well plate, 5 µM-pore filter; Corning
Life Sciences) in a total volume of 150 µl DMEM medium.
Next, 600 ml of the supernatant was concentrated 10-fold, and
placed in the lower chamber of the Transwell. Further control wells
assessed the chemotactic activity of recombinant CXCL12 (rCXCL12)
at concentrations of 100 ng/ml. Cells were harvested from the lower
chamber after 3 h and counted using a hemocytometer.
Annexin V staining
Apoptosis was measured using the Calbiochem assay
kit (Calbiochem, San Diego, CA, USA) according to the
manufacturer's instructions. K562 cells expressing scrambled or
RUNX3-targeting shRNA were incubated with
As2O3. After incubation, cells were washed
with phosphate-buffered saline (PBS) and stained using
phycoerythrin-labeled Annexin V. Numbers of both RFP- and Annexin
V-positive cells were determined by flow cytometry. In some cases,
the Annexin V-propidium iodide staining kit (Calbiochem) was used
instead, according to the manufacturer's protocol.
Western blot analysis
Cells were cultured with the indicated
concentrations of As2O3 for the specified
times, harvested, washed, and lysed using RIPA buffer containing
protease inhibitors. After normalization for total protein content
(60 µg/lane), the samples were subjected to 12% SDS-PAGE and
then transferred to a PVDF membrane. After blocking with 5% non-fat
dry milk and 0.1% Tween-20 in Tris-buffered saline, the membranes
were incubated with the following primary antibodies: Mcl-1 (Cell
Signaling Technology, Inc.), RUNX3 (BD Pharmingen), p21, Bax,
Bcl-2, cleaved caspase-3, cleaved caspase-9, and β-actin (all from
Cell Signaling Technology, Inc.). After extensive rinsing with
TBST, the membranes were incubated with HRP-conjugated anti-mouse
or rabbit secondary antibodies (Cell Signaling Technology, Inc.),
and proteins were detected with the use of an enhanced
chemiluminescence system (EMD Millipore, Billerica, MA, USA) and
captured on a light-sensitive imaging film.
Statistical analysis
All experiments were repeated in triplicate. The
data are reported as the mean ± SEM. Statistical analyses were
performed using Student's t-test or one-way ANOVA. Probability (P)
value of 0.05 was considered to be significant (SPSS v13.0 for
windows).
Results
As2O3 induces
expression of RUNX3 and translocation of RUNX3 into the
nucleus
To determine whether the expression of RUNX3 is
induced by As2O3 treatment, western blot
analysis was performed to detect the level of total RUNX3 and the
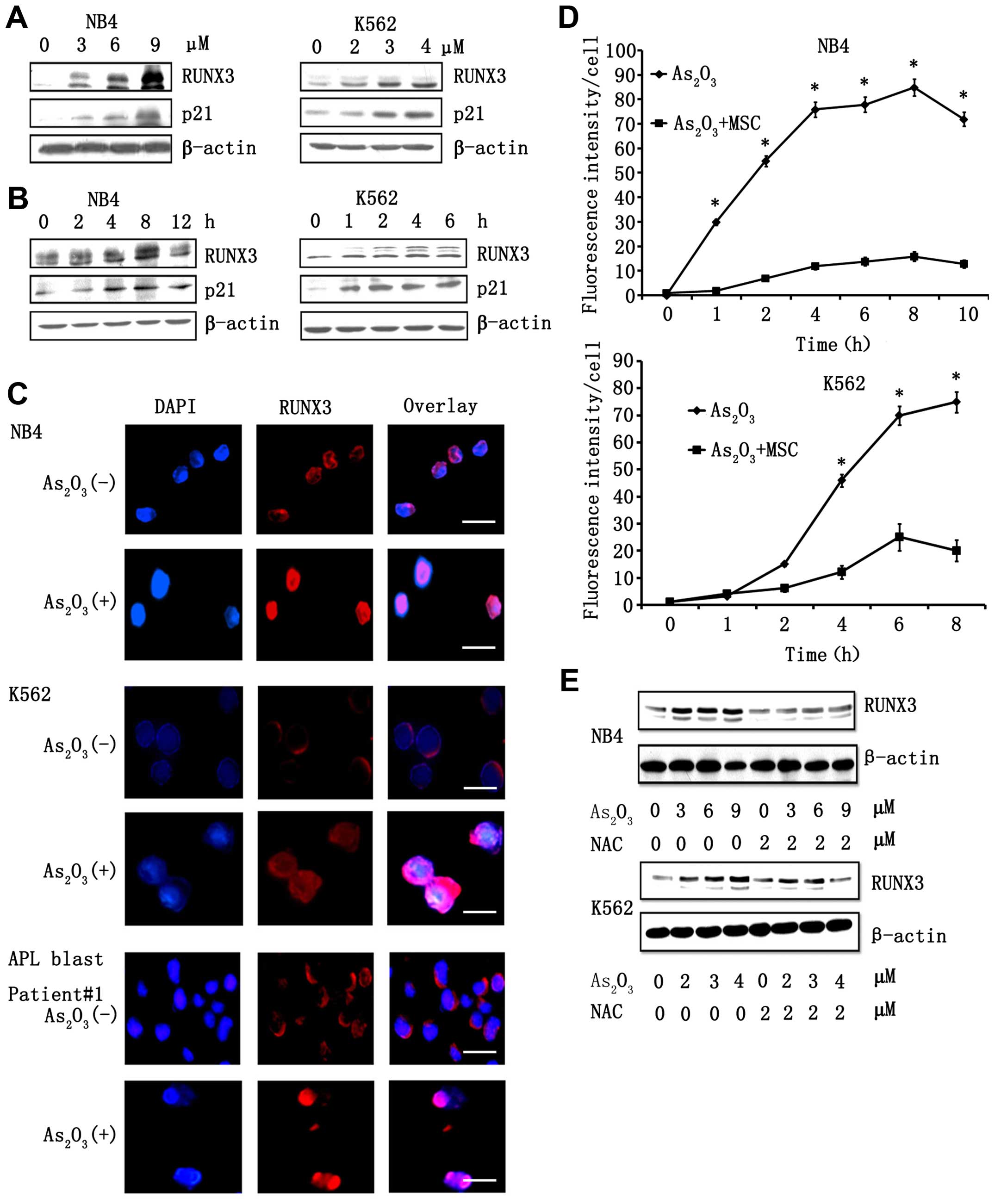
RUNX3 target gene, p21. As shown in (Fig. 1A), As2O3
treatment resulted in the elevation of total RUNX3 in K562 and NB4
cells in a dose-dependent manner. Elevation of RUNX3 levels was
observed at 3 h post-treatment with 5 µM
As2O3 in NB4 cells, and expression levels
reached a peak at 9 h post-treatment. An increase in RUNX3
expression was detected at 1 h following treatment with 3 µM
As2O3 in K562 cells, and expression levels
reached a peak at 4 h post-treatment (Fig. 1B). Similarly, we also observed that
the expression of RUNX3 mRNA was enhanced after
As2O3 treatment in both K562 and NB4 cell
lines. Protein levels of p21 were elevated in a dose- and
time-dependent manner and corresponded to the elevation of RUNX3
observed following As2O3 treatment.
Immunofluorescence staining revealed that RUNX3 was located mainly
in the cytoplasm before As2O3 treatment and
accumulated in the nucleus following As2O3
treatment (Fig. 1C). Consistent
with the results obtained using NB4 cells, our analysis of K562
cells and primary APL blast cells from three patients revealed that
RUNX3 localized in the cytoplasm prior to
As2O3 treatment and translocated into the
nucleus following As2O3 treatment. It has
been previously reported that As2O3 treatment
resulted in elevation in cellular ROS stores and production. In our
studies, treatment of cells with As2O3 also
resulted in the generation of ROS (Fig.
1D). This ROS induction seems to be necessary for
As2O3-dependent elevation of RUNX3 because
pre-treatment of cells with NAC, a scavenger of ROS, resulted in
the inhibition of As2O3-induced expression of
RUNX3 (Fig. 1E).
RUNX3 is required for
As2O3-induced apoptosis
We next determined the role of RUNX3 in
As2O3-induced apoptosis using RNA
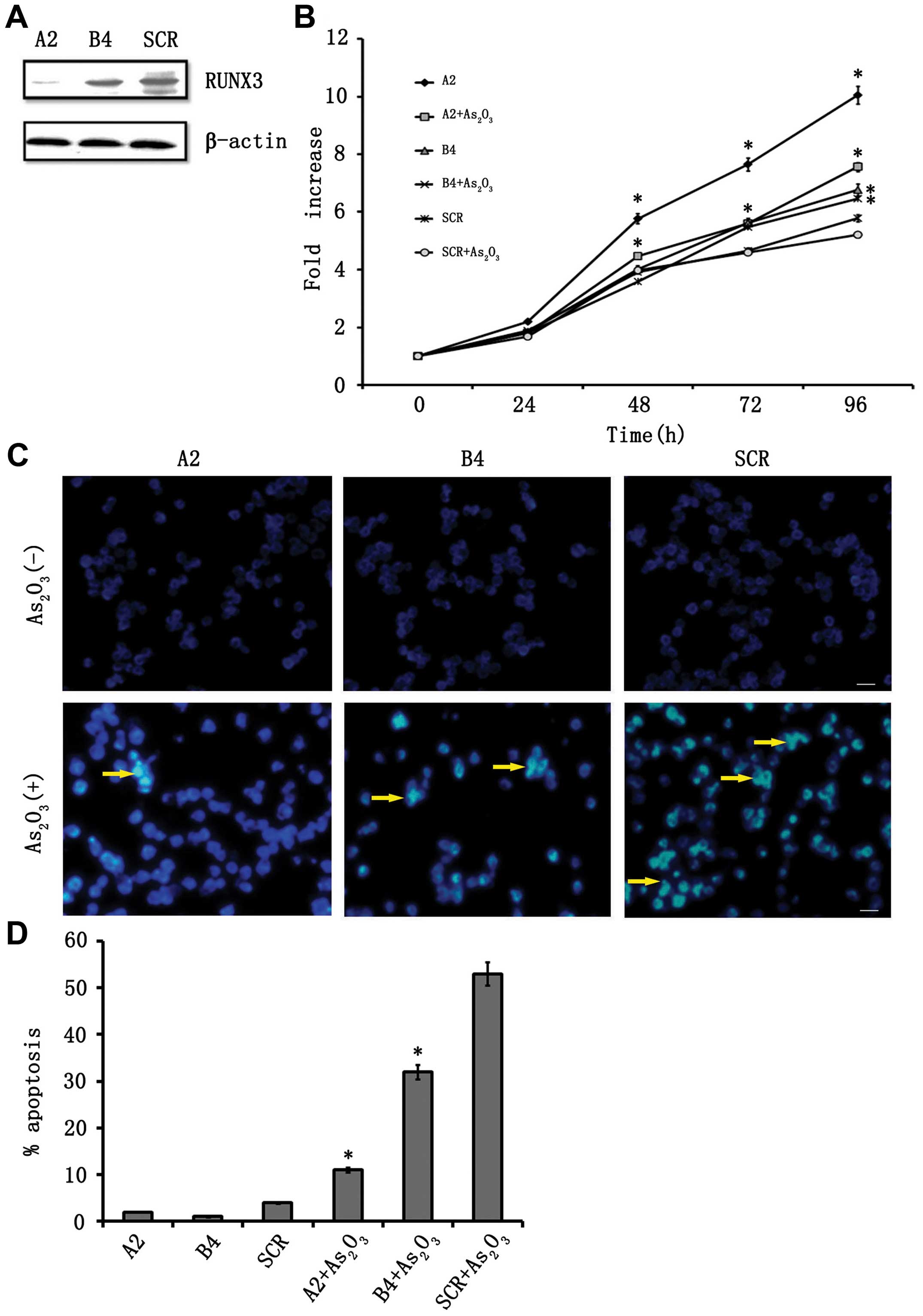
interference to reduce RUNX3 mRNA. We established two stable clones
expressing shRNA specific to RUNX3, designated A2 and B4, and a
scramble shRNA-expressing clone, designated SCR. Western blot
analysis with anti-RUNX3 antibody revealed that endogenous RUNX3
protein levels were markedly diminished in A2 clones and
significantly reduced in the B4 clone compared with SCR (Fig. 2A). Cell growth assays showed that A2
cells exhibited significantly enhanced growth compared with SCR
cells. As2O3 inhibited the growth of SCR
cells, but this growth inhibition was compromised in the RUNX3
shRNA-transduced cells treated with As2O3
(Fig. 2B). Next, we examined the
involvement of RUNX3 in As2O3-induced
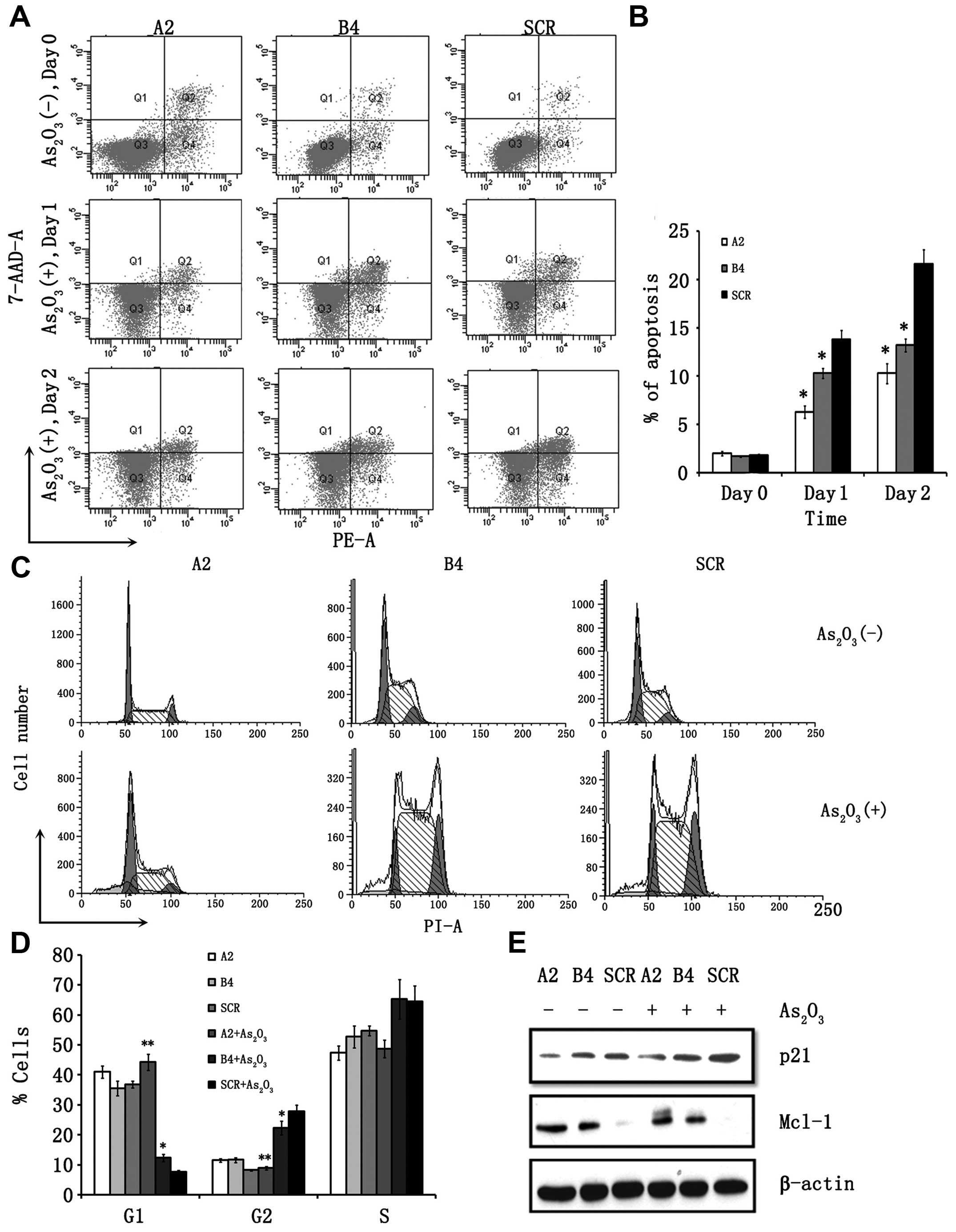
apoptosis and cell cycle arrest. As shown in (Fig. 3A), the ratio of Annexin V and
propidium iodide-positive cells was increased in a time-dependent
manner, and 21.6% of the SCR cells were positive on the second day
after As2O3 treatment. By contrast, apoptosis
was significantly reduced during the observation periods up to day
2 in RUNX3 knockdown clones (Fig. 3A
and B). Similar results were obtained from our Hoechst 33258
staining analysis (Fig. 2C and D).
Fig. 6H shows that re-expression of
RUNX3 enhanced sensitivity of K562 cells to
As2O3 in a dose-dependent manner.
Transduction of control constructs alone did not alter cellular
response to As2O3. The percentages of cells
in G1, S, and G2/M phase are illustrated in Fig. 3C and D. In SCR cells, treatment with
As2O3 increased the fraction of cells in G2/M
progressively from 8.95 to 27.61%. However, suppression of RUNX3
expression by RUNX3 shRNA attenuated
As2O3-induced cell cycle arrest. Consistent
with these results, p21 expression was suppressed in the A2 and B4
clones after As2O3 treatment. A decrease in
the Mcl-1 protein was observed after treatment with
As2O3 in SCR clones, though this reduction in
Mcl-1 levels was compromised in A2 and B4 clones (Fig. 3E).
Mesenchymal stromal stem cells protect
leukemia cells from As2O3-induced
apoptosis
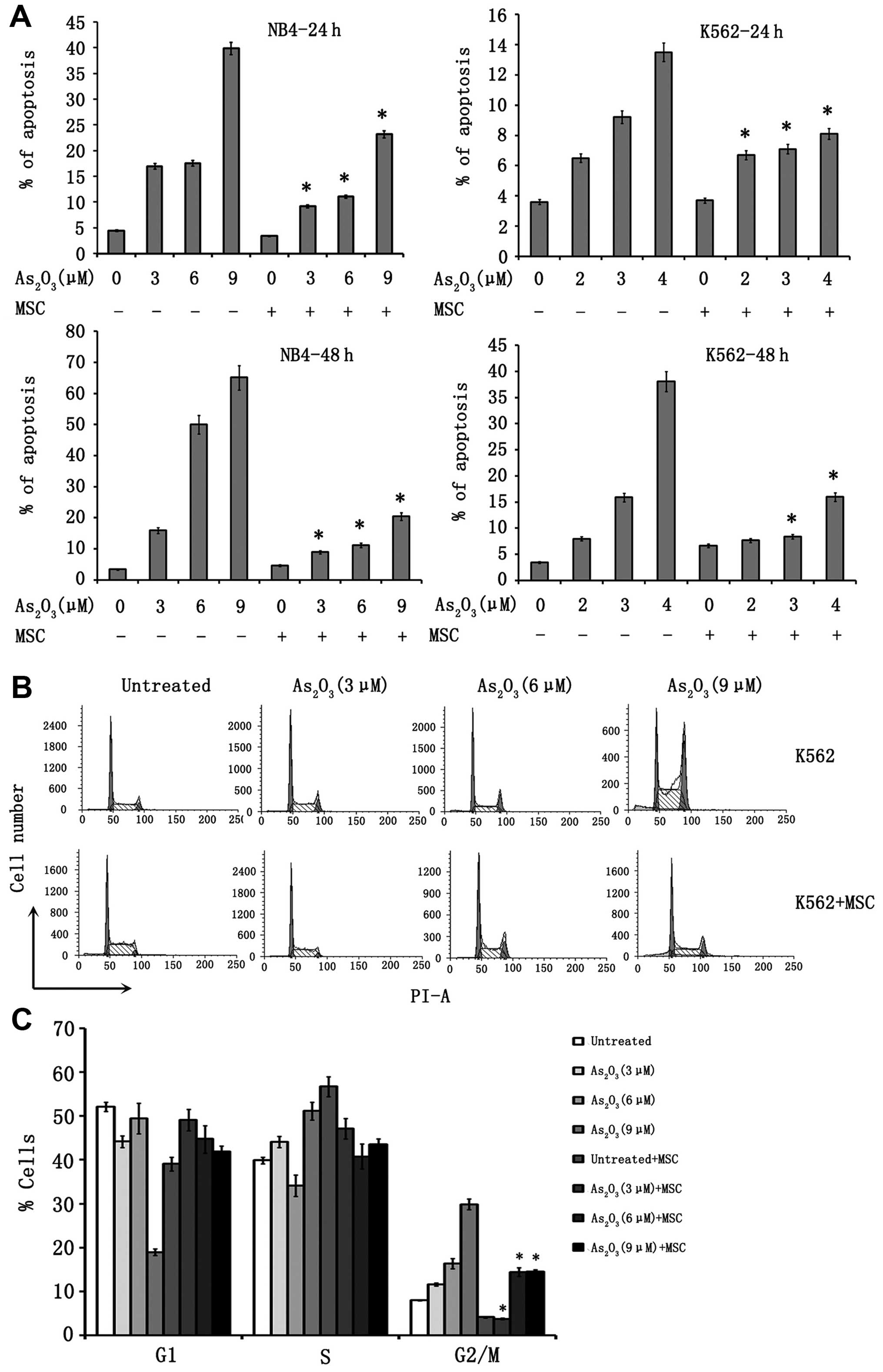
To assess whether MSC could protect leukemia cells
from As2O3-induced apoptosis, K562 and NB4
cells were exposed to a range of As2O3
concentrations in the presence or absence of MSC. After 24 and 48
h, cells were analyzed by probing for Annexin V and propidium
iodide (Fig. 4A). While exposure of
NB4 cells to As2O3 for 24 or 48 h in control
cultures induced apoptosis in a dose-dependent fashion (6.5±2,
9.2±3.5 and 13.5±3.7% of Annexin V/propidium iodide+
cells for 24 h; 8±2.8, 15.9±4.1 and 38.1±5.4% for 48 h for
As2O3 at 3, 6 and 9 µM, respectively),
the proportion of apoptotic NB4 cells in the presence of MSC was
significantly reduced (6.7±1.8, 7.1±3 and 8.1±2.6% for 24 h;
7.7±3.3, 8.4±3.5 and 16±4.2% for 48 h, with
As2O3 at 3, 6 and 9 µM, respectively;
P=0.003, 0.015 and 0.024, NB4 vs. NB4 + MSC with 3, 6 and 9
µM As2O3, respectively) (Fig. 4A). As shown in Fig. 4A, the protective effect of MSC was
also observed for K562 cells. No significant protection was
observed when CML cells were cultured and treated on a monolayer of
endothelial cells.
We also determined whether MSC affect
As2O3-induced cell cycle arrest. As shown in
(Fig. 4B and C),
As2O3 treatment of K562 cells resulted in a
dose-dependent accumulation of cells with 4N and <2N sub-G1 DNA
content, which suggests induction of G2/M-phase cell cycle arrest
and apoptosis. MSC robustly reduced the proportion of G2/M-phase
and sub-G1 cells. MSC did not significantly affect cell cycle
arrest induced by As2O3 in NB4 cells. To
determine if MSC modulate the level of ROS, K562 and NB4 cells
incubated with or without As2O3, either in
the presence or absence of MSC, were analyzed for ROS by DCFDA
staining. As shown in Fig. 1D, the
level of ROS in NB4 cells increased after treatment with 6
µM As2O3 in a time-dependent manner;
however, MSC were able to attenuate the quantity of ROS produced
following As2O3 treatment.
The intrinsic, mitochondrial pathway is the
principal mechanism of As2O3-induced
apoptosis. In the mitochondrial apoptotic pathway, the ratio of
expression of pro-apoptotic Bax to that of anti-apoptotic Bcl-2
ultimately results in either cell death or survival. We determined
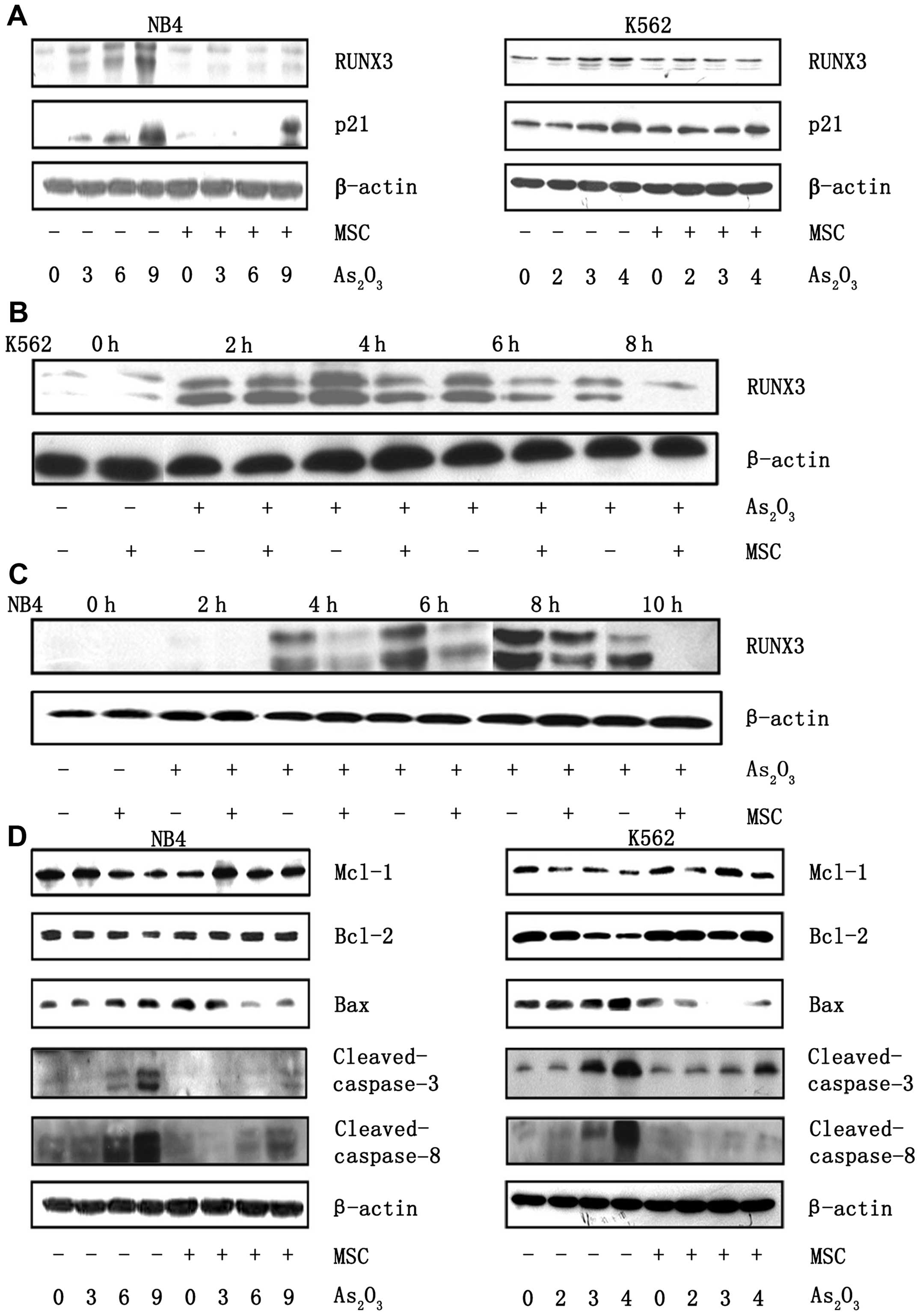
the effect of MSC on the Bax/Bcl-2 ratio. Upregulation of Bax
expression, as well as downregulation of Bcl-2 expression induced
by As2O3, was blocked in the presence of MSC
(Fig. 5D).
MSCs have been found to protect NB4 and K562 cells
from As2O3-induced apoptosis, and this
protection mechanism is related to the expression of apoptotic
proteins. We measured protein levels of cleaved caspase-3 and
cleaved caspase-8. Consistent with the previous report,
As2O3 treatment induced the activation of
caspase-8, whereas this modulation was not noted in the presence of
MSC. As2O3-induced death of cells is
associated with strong activation of caspase-3, which was
significantly inhibited when cells were treated in the presence of
MSC (Fig. 5D). Additionally, there
were low levels of Mcl-1 protein in NB4 and K562 cells treated with
As2O3. However, in the presence of MSC, the
decrease in Mcl-1 levels was compromised (Fig. 5D).
MSC reduce the expression of RUNX3
induced by As2O3 via CXCL12/CXCR4
signaling
We have demonstrated that RUNX3 is required for
As2O3-induced apoptosis. To determine whether
MSC modulate the expression of RUNX3, western blot analysis was
used to detect the level of total RUNX3 in K562 and NB4 cells
treated with As2O3 in the presence or absence
of MSC (Fig. 5A). The presence of
MSC reduced RUNX3 protein expression of cells treated with
different concentrations of As2O3 compared to
the cultures in which the leukemic cells were exposed to
As2O3 without MSC. The expression of p21
induced by As2O3 was also inhibited in the
presence of MSC. We next studied the subcellular distribution of
RUNX3 by performing a western blot analysis on nuclear extracts
from NB4 and K562 cells treated with As2O3
for different time intervals in the presence or in the absence of
MSC. In K562 cells, the expression of RUNX3 was induced in 2 h,
peaking at 4 h. In NB4 cells, RUNX3 expression was induced later
and peaked at 9 h. In the presence of MSC, the level of RUNX3 in
the nuclear fraction was significantly reduced at the indicated
time in NB4 and K562 cells (Fig. 5B and
C).
CXCL12/CXCR4 signaling is an important mediator for
leukemia-stroma interaction, and this interaction has consequences
on tumor survival. Therefore, we determined whether CXCL12/CXCR4
modulates the expression of transcription factor RUNX3 in K562
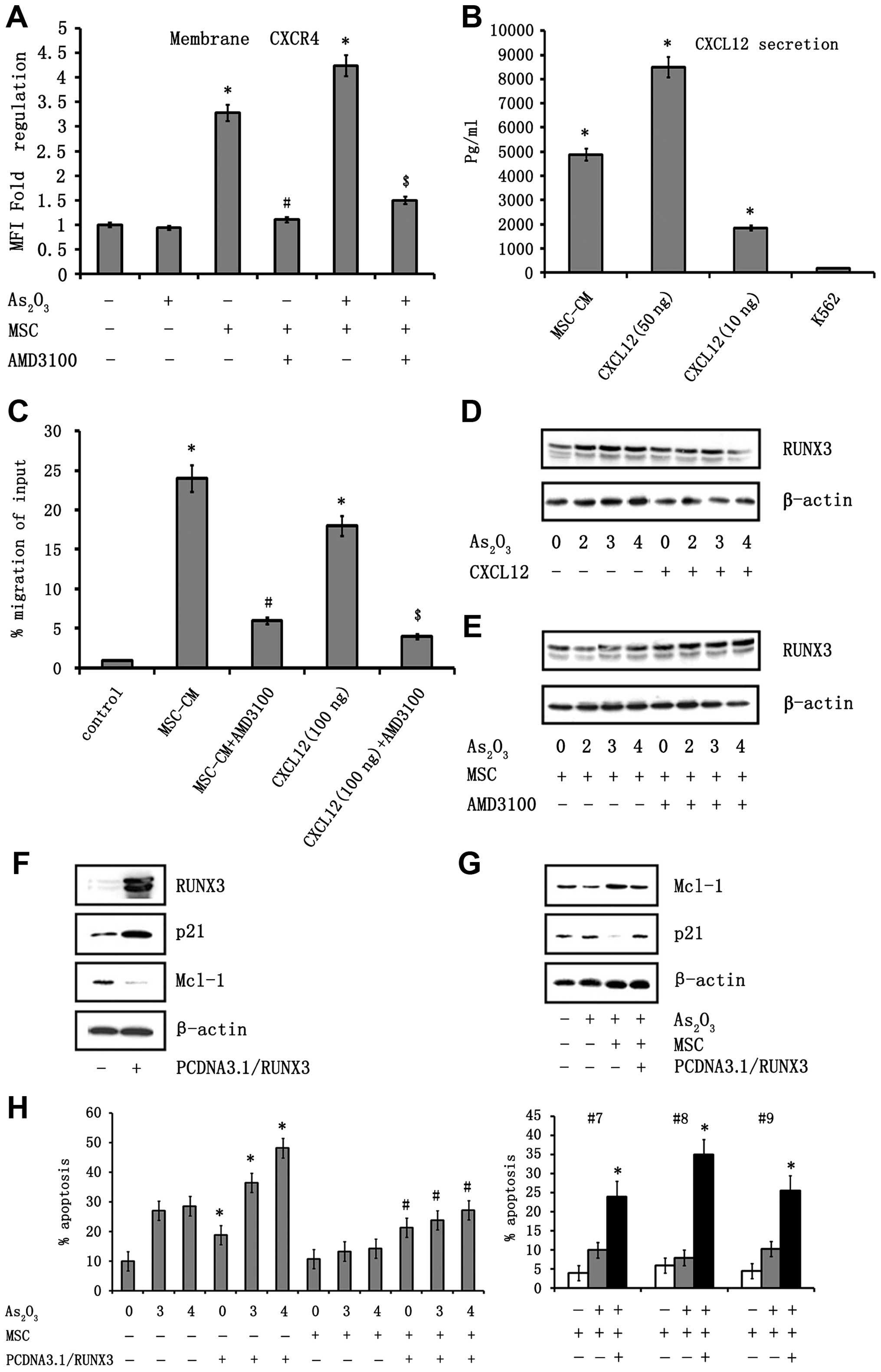
cells. The expression level of CXCR4 was analyzed in K562 cells by
flow cytometry (Fig. 6A). K562
cells showed higher expression of CXCR4 when co-cultured with MSC.
Furthermore, this increase in CXCR4 expression was enhanced in the
presence of As2O3. The expression of CXCR4
peaked 10 min post-treatment with As2O3 when
K562 cells were co-cultured with MSC. Second, the production of
CXCL12 by MSC was analyzed in MSC supernatants by western blot
analysis (Fig. 6B). To determine
whether the CXCL12/CXCR4 interaction had functional activity, we
investigated the chemotactic properties of MSC on K562 cells in a
transmigration assay. The K562 cells migrated toward a CXCL12
gradient, demonstrating that CXCR4 was fully functional. As shown
in Fig. 6C, MSC supernatant
exhibited potent chemoattractive activity on K562 cells, as 24±3.2%
of K562 cells migrated toward undiluted supernatant compared to
0.95±0.06% of cells that migrated in the presence of media
(P=0.013). Blocking CXCR4 led to a significant reduction in
chemoattractive activity in response to MSC, as well as the
chemokine CXCL12 (P=0.014; Fig.
6C). All of these data indicate the presence of a functional
CXCR4/CXCL12 signaling between CML cells and MSC and that this
signaling is important for promoting migration.
To assess whether CXCR4/CXCL12 could modulate the
expression of RUNX3, the expression of RUNX3 was analyzed in the
presence of CXCL12 by western blot analysis. As shown in Fig. 6D, CXCL12 induced a significant
reduction of RUNX3 mRNA expression in K562 cells. Remarkably,
treatment of K562 cells with the CXCR4 antagonist AMD3100 induced
RUNX3 expression (Fig. 6E).
Moreover, the expression of RUNX3 was upregulated after treatment
with AMD3100 in the presence of MSC, indicating that the inhibition
of CXCR4/CXCL12 signaling was rescuing the expression of RUNX3.
Expression of RUNX3 overcomes
MSC-mediated As2O3 resistance
Mesenchymal stromal cells have been found to
protect leukemia cells from As2O3-induced
apoptosis. We tested the hypothesis that the overexpression of
RUNX3 could restore the effect of As2O3 on
K562 cells. K562 cells were transfected with a RUNX3 expression
vector or an empty control vector. The expression of RUNX3 in these
cells was confirmed by western blot analysis (Fig. 6F). When K562 cells transfected with
RUNX3 were co-cultured with MSC, the Annexin V and PI-positive
cells treated with increasing concentrations of
As2O3 was significantly increased compared to
the cultures in which leukemic cells transfected with empty vector
were exposed to As2O3 in the presence of MSC.
Similar results were observed from APL primary blast patients
(Fig. 6H). In addition, we found
that p21 protein expression was significantly increased after
exposure to As2O3 in cells transfected with
the RUNX3 expression vector in the presence of MSC, but not for
cells transfected with control vector (Fig. 6G). The influence of overexpression
of RUNX3 on the expression of anti-apoptotic proteins in the
presence of the MSC was determined by immunoblotting. Mcl-1 protein
upregulation was observed in K562 cells grown with stromal cells.
However, cells transfected with RUNX3 downregulated stroma-induced
Mcl-1 levels in K562 cells (Fig.
6G).
Discussion
In this study, we examined the involvement of RUNX3
in As2O3-induced cellular apoptosis using the
APL-derived, NB4 cell line and the CML-derived, K562 cell line.
As2O3-induced expression of RUNX3 and nuclear
localization of RUNX3 were both observed in NB4 and K562 cells. The
transcriptional activation of RUNX3 following treatment with
As2O3 was confirmed by the upregulation of
p21, a target gene for RUNX3. It is now known that the generation
of ROS is one of the major mechanisms through which
As2O3 affects neoplastic cells. Treatment
with NAC, a scavenger of ROS, resulted in the inhibition of
As2O3-induced expression of RUNX3.
Furthermore, knockdown of endogenous RUNX3 by shRNA blocked
As2O3-induced apoptosis and cell cycle arrest
in the K562 cell lines. Our data strongly suggest that RUNX3 may be
a key molecule in a series of cellular responses induced by
As2O3 treatment during leukemia therapy.
In this study, we provide evidence that a
cell-contact mediated interaction between MSC and leukemia cells
effectively protects leukemia cells from
As2O3-induced cell death. In agreement with
earlier results, we found that the leukemia-stroma interaction and
the consequences that this interaction has on tumor survival
following exposure to As2O3, involve
CXCR4/CXCL12 signaling. However, the precise mechanism by which
CXCR4/CXCL12 signaling guard leukemia cells from
As2O3-induced apoptosis was unclear. In this
study, we identified RUNX3 as a key transcription factor involved
in the protection of leukemia cells through an interaction with MSC
in As2O3-treated cell lines. We show at the
protein level that the presence of CXCL12 or MSC the upregulation
of RUNX3 that occurs during treatment of cells with
As2O3. A CXCR4 antagonist, AMD3100, which
blocks CXCL12/CXCR4 signaling, could efficiently rescue the
expression of RUNX3. Regulation of RUNX3 expression appears to be
the effect of CXCL12/CXCR4 signaling in K562 cells. This finding
suggests that downregulation of RUNX3 expression is involved in the
development of drug-resistance signals derived from CML-MSC
interactions. Taken together with our observation that active RUNX3
restores sensitivity of chronic myeloid leukemia cells to
As2O3, MSC-induced drug-resistance in
leukemia cells may be overcome by forced activation of RUNX3 and
subsequent inhibition of Mcl-1.
Mcl-1 is an early response gene that functions as a
regulator of cell viability (38).
Mcl-1 can undergo rapid upregulation, as well as downregulation
(39), which allows Mcl-1 to
provide an acute protective function from apoptosis induced by
various factors, including DNA damage (40), growth factor withdrawal (41,42)
and treatment with cytotoxic agents (43). Western blot analysis suggested that
Mcl-1 expression decreased in K562/RUNX3 and increased in
K562/siRUNX3 compared with respective control cells. However, this
result did not necessarily mean that RUNX3 was a transcriptional
repressor of Mcl-1. Further study is needed to clarify the
regulatory effects of RUNX3 on Mcl-1 expression. Mcl-1 protein
upregulation was observed in K562 cells grown in the presence of
stromal cells. The overexpression of RUNX3 downregulated endogenous
as well as stroma-induced Mcl-1 levels in K562 cells. Those results
suggest the induction of RUNX3 is associated with the expression of
RUNX3 and bone marrow stromal cells may support K562 cells via
induction of Mcl-1.
In summary, we report that RUNX3 plays a critical
role in As2O3-induced apoptosis and
CXCR4/CXCL12 signaling involved in the leukemia-stroma interaction.
CXCR4/CXCL12 signaling downregulates the expression of the
transcription factor RUNX3. Reduced levels of RUNX3 in turn
compromise the apoptosis induced by As2O3.
The overexpression of RUNX3 can effectively antagonize survival and
drug-resistance signals derived from CML-MSC interactions.
Abbreviations:
|
RUNX3
|
runt-related transcription factor
3
|
|
MSC
|
mesenchymal stem cell
|
|
As2O3
|
arsenic trioxide
|
|
SDF-1
|
stromal cell-derived factor-1 (also
known as CXCL12, C-X-C motif chemokine 12)
|
|
CXCR4
|
C-X-C chemokine receptor type 4
|
|
APL
|
acute promyelocytic leukemia
|
|
ROS
|
reactive oxygen species
|
|
shRNAs
|
short hairpin RNA
|
References
|
1
|
Ding W, Knox TR, Tschumper RC, Wu W,
Schwager SM, Boysen JC, Jelinek DF and Kay NE: Platelet-derived
growth factor (PDGF)-PDGF receptor interaction activates bone
marrow-derived mesenchymal stromal cells derived from chronic
lymphocytic leukemia: implications for an angiogenic switch. Blood.
116:2984–2993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li W, Zhou Y, Yang J, Zhang X, Zhang H,
Zhang T, Zhao S, Zheng P, Huo J and Wu H: Gastric cancer-derived
mesenchymal stem cells prompt gastric cancer progression through
secretion of interleukin-8. J Exp Clin Cancer Res. 34:522015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arai F, Hirao A, Ohmura M, Sato H,
Matsuoka S, Takubo K, Ito K, Koh GY and Suda T: Tie2/angiopoietin-1
signaling regulates hematopoietic stem cell quiescence in the bone
marrow niche. Cell. 118:149–161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang F, Wang M, Yang T, Cai J, Zhang Q,
Sun Z, Wu X, Zhang X, Zhu W, Qian H, et al: Gastric cancer-derived
MSC-secreted PDGF-DD promotes gastric cancer progression. J Cancer
Res Clin Oncol. 140:1835–1848. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vianello F, Villanova F, Tisato V, Lymperi
S, Ho KK, Gomes AR, Marin D, Bonnet D, Apperley J, Lam EW, et al:
Bone marrow mesenchymal stromal cells non-selectively protect
chronic myeloid leukemia cells from imatinib-induced apoptosis via
the CXCR4/CXCL12 axis. Haematologica. 95:1081–1089. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sison EA and Brown P: The bone marrow
microenvironment and leukemia: Biology and therapeutic targeting.
Expert Rev Hematol. 4:271–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Hayre M, Salanga CL, Kipps TJ, Messmer
D, Dorrestein PC and Handel TM: Elucidating the CXCL12/CXCR4
signaling network in chronic lymphocytic leukemia through
phosphopro-teomics analysis. PLoS One. 5:e117162010. View Article : Google Scholar
|
|
8
|
Li X, Guo H, Duan H, Yang Y, Meng J, Liu
J, Wang C and Xu H: Improving chemotherapeutic efficiency in acute
myeloid leukemia treatments by chemically synthesized peptide
interfering with CXCR4/CXCL12 axis. Sci Rep. 5:162282015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han TT, Fan L, Li JY and Xu W: Role of
chemokines and their receptors in chronic lymphocytic leukemia:
Function in microenvironment and targeted therapy. Cancer Biol
Ther. 15:3–9. 2014. View Article : Google Scholar :
|
|
10
|
Möhle R, Failenschmid C, Bautz F and Kanz
L: Overexpression of the chemokine receptor CXCR4 in B cell chronic
lymphocytic leukemia is associated with increased functional
response to stromal cell-derived factor-1 (SDF-1). Leukemia.
13:1954–1959. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
de Lourdes Perim A, Amarante MK,
Guembarovski RL, de Oliveira CE and Watanabe MA: CXCL12/CXCR4 axis
in the pathogenesis of acute lymphoblastic leukemia (ALL): A
possible therapeutic target. Cell Mol Life Sci. 72:1715–1723. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alsayed Y, Ngo H, Runnels J, Leleu X,
Singha UK, Pitsillides CM, Spencer JA, Kimlinger T, Ghobrial JM,
Jia X, et al: Mechanisms of regulation of CXCR4/SDF-1
(CXCL12)-dependent migration and homing in multiple myeloma. Blood.
109:2708–2717. 2007.
|
|
13
|
Nervi B, Ramirez P, Rettig MP, Uy GL, Holt
MS, Ritchey JK, Prior JL, Piwnica-Worms D, Bridger G, Ley TJ, et
al: Chemo-sensitization of acute myeloid leukemia (AML) following
mobilization by the CXCR4 antagonist AMD3100. Blood. 113:6206–6214.
2009. View Article : Google Scholar :
|
|
14
|
Sison EA, Magoon D, Li L, Annesley CE, Rau
RE, Small D and Brown P: Plerixafor as a chemosensitizing agent in
pediatric acute lymphoblastic leukemia: efficacy and potential
mechanisms of resistance to CXCR4 inhibition. Oncotarget.
5:8947–8958. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen ZH, Zeng DF, Ma YY, Zhang X, Zhang C
and Kong PY: Are there any new insights for G-CSF and/or AMD3100 in
chemotherapy of haematological malignants? Med Oncol. 32:2622015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kashyap MK, Kumar D, Jones H,
Amaya-Chanaga CI, Choi MY, Melo-Cardenas J, Ale-Ali A, Kuhne MR,
Sabbatini P, Cohen LJ, et al: Ulocuplumab (BMS-936564/MDX1338): A
fully human anti-CXCR4 antibody induces cell death in chronic
lymphocytic leukemia mediated through a reactive oxygen species-
dependent pathway. Oncotarget. 7:2809–2822. 2016.
|
|
17
|
Niedermeier M, Hennessy BT, Knight ZA,
Henneberg M, Hu J, Kurtova AV, Wierda WG, Keating MJ, Shokat KM and
Burger JA: Isoform-selective phosphoinositide 3′-kinase inhibitors
inhibit CXCR4 signaling and overcome stromal cell-mediated drug
resistance in chronic lymphocytic leukemia: A novel therapeutic
approach. Blood. 113:5549–5557. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu CC, Wang H, Wang WD, Zhu MY, Geng QR
and Lu Y: Consolidation therapy of arsenic trioxide alternated with
chemotherapy achieves remarkable efficacy in newly diagnosed acute
promyelocytic leukemia. Onco Targets Ther. 8:3297–3303.
2015.PubMed/NCBI
|
|
19
|
Soignet SL, Frankel SR, Douer D, Tallman
MS, Kantarjian H, Calleja E, Stone RM, Kalaycio M, Scheinberg DA,
Steinherz P, et al: United States multicenter study of arsenic
trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol.
19:3852–3860. 2001.PubMed/NCBI
|
|
20
|
Shen Y, Shen ZX, Yan H, Chen J, Zeng XY,
Li JM, Li XS, Wu W, Xiong SM, Zhao WL, et al: Studies on the
clinical efficacy and pharmacokinetics of low-dose arsenic trioxide
in the treatment of relapsed acute promyelocytic leukemia: A
comparison with conventional dosage. Leukemia. 15:735–741. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chendamarai E, Ganesan S, Alex AA, Kamath
V, Nair SC, Nellickal AJ, Janet NB, Srivastava V, Lakshmi KM,
Viswabandya A, et al: Comparison of newly diagnosed and relapsed
patients with acute promyelocytic leukemia treated with arsenic
trioxide: Insight into mechanisms of resistance. PLoS One.
10:e01219122015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lengfelder E, Lo-Coco F, Ades L,
Montesinos P, Grimwade D, Kishore B, Ramadan SM, Pagoni M, Breccia
M, Huerta AJ, et al European LeukemiaNet: Arsenic trioxide-based
therapy of relapsed acute promyelocytic leukemia: Registry results
from the European Leukemia Net. Leukemia. 29:1084–1091. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Salmon JM, Bots M, Vidacs E, Stanley KL,
Atadja P, Zuber J and Johnstone RW: Combining the differentiating
effect of panobinostat with the apoptotic effect of arsenic
trioxide leads to significant survival benefit in a model of
t(8;21) acute myeloid leukemia. Clin Epigenetics. 7:22015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oancea C, Rüster B, Brill B, Roos J,
Heinssmann M, Bug G, Mian AA, Guillen NA, Kornblau SM, Henschler R,
et al: STAT activation status differentiates leukemogenic from
non-leukemogenic stem cells in AML and is suppressed by arsenic in
t(6;9)-positive AML. Genes Cancer. 5:378–392. 2014.
|
|
25
|
Evens AM, Tallman MS and Gartenhaus RB:
The potential of arsenic trioxide in the treatment of malignant
disease: Past, present, and future. Leuk Res. 28:891–900. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J,
Cai X, Han ZG, Ni JH, Shi GY, Jia PM, et al: Use of arsenic
trioxide (As2O3) in the treatment of acute
promyelocytic leukemia (APL): I. As2O3 exerts
dose-dependent dual effects on APL cells. Blood. 89:3345–3353.
1997.PubMed/NCBI
|
|
27
|
Lallemand-Breitenbach V, Zhu J, Chen Z and
de Thé H: Curing APL through PML/RARA degradation by
As2O3. Trends Mol Med. 18:36–42. 2012.
View Article : Google Scholar
|
|
28
|
Cai X, Shen YL, Zhu Q, Jia PM, Yu Y, Zhou
L, Huang Y, Zhang JW, Xiong SM, Chen SJ, et al: Arsenic
trioxide-induced apoptosis and differentiation are associated
respectively with mitochondrial transmembrane potential collapse
and retinoic acid signaling pathways in acute promyelocytic
leukemia. Leukemia. 14:262–270. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guan L, Han B, Li Z, Hua F, Huang F, Wei
W, Yang Y and Xu C: Sodium selenite induces apoptosis by
ROS-mediated endoplasmic reticulum stress and mitochondrial
dysfunction in human acute promyelocytic leukemia NB4 cells.
Apoptosis. 14:218–225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Menheniott TR, Judd LM and Giraud AS:
RUNX3 methylation and anti-tumor immunity. Oncoscience. 2:789–790.
2015.PubMed/NCBI
|
|
31
|
Ito K, Liu Q, Salto-Tellez M, Yano T, Tada
K, Ida H, Huang C, Shah N, Inoue M, Rajnakova A, et al: RUNX3, a
novel tumor suppressor, is frequently inactivated in gastric cancer
by protein mislocalization. Cancer Res. 65:7743–7750.
2005.PubMed/NCBI
|
|
32
|
Yano T, Ito K, Fukamachi H, Chi XZ, Wee
HJ, Inoue K, Ida H, Bouillet P, Strasser A, Bae SC, et al: The
RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells
undergoing transforming growth factor beta-induced apoptosis. Mol
Cell Biol. 26:4474–4488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chi XZ, Yang JO, Lee KY, Ito K, Sakakura
C, Li QL, Kim HR, Cha EJ, Lee YH, Kaneda A, et al: RUNX3 suppresses
gastric epithelial cell growth by inducing p21(WAF1/Cip1)
expression in cooperation with transforming growth factor
{beta}-activated SMAD. Mol Cell Biol. 25:8097–8107. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ito K, Lim AC, Salto-Tellez M, Motoda L,
Osato M, Chuang LS, Lee CW, Voon DC, Koo JK, Wang H, et al: RUNX3
attenuates β-catenin/T cell factors in intestinal tumorigenesis.
Cancer Cell. 14:226–237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pande S, Ali SA, Dowdy C, Zaidi SK, Ito K,
Ito Y, Montecino MA, Lian JB, Stein JL, van Wijnen AJ, et al:
Subnuclear targeting of the Runx3 tumor suppressor and its
epigenetic association with mitotic chromosomes. J Cell Physiol.
218:473–479. 2009. View Article : Google Scholar
|
|
36
|
Okorokov AL, Rubbi CP, Metcalfe S and
Milner J: The interaction of p53 with the nuclear matrix is
mediated by F-actin and modulated by DNA damage. Oncogene.
21:356–367. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bae SC, Takahashi E, Zhang YW, Ogawa E,
Shigesada K, Namba Y, Satake M and Ito Y: Cloning, mapping and
expression of PEBP2 alpha C, a third gene encoding the mammalian
Runt domain. Gene. 159:245–248. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Beekman AM and Howell LA: Small-molecule
and peptide inhibitors of the pro-survival protein Mcl-1. Chem Med
Chem. 11:802–813. 2016. View Article : Google Scholar
|
|
39
|
Belmar J and Fesik SW: Small molecule
Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther.
145:76–84. 2015. View Article : Google Scholar :
|
|
40
|
Cuconati A, Mukherjee C, Perez D and White
E: DNA damage response and MCL-1 destruction initiate apoptosis in
adenovirus-infected cells. Genes Dev. 17:2922–2932. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bose P and Grant S: Mcl-1 as a Therapeutic
Target in Acute Myelogenous Leukemia (AML). Leuk Res Rep. 2:12–14.
2013.PubMed/NCBI
|
|
42
|
Lee WS, Park YL, Kim N, Oh HH, Son DJ, Kim
MY, Oak CY, Chung CY, Park HC, Kim JS, et al: Myeloid cell
leukemia-1 is associated with tumor progression by inhibiting
apoptosis and enhancing angiogenesis in colorectal cancer. Am J
Cancer Res. 5:101–113. 2014.
|
|
43
|
Lee JS, Tang SS, ortiz V, Vo TT and Fruman
DA: MCL-1-independent mechanisms of synergy between dual PI3K/mTOR
and BCL-2 inhibition in diffuse large B cell lymphoma. Oncotarget.
6:35202–35217. 2015.PubMed/NCBI
|