Visceral obesity or, in its broader terminology,
metabolic syndrome is an emerging lifestyle-related health problem
that has globally increased in prevalence in recent decades
(1–5). Metabolic syndrome is clinically
defined as a combination of visceral obesity, increased blood
pressure, insulin resistance and dyslipidemia (6,7). This
condition has been extensively investigated due to its correlation
with various health problems in humans, including cardiovascular
diseases, type 2 diabetes (T2D) and non-alcoholic fatty liver
disease. In addition to these diseases, which are directly related
to visceral adiposity, recent studies have also explored other
consequences of metabolic syndrome, such as thromboembolic
conditions, hypogonadism and various cancers (8–11).
Recent studies have suggested that the pathophysiology of metabolic
syndrome is partly associated with dysfunction of the intestinal
flora (12–14), a condition called dysbiosis
(15). Both brain functions and gut
microbiota influence fat intake and increased body mass, which in
turn is a source of cytokines that induce chronic, low-grade
inflammation and carcinogenesis (16–18).
Additionally, obesity itself influences the ecology of the gut
microbiota population (19).
Colorectal cancer (CRC) is one the cancers that has
been associated with metabolic syndrome. CRC is an epithelial
cell-derived adenocarcinoma that develops through a multistep
carcinogenic process known as the adenoma-carcinoma sequence.
Epidemiological studies have demonstrated that colorectal adenomas
and CRC risk are higher in individuals with metabolic syndrome or
any of its components (20–22). A recent meta-analysis of cohort
studies reported that metabolic syndrome was associated with higher
relative risks of CRC in men at 1.25 (95% confidence interval;
1.19–1.32) and in women at 1.34 (1.09–1.64) (22). In recent decades, partly because of
the improved resolution of high-throughput sequencing technology,
the gut microbiome has been extensively investigated. Studies have
suggested a link between diet-induced microbiota dysbiosis and
colorectal carcinogenesis (22).
This review aims to update the current understanding
of metabolic syndrome and its role as a pathogenic factor in
colorectal pathogenesis.
Obesity is a major risk factor for several types of
cardiovascular and metabolic diseases, such as hyperglycemia, T2D,
hypertension and atherosclerosis (23). Individuals with more than three of
these symptoms with or without obesity are diagnosed with
‘metabolic syndrome’. Although obesity and metabolic syndrome are
generally considered a problem of the gut, neurobiological studies
have shown that the brain also plays an important role since
neurobiological diseases, such as stroke, dementia, intracranial
hypertension and sleep disorders are associated with obesity
(24). Moreover, obesity can be
initiated by pathological conditions of the brain and several
neuropsychiatric drugs (25,26).
Therefore, the brain appears to be a central regulator that directs
energy fluxes within an organism.
Obesity development may be caused by this allocative
defect, in which the brain requires energy from food consumption
instead of blood sugar (28). The
mechanism of this failure may result from an abnormality in various
regions of the brain, such as the hypothalamus, amygdala or
hippocampus. Clinical studies dating back more than a century have
demonstrated that hyperphagia and obesity can be induced by lesions
of the ventromedial hypothalamus (VMH), a part of the brain that
plays a role in preserving body mass (28,33).
In animal studies, lesions of the VMH have caused weight gain in
dogs, while VMH stimulation led to satiety (28,34).
It is possible that lesions of the VMH lead to increased
parasympathetic activity, resulting in an inadequate allocation of
glucose to the brain. Then, the animal becomes hyperinsulinemic,
finally resulting in hypoglycemia, neuroglycopenia and excessive
body mass (35–37). In addition, several defective
signaling pathways in the hypothalamus have been reported to be
involved in obesity development. For example, studies have shown
that loss of the leptin receptors in the hypothalamus resulted in
obesity, insulin resistance and diabetes in mice (38,39).
Leptin signaling abnormalities have also been found in obese
patients and were caused by a monogenetic defect or a leptin
receptor mutation in the hypothalamus (40,41).
A regulator of insulin and leptin signaling, protein
tyrosine phosphatase 1B, has also been associated with leptin and
insulin resistance in hypothalamic neurons (42,43) as
neuronal protein tyrosine phosphatase 1B-knockout mice showed
improved glucose tolerance following chronic high-fat feeding
(44). Moreover, VMH lesions may be
associated with an unusual vagus nerve-hypothalamus interaction
pathway leading to miscommunication between the adipose tissue and
β-cells (45). Other factors that
result in obesity include a lack of α-melanocyte-stimulating
hormone (46), insulin (47) or interleukin-6 (IL-6) (48) or bilateral lesions of the amygdala
(36,49). However, not all obese individuals
develop obesity due to lesions of the brain. The core concept of
the selfish brain theory, the brain-pull hypothesis, posits that
‘brain-pull’ is the force with which the brain actively demands
energy from the body (50),
indicating that the brain has an active approach associated with
specific brain-pull mechanisms (51). Stress has been suggested as the
major mechanism that causes the brain to enter a hypervigilant
state. This leads to an increased requirement for cerebral energy
and initiation of brain-pull mechanisms (52,53),
such as ‘cerebral insulin suppression’ that over time results in
the loss of subcutaneous adipose tissue, the accumulation of
visceral fat and an increased cerebral glucose supply (51).
Metabolic syndrome and obesity are related to a
chronic, low-grade systemic inflammatory condition characterized by
increasing levels of circulating inflammatory cytokines and free
fatty acids (FFAs) (54). In
contrast to classical inflammation, this ‘metabolic inflammation’
(55,56) does not necessarily involve pathogens
but occurs as a result of metabolic homeostatic abnormalities.
Nowadays, obesity has been associated with inflammatory adipose
tissue, which may be caused by higher immune cell accumulation in
the fatty tissue region (57). The
immune cells associated with this metabolic inflammation include
macrophage, neutrophil and lymphocyte.
ATMs appear to play a major role in the regulation
of obesity-induced inflammation as ATMs generally play a key role
in most cases of chronic inflammation, and these cells can comprise
up to 40% of the cells in the adipose tissue (58). It is possible that obesity-induced
inflammation is regulated by macrophages (59), and ATM accumulation can be found
after as little as 1 week on a high-fat diet (60). Moreover, numerous studies have
indicated that accumulation of monocytes and macrophages is
positively linked to subcutaneous fat accumulation and weight gain
(61). Elevated monocyte levels can
be found in the circulation of obese individuals, and the levels
decrease when the fat is lost (62,63).
These circulating monocytes can be attracted to recruit into the
adipose tissue by macrophage chemoattractant protein-1 (MCP-1)
(also as known as CCL2), which is secreted by enlarging adipocytes
(64,65). Interestingly, mice that lack CCR2,
the receptor for MCP-1, showed a decreased food intake and obesity
development and increased eosinophil accumulation, which highly
increase the expression of IL-4 and IL-13 in the adipose tissue
during high-fat diet feeding, leading to the predominance of M2
macrophages (66,67). In addition to CCL2, other studies
have found that CXCL12, CXCL14 and CCL5 also play roles in
macrophage migration (68–70).
In the tissues, macrophages can be subgrouped into 2
types: the classically activated macrophages, termed M1 and the
alternatively activated macrophages, termed M2 (71). The M1 macrophage is a
CD11c+ pro-inflammatory cell that produces inducible
nitric oxide synthase and inflammatory cytokines, such as IL-6 and
tumor necrosis factor-α (TNF-α), while the M2 macrophages, which
can be identified by the markers CD206, CD209 and CD301 (Mgl1/2),
express arginase 1 (Arg1) and immunosuppressive cytokines,
including IL-10, transforming growth factor-β (TGF-β), IL-1
receptor antagonist (IL-1RA)-α and IL-4 (59). Interestingly, in obese adipose
tissue, the production of anti-inflammatory IL-10 is decreased, and
increased levels of inflammatory TNF-α are observed (72). This may be the obvious evidence
showing the dominance of the pro-inflammatory macrophage in adipose
tissue of obese person. However, recent study demonstrated that
inflammatory ATMs found in obese adipose tissue have a uniquely
distinct phenotype from M1 macrophages called ‘metabolically
activated (MMe) macrophages’ (73),
and this phenotype can be induced in vitro by treating
macrophages with palmitate, insulin and glucose (metabolic
activation) (73). While M1
macrophages display cellular markers CD319, CD274 and CD38, ATMs
from obese individuals express lipid transport proteins ATP-binding
cassette member 1 (ABCA1) and fatty acid translocase (CD36)
(73). Although, both types of
macrophages have different phynotypes, they still shared the
pro-inflammatory properties by producing TNF-α, IL-6 and IL-1β
(73).
Neutrophils are granulocytes that carry a large
number of granules (intracellular vesicles), which contain
anti-microbial agents, including lysozyme, neutrophil elastase
(NE), myeloperoxidase (MPO) and defensins (59). Although neutrophils are known to
play a role in inflammation, it is still unclear whether this type
of immune cells is involved in obesity-induced inflammation.
Several studies have found that neutrophil migration into the
adipose tissue and classical inflammation occurred within 3 days of
beginning a high-fat diet in mice (74,75).
However, another study found that the neutrophils decreased after 1
week of high-fat feeding, which suggests that neutrophils may play
different roles in different stages of obesity development. In this
study, they appeared to induce early polarization of ATMs toward
the M1 phenotype with corresponding increases in TNF-α and IL-6
(74). Increased percentages of
peripheral blood neutrophils have also been found in obese males
(76).
Lymphocytes are strongly associated with
inflammatory processes in obesity. Although there are several
lymphocyte cell types that are related to obesity and metabolic
syndrome, pro-inflammatory Th1, Th17 and CD8+ T cells
predominate over anti-inflammatory regulatory T cells (Treg) and
Th2 cells, which are found in higher proportions in lean adipose
tissue (77,78). One study found that mice fed a
high-fat diet displayed more Th1 polarization and interferon-γ
(IFN-γ)production, which occurred several months after macrophage
accumulation and insulin resistance (79). However, the number of Treg cells was
decreased in the adipose tissue of obese mice, but insulin
sensitivity can also be improved when these cells are increased
(78). An increased number of Th17
cells in the spleen has been found in mice fed a high-fat diet, but
this cell was absent in IL-6−/− mice fed the same
high-fat diet, indicating that IL-6 is important for Th17 expansion
in obesity (80).
In addition to T cells, B cells have also shown a
positive association with obesity in high-fat diet-fed mice because
infiltration of B cells can be found in the adipose tissue of obese
mice, and these cells can activate T cells and polarize macrophages
toward the M1 type (83). One study
found that the number of IgG-associated B cells increased rapidly
after 4 weeks on a high-fat diet (84), and the cell numbers then remained
consistent during the weight gain (83,85).
By using neutralizing antibodies against the B cells, other studies
have found that obesity-induced insulin resistance in mice could be
improved, and a reduction in M1 macrophages and activation of
CD8+ T cells along with an increase in Treg numbers and
M2 signatures could be observed in the adipose tissue (83,86).
Although B cells are associated with the inflammatory condition in
obesity, regulatory B cells, which produce IL-10, have been shown
to improve insulin resistance and reduce pro-inflammatory cytokine
production in the skeletal muscles of mice fed a high-fat diet
(83). These results support the
findings from other studies, which found that low levels of IL-10
were associated with metabolic syndrome and T2D (87,88).
The human intestine contains a unique group of
microorganisms comprising up to 1,000 different species with ~100
trillion cells of bacteria, yeasts and parasites (89). There are over 50 bacterial phyla
that comprise the gastrointestinal (GI) microbiota, but ~90%
consists of Bacteroidetes and Firmicutes (90,91),
and diet is clearly seemed to be a major influence on microbiota
compositions.
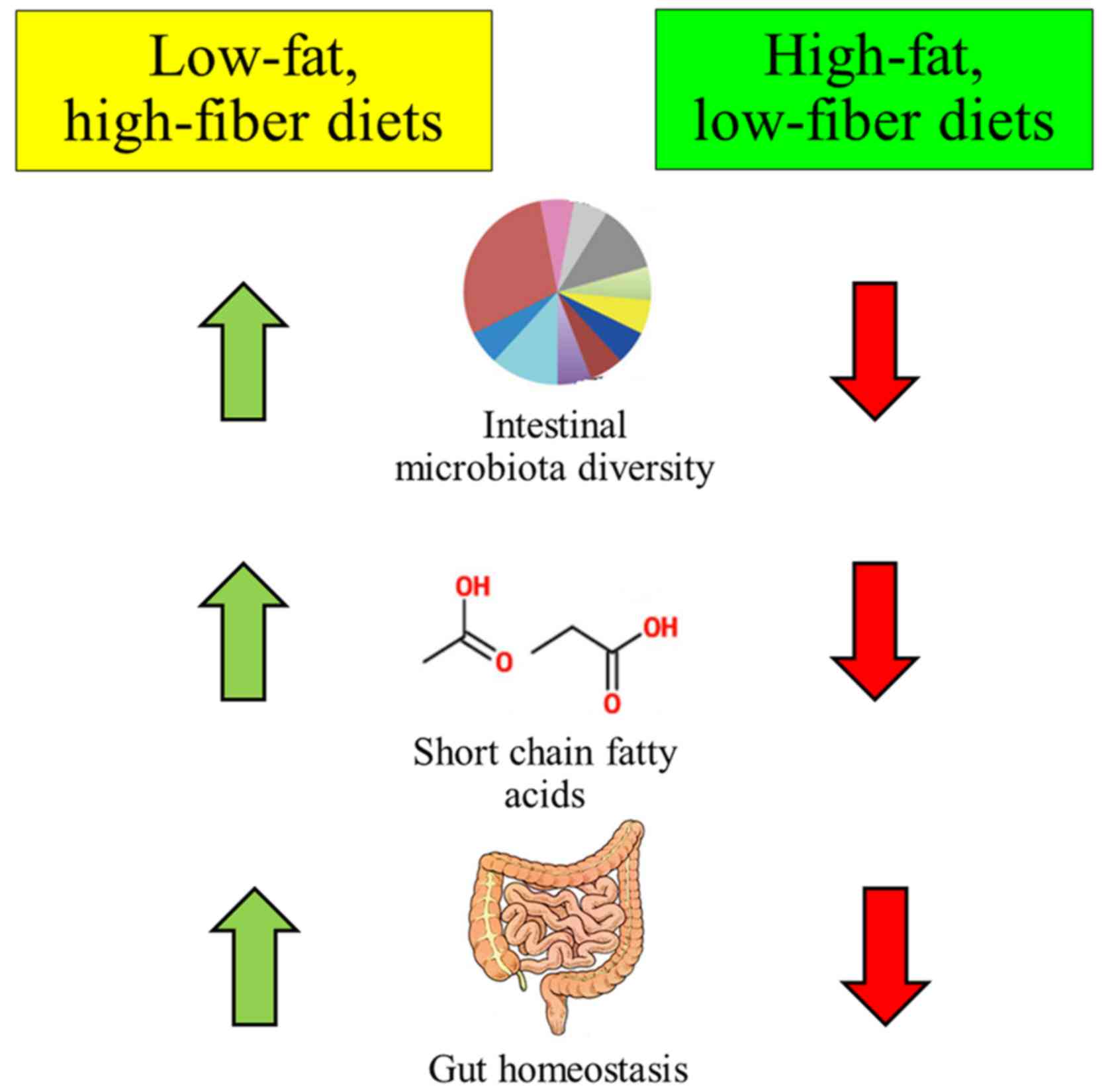
Various evidences have shown that alteration of the
diet can result in changes in the bacterial composition that also
affected gut metabolic activity, especially the production of
short-chain fatty acids (Fig. 1)
(92). This scenario can be found
in obese individuals (93) as
high-fat diets or diets low in fiber have been associated with a
higher abundance of Firmicutes (94). Studies comparing obese individuals
and their lean twins have also shown a higher predominance of
Firmicutes and lower abundance of Bacteroidetes
(93,95) in the obese subjects. Other studies,
however, have not found a difference (96,97).
Alternatively, the association studies between gut
microbiota in obesity and metabolic syndrome have been performed by
using germ-free mice. Researchers found that conventionalization of
germ-free C57BL/6 mice with normal microbiota harvested from
conventionally raised, genetically obese mice resulted in 60%
higher levels of body fat and the development of insulin resistance
in 2 weeks, despite reduced food consumption (98). Furthermore, a large human study
found that low bacterial richness showed a positive correlation
with higher weight gain supporting the concept that some bacteria
may be more abundant than other types in obese individuals
(99). Another study also found
that obesity could be transmissible because microbiota derived from
discordant obese twins caused germ-free mice to become obese when
they were transplanted with these microbiota (100). This study also demonstrated that
diet played a major role in gut microbiota diversity because the
gut of low-fat, high-fiber diet-fed obese mice was finally
dominated by the lean microbiota, which prevented increased
adiposity, even when the mice were cohoused with mice with obese
microbiota, suggesting that diet is responsible for the development
of this phenotype (100).
It is clear that the incidence of CRC is higher in
developed countries, but in recent years, it has also become a
public health problem in developing countries due to economic
growth and an increasingly westernized lifestyle, including high
consumption of red meats and high-calorie diets, which are
associated with an increased incidence of CRC (103,104). This dietary pattern of higher
consumption of animal protein (Western diet) that is related to the
epidemiology of CRC demonstrates the role of diet in CRC
development.
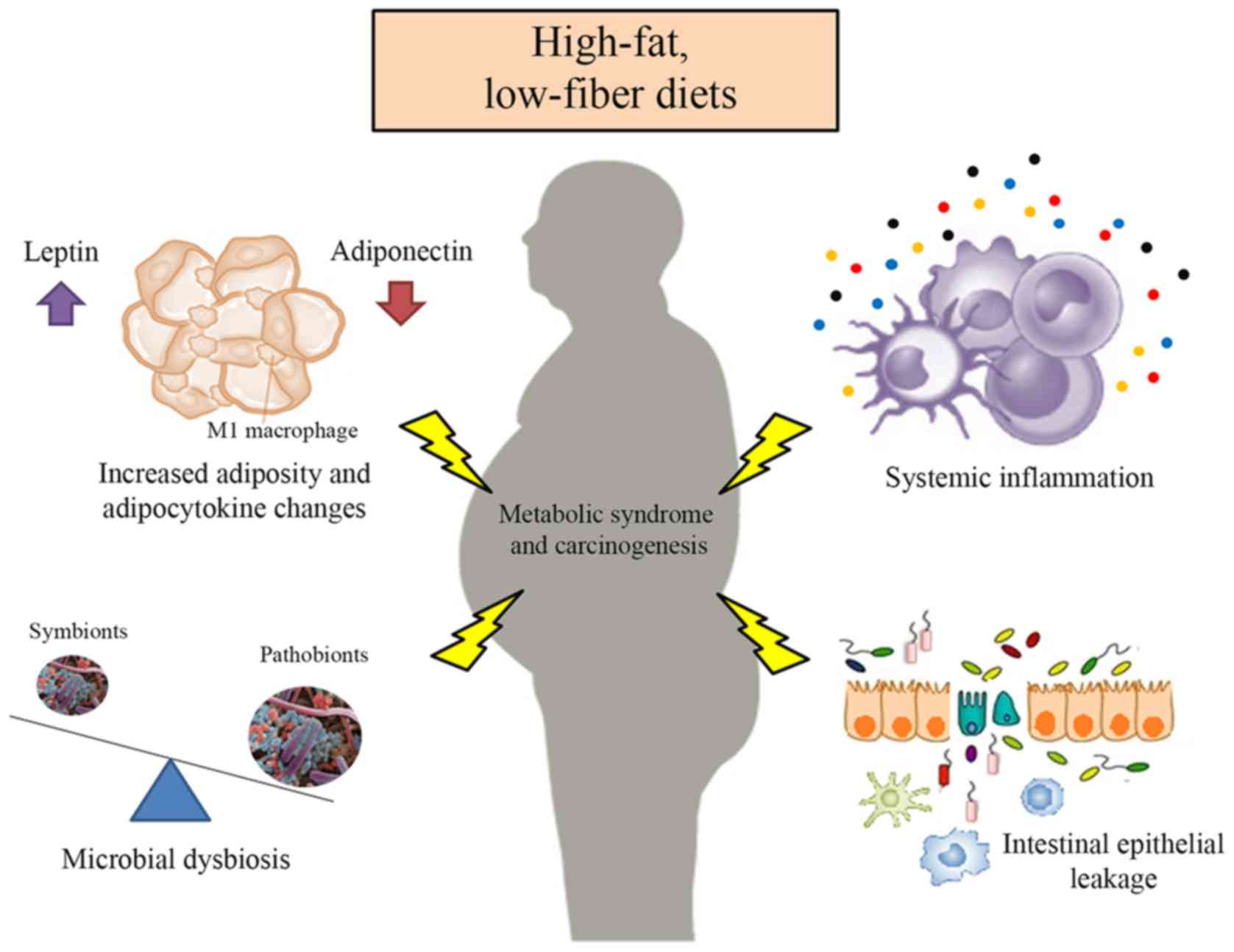
The composition and activity of the intestinal
microbiota can be influenced by diet (105), and there is considerable evidence
linking diet-induced microbiota dysbiosis to CRC tumorigenesis
(106) (Fig. 2). This relationship may be mediated
by metabolites and inflammatory induction, which induce genetic
mutations and inhibit apoptosis or stimulate angiogenesis and cell
proliferation (107). One study
found that the increased consumption of red meat was associated
with higher levels of sulfur, nitrates, ammonia, amines,
branched-chain amino acids, and H2S in the colon
(108), which all have been shown
to be pro-inflammatory and genotoxic, that in turn facilitates
colon tumorigenesis by causing DNA damage and inhibiting the
oxidation of butyrate (109,110). Moreover, several species of
Bacteroides and some Firmicutes have shown the
ability to ferment aromatic amino acids, leading to the production
of toxic compounds, such as phenylacetic acid, phenols, indoles,
and p-cresol, that promote colon carcinogenesis by DNA alkylation
and mutations (111).
Lipid metabolism byproducts, such as bile acids,
appear to be associated with the development of CRCs. Secondary
bile acids, such as deoxycholic acid (DCA), are involved in the
higher levels of reactive oxygen species (ROS), DNA damage, genomic
instability and tumor growth (112,113). Moreover, an increased risk of CRC
via the higher secondary bile acid production and ROS has been
found in individuals consuming a high-fat, low-fiber diet who have
a higher composition of inflammatory 7α-dehydroxylating bacteria
and sulfur-reducing bacteria, which produce H2S and
secondary bile acids, respectively (109). Nitric oxide is another compound
produced by specific types of bacteria. Its secondary reactive
species can activate inflammation and damage DNA. An investigation
by Sobko et al demonstrated that nitrite and
nitrate-supplemented diets are sources for nitric oxide production
by Lactobacilli and Bifidobacteria, leading to
colonic mucosal inflammation and DNA damage (114,115).
The barrier dysfunction mediated by microbial
products during CRC development also leads to adenoma invasion by
stimulating inflammatory cytokines, such as IL-17 and IL-23, which,
in turn, support tumor growth (116). The colonic inflammation model has
also demonstrated the loss of the mucosal barrier in dextran
sulfate sodium (DSS)-induced colitis. However, when feeding rats
with cheese whey protein, colitis markers were reduced, and fecal
mucin, a family of glycosylated proteins that play a role in the
intestinal mucus layer, and fecal Lactobacilli and
Bifidobacteria counts were then increased (117).
Numerous growth factors, hormones and cytokines,
known as adipocytokines, can be secreted by adipose tissue and have
pleiotropic effects, including regulation of food intake and
metabolism, crosstalk with insulin signaling and inflammatory
pathways, and promotion of angiogenesis and cellular proliferation
(118,119). These adipocytokines include
leptin, resistin, adiponectin and various cytokines, such as TNF-α
and IL-6. In obese patients, the levels of these adipocytokines are
altered, further contributing to an increased risk of CRC (120,121) (Fig.
2).
Adiponectin, also known as Acrp30, ADIPOQ, apM1 or
GBP28, is a 30 kDa protein that shares homologies with complement
factors and TNF-α, and is predominantly secreted by adipocytes
(122–124). This protein stimulates insulin
secretion, increases fatty acid metabolism, activates energy
consumption, and is also believed to have an anti-inflammatory role
(124,125). The results from epidemiological
and preclinical studies have indicated a protective role of
adiponectin against tumorigenesis because the levels of adiponectin
were negatively correlated with the risk of CRC development
(126,127). Recent meta-analyses have shown
that, in men, a 1 µg/ml increase in adiponectin levels was
associated with a 2% reduced risk of CRC (128–130). Studies in animal models have also
shown that adiponectin is involved in the suppression of colon
carcinogenesis. High-fat diet-fed mice had significantly more
polyps when the adiponectin gene was suppressed (131). Moreover, in vitro
experiment showed that the growth of CRC cells can be inhibited by
adiponectin through the activation of adenosine
monophosphate-activated protein kinase (AMPK) and suppression of
mammalian target of rapamycin (mTOR) pathways (132). As anti-angiogenic function,
neovascularization suppression can be found in adiponectin-treated
mice fed a high-fat diet, resulting in significant inhibition of
CRC cell growth (133).
Adiponectin also showed immunoregulatory functions because this
protein induces macrophage infiltration in tumor area (134).
TNF-α levels in systemic and adipose tissue are
higher in obese individuals than in lean subjects (145). Although this cytokine has a
functional role in apoptosis, cell necrosis, tumorigenesis
inhibition, and appetite reduction (143), its pro-inflammatory role now has
been associated with all steps of tumorigenesis (145). High levels of TNF-α in the colonic
mucosa of high-fat diet mice resulted in β-catenin activation,
leading to increased transcription of c-Myc, a downstream
molecule in the Wnt signaling pathway (146). Moreover, this pro-inflammatory
cytokine can activate NF-κB, a transcription factor associated with
cell proliferation, apoptosis inhibition, tissue invasion,
angiogenesis and metastasis, implying that this stimulation is
involved in the progression of carcinogenesis (147,148).
Although insulin and IGFs are produced by β-cells in
the pancreas and liver, respectively, not in adipose tissue, these
factors still play a role in CRC cell proliferation and the
progression of cancer both in vitro and in vivo.
Circulating total IGF-1 has been positively associated with obesity
and thus also contributes to an increased CRC risk (149); one study showed that IGF receptors
were found in CRC cells (150).
Importantly, several investigations have demonstrated that by
binding to these receptors, insulin stimulates proliferation of
cultured colonocytes and CRC cells directly, leading to the
activation of the MAPK pathway (151,152). This scenario was supported in a
study that used monoclonal antibodies against IGF1 and found
inhibition of CRC stem cells in mice (153).
Over the last centuries, diets have been shown to
affect the pathophysiological development of metabolic syndrome and
CRC. Although the precise mechanisms have not been clearly
elucidated, it is believed that intestinal inflammation and
alteration of adipokines, which may result from a high-fat,
low-fiber diet, plays a key role in these processes. This type of
diet influences the metabolic activity of the gut and changes the
composition of the microbiota, resulting in inflammation and
carcinogenesis. Importantly, further investigations are needed for
a deeper understanding of the effect of these abnormalities on
metabolic imbalance and CRC development. This may lead to the
development of new strategies that improve or even treat metabolic
syndrome and CRC.
|
1
|
Balkau B, Deanfield JE, Després JP,
Bassand JP, Fox KA, Smith SC Jr, Barter P, Tan CE, Van Gaal L,
Wittchen HU, et al: International Day for the Evaluation of
Abdominal Obesity (IDEA): a study of waist circumference,
cardiovascular disease, and diabetes mellitus in 168,000 primary
care patients in 63 countries. Circulation. 116:1942–1951. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grundy SM: Metabolic syndrome pandemic.
Arterioscler Thromb Vasc Biol. 28:629–636. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prasad DS, Kabir Z, Dash AK and Das BC:
Prevalence and risk factors for metabolic syndrome in Asian
Indians: a community study from urban Eastern India. J Cardiovasc
Dis Res. 3:204–211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gu D, Reynolds K, Wu X, Chen J, Duan X,
Reynolds RF, Whelton PK and He J: InterASIA Collaborative Group:
Prevalence of the metabolic syndrome and overweight among adults in
China. Lancet. 365:1398–1405. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reynolds K and He J: Epidemiology of the
metabolic syndrome. Am J Med Sci. 330:273–279. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilson PW: Metabolic risk factors for
coronary heart disease: current and future prospects. Curr Opin
Cardiol. 14:176–185. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miranda PJ, DeFronzo RA, Califf RM and
Guyton JR: Metabolic syndrome: definition, pathophysiology, and
mechanisms. Am Heart J. 149:33–45. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dentali F, di Minno MN, Gianni M, di Minno
G, Squizzato A and Ageno W: The role of the metabolic syndrome in
patients with provoked venous thromboembolic events. Thromb
Haemost. 109:759–761. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Rooy MJ, Duim W, Ehlers R, Buys AV and
Pretorius E: Platelet hyperactivity and fibrin clot structure in
transient ischemic attack individuals in the presence of metabolic
syndrome: a microscopy and thromboelastography study. Cardiovasc
Diabetol. 14:862015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ebrahimi F and Christ-Crain M: Metabolic
syndrome and hypogonadism - two peas in a pod. Swiss Med Wkly.
146:w142832016.PubMed/NCBI
|
|
11
|
Uzunlulu M, Telci Caklili O and Oguz A:
Association between metabolic syndrome and cancer. Ann Nutr Metab.
68:173–179. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen J, He X and Huang J: Diet effects in
gut microbiome and obesity. J Food Sci. 79:R442–R451. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Festi D, Schiumerini R, Eusebi LH, Marasco
G, Taddia M and Colecchia A: Gut microbiota and metabolic syndrome.
World J Gastroenterol. 20:16079–16094. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Parekh PJ, Balart LA and Johnson DA: The
Influence of the gut microbiome on obesity, metabolic syndrome and
gastrointestinal disease. Clin Transl Gastroenterol. 6:e912015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marchesi JR, Adams DH, Fava F, Hermes GD,
Hirschfield GM, Hold G, Quraishi MN, Kinross J, Smidt H, Tuohy KM,
et al: The gut microbiota and host health: a new clinical frontier.
Gut. 65:330–339. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pigrau M, Rodiño-Janeiro BK, Casado-Bedmar
M, Lobo B, Vicario M, Santos J and Alonso-Cotoner C: The joint
power of sex and stress to modulate brain-gut-microbiota axis and
intestinal barrier homeostasis: implications for irritable bowel
syndrome. Neurogastroenterol Motil. 28:463–486. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aballay LR, Eynard AR, Díaz M, del P,
Navarro A and Muñoz SE: Overweight and obesity: a review of their
relationship to metabolic syndrome, cardiovascular disease, and
cancer in South America. Nutr Rev. 71:168–179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Welty FK, Alfaddagh A and Elajami TK:
Targeting inflammation in metabolic syndrome. Transl Res.
167:257–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ley RE, Bäckhed F, Turnbaugh P, Lozupone
CA, Knight RD and Gordon JI: Obesity alters gut microbial ecology.
Proc Natl Acad Sci USA. 102:11070–11075. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pischon T, Lahmann PH, Boeing H,
Friedenreich C, Norat T, Tjønneland A, Halkjaer J, Overvad K,
Clavel-Chapelon F, Boutron-Ruault MC, et al: Body size and risk of
colon and rectal cancer in the European Prospective Investigation
Into Cancer and Nutrition (EPIC). J Natl Cancer Inst. 98:920–931.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pelucchi C, Negri E, Talamini R, Levi F,
Giacosa A, Crispo A, Bidoli E, Montella M, Franceschi S and La
Vecchia C: Metabolic syndrome is associated with colorectal cancer
in men. Eur J Cancer. 46:1866–1872. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Esposito K, Chiodini P, Colao A, Lenzi A
and Giugliano D: Metabolic syndrome and risk of cancer: a
systematic review and meta-analysis. Diabetes Care. 35:2402–2411.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kyrou I, Randeva HS and Weickert MO:
Clinical problems caused by obesity: Endotext (Internet). De Groot
LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM,
Koch C, Korbonits M, McLachlan R, New M, et al: MDText.com, Inc.;
South Dartmouth, MA, USA: 2000 https://www.endotext.orgApril 24–2014
|
|
24
|
Knecht S, Ellger T and Levine JA: Obesity
in neurobiology. Prog Neurobiol. 84:85–103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sheth RD: Metabolic concerns associated
with antiepileptic medications. Neurology. 63 Suppl 4:S24–S29.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Broberger C: Brain regulation of food
intake and appetite: molecules and networks. J Intern Med.
258:301–327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peters A, Schweiger U, Pellerin L, Hubold
C, Oltmanns KM, Conrad M, Schultes B, Born J and Fehm HL: The
selfish brain: competition for energy resources. Neurosci Biobehav
Rev. 28:143–180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peters A, Pellerin L, Dallman MF, Oltmanns
KM, Schweiger U, Born J and Fehm HL: Causes of obesity: looking
beyond the hypothalamus. Prog Neurobiol. 81:61–88. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Woods SC, Stein LJ, McKay LD and D Jr
Porte: Suppression of food intake by intravenous nutrients and
insulin in the baboon. Am J Physiol. 247:R393–R401. 1984.PubMed/NCBI
|
|
30
|
Ahima RS, Prabakaran D, Mantzoros C, Qu D,
Lowell B, Maratos-Flier E and Flier JS: Role of leptin in the
neuroendocrine response to fasting. Nature. 382:250–252. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kreier F, Kap YS, Mettenleiter TC, van
Heijningen C, van der Vliet J, Kalsbeek A, Sauerwein HP, Fliers E,
Romijn JA and Buijs RM: Tracing from fat tissue, liver, and
pancreas: a neuroanatomical framework for the role of the brain in
type 2 diabetes. Endocrinology. 147:1140–1147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Uno K, Katagiri H, Yamada T, Ishigaki Y,
Ogihara T, Imai J, Hasegawa Y, Gao J, Kaneko K, Iwasaki H, et al:
Neuronal pathway from the liver modulates energy expenditure and
systemic insulin sensitivity. Science. 312:1656–1659. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dhillon H, Zigman JM, Ye C, Lee CE,
McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein
EA, et al: Leptin directly activates SF1 neurons in the VMH, and
this action by leptin is required for normal body-weight
homeostasis. Neuron. 49:191–203. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brown FD, Fessler RG, Rachlin JR and
Mullan S: Changes in food intake with electrical stimulation of the
ventromedial hypothalamus in dogs. J Neurosurg. 60:1253–1257. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choi S, Sparks R, Clay M and Dallman MF:
Rats with hypothalamic obesity are insensitive to central leptin
injections. Endocrinology. 140:4426–4433. 1999. View Article : Google Scholar
|
|
36
|
Grundmann SJ, Pankey EA, Cook MM, Wood AL,
Rollins BL and King BM: Combination unilateral amygdaloid and
ventromedial hypothalamic lesions: evidence for a feeding pathway.
Am J Physiol Regul Integr Comp Physiol. 288:R702–R707. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Majdic G, Young M, Gomez-Sanchez E,
Anderson P, Szczepaniak LS, Dobbins RL, McGarry JD and Parker KL:
Knockout mice lacking steroidogenic factor 1 are a novel genetic
model of hypothalamic obesity. Endocrinology. 143:607–614. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bingham NC, Anderson KK, Reuter AL,
Stallings NR and Parker KL: Selective loss of leptin receptors in
the ventromedial hypothalamic nucleus results in increased
adiposity and a metabolic syndrome. Endocrinology. 149:2138–2148.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Coleman DL: Obese and diabetes: two mutant
genes causing diabetes-obesity syndromes in mice. Diabetologia.
14:141–148. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Clément K, Vaisse C, Lahlou N, Cabrol S,
Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM,
et al: A mutation in the human leptin receptor gene causes obesity
and pituitary dysfunction. Nature. 392:398–401. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Montague CT, Farooqi IS, Whitehead JP,
Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst
JA, et al: Congenital leptin deficiency is associated with severe
early-onset obesity in humans. Nature. 387:903–908. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kaszubska W, Falls HD, Schaefer VG, Haasch
D, Frost L, Hessler P, Kroeger PE, White DW, Jirousek MR and
Trevillyan JM: Protein tyrosine phosphatase 1B negatively regulates
leptin signaling in a hypothalamic cell line. Mol Cell Endocrinol.
195:109–118. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Egawa K, Maegawa H, Shimizu S, Morino K,
Nishio Y, Bryer-Ash M, Cheung AT, Kolls JK, Kikkawa R and Kashiwagi
A: Protein-tyrosine phosphatase-1B negatively regulates insulin
signaling in l6 myocytes and Fao hepatoma cells. J Biol Chem.
276:10207–10211. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bence KK, Delibegovic M, Xue B, Gorgun CZ,
Hotamisligil GS, Neel BG and Kahn BB: Neuronal PTP1B regulates body
weight, adiposity and leptin action. Nat Med. 12:917–924. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cox JE and Powley TL: Prior vagotomy
blocks VMH obesity in pair-fed rats. Am J Physiol. 240:E573–E583.
1981.PubMed/NCBI
|
|
46
|
Brüning JC, Gautam D, Burks DJ, Gillette
J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D and
Kahn CR: Role of brain insulin receptor in control of body weight
and reproduction. Science. 289:2122–2125. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Volkow ND and Wise RA: How can drug
addiction help us understand obesity? Nat Neurosci. 8:555–560.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wallenius V, Wallenius K, Ahrén B, Rudling
M, Carlsten H, Dickson SL, Ohlsson C and Jansson JO:
Interleukin-6-deficient mice develop mature-onset obesity. Nat Med.
8:75–79. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
King BM: Amygdaloid lesion-induced
obesity: relation to sexual behavior, olfaction, and the
ventromedial hypothalamus. Am J Physiol Regul Integr Comp Physiol.
291:R1201–R1214. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Peters A: The selfish brain: competition
for energy resources. Am J Hum Biol. 23:29–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Peters A and McEwen BS: Stress
habituation, body shape and cardiovascular mortality. Neurosci
Biobehav Rev. 56:139–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hitze B, Hubold C, van Dyken R,
Schlichting K, Lehnert H, Entringer S and Peters A: How the selfish
brain organizes its supply and demand. Front Neuroenergetics.
2:72010.PubMed/NCBI
|
|
53
|
Kubera B, Hubold C, Zug S, Wischnath H,
Wilhelm I, Hallschmid M, Entringer S, Langemann D and Peters A: The
brain's supply and demand in obesity. Front Neuroenergetics.
4:42012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Khodabandehloo H, Gorgani-Firuzjaee S,
Panahi G and Meshkani R: Molecular and cellular mechanisms linking
inflammation to insulin resistance and β-cell dysfunction. Transl
Res. 167:228–256. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cai D and Liu T: Hypothalamic
inflammation: a double-edged sword to nutritional diseases. Ann N Y
Acad Sci. 1243:E1–E39. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cai D and Liu T: Inflammatory cause of
metabolic syndrome via brain stress and NF-κB. Aging (Albany, NY).
4:98–115. 2012. View Article : Google Scholar
|
|
57
|
Gregor MF and Hotamisligil GS:
Inflammatory mechanisms in obesity. Annu Rev Immunol. 29:415–445.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Weisberg SP, McCann D, Desai M, Rosenbaum
M, Leibel RL and Ferrante AW Jr: Obesity is associated with
macrophage accumulation in adipose tissue. J Clin Invest.
112:1796–1808. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lee BC and Lee J: Cellular and molecular
players in adipose tissue inflammation in the development of
obesity-induced insulin resistance. Biochim Biophys Acta.
1842:446–462. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lynch L, Nowak M, Varghese B, Clark J,
Hogan AE, Toxavidis V, Balk SP, O'Shea D, O'Farrelly C and Exley
MA: Adipose tissue invariant NKT cells protect against diet-induced
obesity and metabolic disorder through regulatory cytokine
production. Immunity. 37:574–587. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yoshimura A, Ohnishi S, Orito C, Kawahara
Y, Takasaki H, Takeda H, Sakamoto N and Hashino S: Association of
peripheral total and differential leukocyte counts with
obesity-related complications in young adults. Obes Facts. 8:1–16.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Krinninger P, Ensenauer R, Ehlers K, Rauh
K, Stoll J, Krauss-Etschmann S, Hauner H and Laumen H: Peripheral
monocytes of obese women display increased chemokine receptor
expression and migration capacity. J Clin Endocrinol Metab.
99:2500–2509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Poitou C, Dalmas E, Renovato M, Benhamo V,
Hajduch F, Abdennour M, Kahn JF, Veyrie N, Rizkalla S, Fridman WH,
et al: CD14dimCD16+ and CD14+CD16+ monocytes in obesity and during
weight loss: relationships with fat mass and subclinical
atherosclerosis. Arterioscler Thromb Vasc Biol. 31:2322–2330. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Christiansen T, Richelsen B and Bruun JM:
Monocyte chemoattractant protein-1 is produced in isolated
adipocytes, associated with adiposity and reduced after weight loss
in morbid obese subjects. Int J Obes. 29:146–150. 2005. View Article : Google Scholar
|
|
65
|
Kanda H, Tateya S, Tamori Y, Kotani K,
Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, et
al: MCP-1 contributes to macrophage infiltration into adipose
tissue, insulin resistance, and hepatic steatosis in obesity. J
Clin Invest. 116:1494–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Weisberg SP, Hunter D, Huber R, Lemieux J,
Slaymaker S, Vaddi K, Charo I, Leibel RL and Ferrante AW Jr: CCR2
modulates inflammatory and metabolic effects of high-fat feeding. J
Clin Invest. 116:115–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bolus WR, Gutierrez DA, Kennedy AJ,
Anderson-Baucum EK and Hasty AH: CCR2 deficiency leads to increased
eosinophils, alternative macrophage activation, and type 2 cytokine
expression in adipose tissue. J Leukoc Biol. 98:467–477. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kim D, Kim J, Yoon JH, Ghim J, Yea K, Song
P, Park S, Lee A, Hong CP, Jang MS, et al: CXCL12 secreted from
adipose tissue recruits macrophages and induces insulin resistance
in mice. Diabetologia. 57:1456–1465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nara N, Nakayama Y, Okamoto S, Tamura H,
Kiyono M, Muraoka M, Tanaka K, Taya C, Shitara H, Ishii R, et al:
Disruption of CXC motif chemokine ligand-14 in mice ameliorates
obesity-induced insulin resistance. J Biol Chem. 282:30794–30803.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kitade H, Sawamoto K, Nagashimada M, Inoue
H, Yamamoto Y, Sai Y, Takamura T, Yamamoto H, Miyamoto K, Ginsberg
HN, et al: CCR5 plays a critical role in obesity-induced adipose
tissue inflammation and insulin resistance by regulating both
macrophage recruitment and M1/M2 status. Diabetes. 61:1680–1690.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Dalmas E, Clément K and Guerre-Millo M:
Defining macrophage phenotype and function in adipose tissue.
Trends Immunol. 32:307–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lumeng CN, Bodzin JL and Saltiel AR:
Obesity induces a phenotypic switch in adipose tissue macrophage
polarization. J Clin Invest. 117:175–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kratz M, Coats BR, Hisert KB, Hagman D,
Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS,
et al: Metabolic dysfunction drives a mechanistically distinct
proinflammatory phenotype in adipose tissue macrophages. Cell
Metab. 20:614–625. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Elgazar-Carmon V, Rudich A, Hadad N and
Levy R: Neutrophils transiently infiltrate intra-abdominal fat
early in the course of high-fat feeding. J Lipid Res. 49:1894–1903.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Talukdar S, Oh DY, Bandyopadhyay G, Li D,
Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al: Neutrophils
mediate insulin resistance in mice fed a high-fat diet through
secreted elastase. Nat Med. 18:1407–1412. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Xu X, Su S and Wang X, Barnes V, De Miguel
C, Ownby D, Pollock J, Snieder H, Chen W and Wang X: Obesity is
associated with more activated neutrophils in African American male
youth. Int J Obes (Lond). 39:26–32. 2015.PubMed/NCBI
|
|
77
|
Sell H, Habich C and Eckel J: Adaptive
immunity in obesity and insulin resistance. Nat Rev Endocrinol.
8:709–716. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Feuerer M, Herrero L, Cipolletta D, Naaz
A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, et
al: Lean, but not obese, fat is enriched for a unique population of
regulatory T cells that affect metabolic parameters. Nat Med.
15:930–939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Strissel KJ, DeFuria J, Shaul ME, Bennett
G, Greenberg AS and Obin MS: T-cell recruitment and Th1
polarization in adipose tissue during diet-induced obesity in
C57BL/6 mice. Obesity (Silver Spring). 18:1918–1925. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Winer S, Paltser G, Chan Y, Tsui H,
Engleman E, Winer D and Dosch HM: Obesity predisposes to Th17 bias.
Eur J Immunol. 39:2629–2635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Nishimura S, Manabe I, Nagasaki M, Eto K,
Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al:
CD8+ effector T cells contribute to macrophage recruitment and
adipose tissue inflammation in obesity. Nat Med. 15:914–920. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wu H, Ghosh S, Perrard XD, Feng L, Garcia
GE, Perrard JL, Sweeney JF, Peterson LE, Chan L, Smith CW, et al:
T-cell accumulation and regulated on activation, normal T cell
expressed and secreted upregulation in adipose tissue in obesity.
Circulation. 115:1029–1038. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Winer DA, Winer S, Shen L, Wadia PP,
Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al: B
cells promote insulin resistance through modulation of T cells and
production of pathogenic IgG antibodies. Nat Med. 17:610–617. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sun S, Ji Y, Kersten S and Qi L:
Mechanisms of inflammatory responses in obese adipose tissue. Annu
Rev Nutr. 32:261–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Duffaut C, Galitzky J, Lafontan M and
Bouloumié A: Unexpected trafficking of immune cells within the
adipose tissue during the onset of obesity. Biochem Biophys Res
Commun. 384:482–485. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
DeFuria J, Belkina AC, Jagannathan-Bogdan
M, Snyder-Cappione J, Carr JD, Nersesova YR, Markham D, Strissel
KJ, Watkins AA, Zhu M, et al: B cells promote inflammation in
obesity and type 2 diabetes through regulation of T-cell function
and an inflammatory cytokine profile. Proc Natl Acad Sci USA.
110:5133–5138. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jagannathan M, McDonnell M, Liang Y,
Hasturk H, Hetzel J, Rubin D, Kantarci A, Van Dyke TE, Ganley-Leal
LM and Nikolajczyk BS: Toll-like receptors regulate B cell cytokine
production in patients with diabetes. Diabetologia. 53:1461–1471.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
van Exel E, Gussekloo J, De Craen AJM,
Frölich M, Bootsma-Van Der Wiel A and Westendorp RGJ: Leiden 85
Plus Study: Low production capacity of interleukin-10 associates
with the metabolic syndrome and type 2 diabetes: The Leiden 85-Plus
Study. Diabetes. 51:1088–1092. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Gomes AC, Bueno AA, de Souza RGM and Mota
JF: Gut microbiota, probiotics and diabetes. Nutr J. 13:602014.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Bajzer M and Seeley RJ: Physiology:
obesity and gut flora. Nature. 444:1009–1010. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sekirov I, Russell SL, Antunes LCM and
Finlay BB: Gut microbiota in health and disease. Physiol Rev.
90:859–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
David LA, Maurice CF, Carmody RN,
Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y,
Fischbach MA, et al: Diet rapidly and reproducibly alters the human
gut microbiome. Nature. 505:559–563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Turnbaugh PJ, Hamady M, Yatsunenko T,
Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA,
Affourtit JP, et al: A core gut microbiome in obese and lean twins.
Nature. 457:480–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Trompette A, Gollwitzer ES, Yadava K,
Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod
LP, Harris NL, et al: Gut microbiota metabolism of dietary fiber
influences allergic airway disease and hematopoiesis. Nat Med.
20:159–166. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ley RE, Turnbaugh PJ, Klein S and Gordon
JI: Microbial ecology: human gut microbes associated with obesity.
Nature. 444:1022–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Schwiertz A, Taras D, Schäfer K, Beijer S,
Bos NA, Donus C and Hardt PD: Microbiota and SCFA in lean and
overweight healthy subjects. Obesity (Silver Spring). 18:190–195.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Duncan SH, Lobley GE, Holtrop G, Ince J,
Johnstone AM, Louis P and Flint HJ: Human colonic microbiota
associated with diet, obesity and weight loss. Int J Obes.
32:1720–1724. 2008. View Article : Google Scholar
|
|
98
|
Bäckhed F, Ding H, Wang T, Hooper LV, Koh
GY, Nagy A, Semenkovich CF and Gordon JI: The gut microbiota as an
environmental factor that regulates fat storage. Proc Natl Acad Sci
USA. 101:15718–15723. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Le Chatelier E, Nielsen T, Qin J, Prifti
E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy
S, et al: MetaHIT consortium: Richness of human gut microbiome
correlates with metabolic markers. Nature. 500:541–546. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ridaura VK, Faith JJ, Rey FE, Cheng J,
Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et
al: Gut microbiota from twins discordant for obesity modulate
metabolism in mice. Science. 341:12412142013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sears CL and Garrett WS: Microbes,
microbiota, and colon cancer. Cell Host Microbe. 15:317–328. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Center MM, Jemal A and Ward E:
International trends in colorectal cancer incidence rates. Cancer
Epidemiol Biomarkers Prev. 18:1688–1694. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Center MM, Jemal A, Smith RA and Ward E:
Worldwide variations in colorectal cancer. CA Cancer J Clin.
59:366–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Duncan SH, Belenguer A, Holtrop G,
Johnstone AM, Flint HJ and Lobley GE: Reduced dietary intake of
carbohydrates by obese subjects results in decreased concentrations
of butyrate and butyrate-producing bacteria in feces. Appl Environ
Microbiol. 73:1073–1078. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Keku TO, Dulal S, Deveaux A, Jovov B and
Han X: The gastrointestinal microbiota and colorectal cancer. Am J
Physiol Gastrointest Liver Physiol. 308:G351–G363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Medzhitov R: Origin and physiological
roles of inflammation. Nature. 454:428–435. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Nyangale EP, Mottram DS and Gibson GR: Gut
microbial activity, implications for health and disease: the
potential role of metabolite analysis. J Proteome Res.
11:5573–5585. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Greer JB and O'Keefe SJ: Microbial
induction of immunity, inflammation, and cancer. Front Physiol.
1:1682011. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Módis K, Coletta C, Erdélyi K,
Papapetropoulos A and Szabo C: Intramitochondrial hydrogen sulfide
production by 3-mercaptopyruvate sulfurtransferase maintains
mitochondrial electron flow and supports cellular bioenergetics.
FASEB J. 27:601–611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Nistal E, Fernández-Fernández N, Vivas S
and Olcoz JL: Factors determining colorectal cancer: the role of
the intestinal microbiota. Front Oncol. 5:2202015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Dvorak K, Payne CM, Chavarria M, Ramsey L,
Dvorakova B, Bernstein H, Holubec H, Sampliner RE, Guy N, Condon A,
et al: Bile acids in combination with low pH induce oxidative
stress and oxidative DNA damage: relevance to the pathogenesis of
Barrett's oesophagus. Gut. 56:763–771. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Gill CI and Rowland IR: Diet and cancer:
assessing the risk. Br J Nutr. 88 Suppl 1:S73–S87. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sobko T, Huang L, Midtvedt T, Norin E,
Gustafsson LE, Norman M, Jansson EA and Lundberg JO: Generation of
NO by probiotic bacteria in the gastrointestinal tract. Free Radic
Biol Med. 41:985–991. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sobko T, Reinders CI, Jansson E, Norin E,
Midtvedt T and Lundberg JO: Gastrointestinal bacteria generate
nitric oxide from nitrate and nitrite. Nitric Oxide. 13:272–278.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Grivennikov SI, Wang K, Mucida D, Stewart
CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung
KE, et al: Adenoma-linked barrier defects and microbial products
drive IL-23/IL-17-mediated tumour growth. Nature. 491:254–258.
2012.PubMed/NCBI
|
|
117
|
Sprong RC, Schonewille AJ and van der Meer
R: Dietary cheese whey protein protects rats against mild dextran
sulfate sodium-induced colitis: role of mucin and microbiota. J
Dairy Sci. 93:1364–1371. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Hursting SD, Digiovanni J, Dannenberg AJ,
Azrad M, Leroith D, Demark-Wahnefried W, Kakarala M, Brodie A and
Berger NA: Obesity, energy balance, and cancer: new opportunities
for prevention. Cancer Prev Res (Phila). 5:1260–1272. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Tilg H and Moschen AR: Adipocytokines:
mediators linking adipose tissue, inflammation and immunity. Nat
Rev Immunol. 6:772–783. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Bardou M, Barkun AN and Martel M: Obesity
and colorectal cancer. Gut. 62:933–947. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Barb D, Williams CJ, Neuwirth AK and
Mantzoros CS: Adiponectin in relation to malignancies: a review of
existing basic research and clinical evidence. Am J Clin Nutr.
86:s858–s866. 2007.PubMed/NCBI
|
|
122
|
Scherer PE, Williams S, Fogliano M,
Baldini G and Lodish HF: A novel serum protein similar to C1q,
produced exclusively in adipocytes. J Biol Chem. 270:26746–26749.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kadowaki T, Yamauchi T, Kubota N, Hara K,
Ueki K and Tobe K: Adiponectin and adiponectin receptors in insulin
resistance, diabetes, and the metabolic syndrome. J Clin Invest.
116:1784–1792. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Nakashima R, Kamei N, Yamane K, Nakanishi
S, Nakashima A and Kohno N: Decreased total and high molecular
weight adiponectin are independent risk factors for the development
of type 2 diabetes in Japanese-Americans. J Clin Endocrinol Metab.
91:3873–3877. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Renehan AG, Zwahlen M and Egger M:
Adiposity and cancer risk: new mechanistic insights from
epidemiology. Nat Rev Cancer. 15:484–498. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Guffey CR, Fan D, Singh UP and Murphy EA:
Linking obesity to colorectal cancer: recent insights into
plausible biological mechanisms. Curr Opin Clin Nutr Metab Care.
16:595–600. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Hillenbrand A, Fassler J, Huber N, Xu P,
Henne-Bruns D, Templin M, Schrezenmeier H, Wolf AM and Knippschild
U: Changed adipocytokine concentrations in colorectal tumor
patients and morbidly obese patients compared to healthy controls.
BMC Cancer. 12:5452012. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Xu XT, Xu Q, Tong JL, Zhu MM, Huang ML,
Ran ZH and Xiao SD: Meta-analysis: circulating adiponectin levels
and risk of colorectal cancer and adenoma. J Dig Dis. 12:234–244.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
An W, Bai Y, Deng SX, Gao J, Ben QW, Cai
QC, Zhang HG and Li ZS: Adiponectin levels in patients with
colorectal cancer and adenoma: a meta-analysis. Eur J Cancer Prev.
21:126–133. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Gulcelik MA, Colakoglu K, Dincer H, Dogan
L, Yenidogan E and Gulcelik NE: Associations between adiponectin
and two different cancers: breast and colon. Asian Pac J Cancer
Prev. 13:395–398. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Fujisawa T, Endo H, Tomimoto A, Sugiyama
M, Takahashi H, Saito S, Inamori M, Nakajima N, Watanabe M, Kubota
N, et al: Adiponectin suppresses colorectal carcinogenesis under
the high-fat diet condition. Gut. 57:1531–1538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Sugiyama M, Takahashi H, Hosono K, Endo H,
Kato S, Yoneda K, Nozaki Y, Fujita K, Yoneda M, Wada K, et al:
Adiponectin inhibits colorectal cancer cell growth through the
AMPK/mTOR pathway. Int J Oncol. 34:339–344. 2009.PubMed/NCBI
|
|
133
|
Moon HS, Liu X, Nagel JM, Chamberland JP,
Diakopoulos KN, Brinkoetter MT, Hatziapostolou M, Wu Y, Robson SC,
Iliopoulos D, et al: Salutary effects of adiponectin on colon
cancer: in vivo and in vitro studies in mice. Gut. 62:561–570.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Sun Y and Lodish HF: Adiponectin
deficiency promotes tumor growth in mice by reducing macrophage
infiltration. PLoS One. 5:e119872010. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Vansaun MN: Molecular pathways:
adiponectin and leptin signaling in cancer. Clin Cancer Res.
19:1926–1932. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Friedman JM and Halaas JL: Leptin and the
regulation of body weight in mammals. Nature. 395:763–770. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Ryan AS, Berman DM, Nicklas BJ, Sinha M,
Gingerich RL, Meneilly GS, Egan JM and Elahi D: Plasma adiponectin
and leptin levels, body composition, and glucose utilization in
adult women with wide ranges of age and obesity. Diabetes Care.
26:2383–2388. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Tamakoshi K, Toyoshima H, Wakai K, Kojima
M, Suzuki K, Watanabe Y, Hayakawa N, Yatsuya H, Kondo T, Tokudome
S, et al: Leptin is associated with an increased female colorectal
cancer risk: a nested case-control study in Japan. Oncology.
68:454–461. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Delort L, Rossary A, Farges MC, Vasson MP
and Caldefie-Chézet F: Leptin, adipocytes and breast cancer: focus
on inflammation and anti-tumor immunity. Life Sci. 140:37–48. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Wang D, Chen J, Chen H, Duan Z, Xu Q, Wei
M, Wang L and Zhong M: Leptin regulates proliferation and apoptosis
of colorectal carcinoma through PI3K/Akt/mTOR signalling pathway. J
Biosci. 37:91–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Padidar S, Farquharson AJ, Williams LM,
Kearney R, Arthur JR and Drew JE: High-fat diet alters gene
expression in the liver and colon: links to increased development
of aberrant crypt foci. Dig Dis Sci. 57:1866–1874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Endo H, Hosono K, Uchiyama T, Sakai E,
Sugiyama M, Takahashi H, Nakajima N, Wada K, Takeda K, Nakagama H,
et al: Leptin acts as a growth factor for colorectal tumours at
stages subsequent to tumour initiation in murine colon
carcinogenesis. Gut. 60:1363–1371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Braun S, Bitton-Worms K and LeRoith D: The
link between the metabolic syndrome and cancer. Int J Biol Sci.
7:1003–1015. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Grossmann ME, Mizuno NK, Bonorden MJL, Ray
A, Sokolchik I, Narasimhan ML and Cleary MP: Role of the
adiponectin leptin ratio in prostate cancer. Oncol Res. 18:269–277.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Sharma D, Wang J, Fu PP, Sharma S,
Nagalingam A, Mells J, Handy J, Page AJ, Cohen C, Anania FA, et al:
Adiponectin antagonizes the oncogenic actions of leptin in
hepatocellular carcinogenesis. Hepatology. 52:1713–1722. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Liu Z, Brooks RS, Ciappio ED, Kim SJ,
Crott JW, Bennett G, Greenberg AS and Mason JB: Diet-induced
obesity elevates colonic TNF-α in mice and is accompanied by an
activation of Wnt signaling: a mechanism for obesity-associated
colorectal cancer. J Nutr Biochem. 23:1207–1213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Dolcet X, Llobet D, Pallares J and
Matias-Guiu X: NF-κB in development and progression of human
cancer. Virchows Arch. 446:475–482. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Pais R, Silaghi H, Silaghi AC, Rusu ML and
Dumitrascu DL: Metabolic syndrome and risk of subsequent colorectal
cancer. World J Gastroenterol. 15:5141–5148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Clayton PE, Banerjee I, Murray PG and
Renehan AG: Growth hormone, the insulin-like growth factor axis,
insulin and cancer risk. Nat Rev Endocrinol. 7:11–24. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Singh P and Rubin N: Insulinlike growth
factors and binding proteins in colon cancer. Gastroenterology.
105:1218–1237. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Giovannucci E: Nutrition, insulin,
insulin-like growth factors and cancer. Horm Metab Res. 35:694–704.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Komninou D, Ayonote A, Richie JP Jr and
Rigas B: Insulin resistance and its contribution to colon
carcinogenesis. Exp Biol Med (Maywood). 228:396–405.
2003.PubMed/NCBI
|
|
153
|
Hart LS, Dolloff NG, Dicker DT, Koumenis
C, Christensen JG, Grimberg A and El-Deiry WS: Human colon cancer
stem cells are enriched by insulin-like growth factor-1 and are
sensitive to figitumumab. Cell Cycle. 10:2331–2338. 2011.
View Article : Google Scholar : PubMed/NCBI
|