Introduction
Gene fusion commonly occurs through chromosome
rearrangement and translocation during tumorigenesis (1,2). They
are powerful ‘gain of function’ mutations in cancers that alter
protein expression, remove regulatory domains, force
oligomerization, change subcellular localization or fuse to a new
domain (3). Moreover, fusion genes
are tumor-specific and have been used as diagnostic markers and
therapeutic targets (3). For
example, ~98% of patients with acute promyelocytic leukemia carry a
translocation of chromosomes 15 and 17, which creates the fusion
for retinoic acid receptor α (RARα) and promyelocytic leukemia
(PML) protein. More than 80% of patients who carry the RARα-PML
fusion achieve prolonged remission after treatment with
all-trans retinoic acid (4).
Historically, fusion genes have been mainly
associated with hematological and mesenchymal malignancies
(2,5). Although, over 440 gene fusions have
been identified in benign tumors and cancers, only ~15% are found
in epithelial tumors (6). Discovery
of fusion genes has traditionally relied on the detection of
translocations using cytogenetic techniques. However, the low
resolution of cytogenetic techniques and the complex karyotypes of
epithelial tumors make it difficult to identify these
translocations in epithelial tumors (3). In addition, gene fusion can also be
produced by small intra-chromosomal rearrangements such as
deletions, amplifications or insertions that cannot be detected
using cytogenetic techniques (3).
In recent years, several new non-cytogenetic technologies have been
developed to resolve this problem. For example, pair-end sequencing
of cancer cDNA using new high-throughput sequencing platforms,
high-resolution single nucleotide polymorphism (SNP) arrays and
array comparative genomic hybridization (aCGH) can detect small
copy number aberrations as candidate fusion loci and be used to
suggest the potential breakpoints in cancer genomes (7,8).
In our previous whole-genome screening study using
500K SNP arrays, we identified 57 homozygous deletions (HDs) and
653 amplicons in 23 cancer cell lines (9). These aberrations were produced by
either inter-chromosome or intra-chromosome rearrangements, and
also created many chromosome breakpoints. Gene fusion can occur if
two breakpoints are both located in the gene region. In the present
study, we applied the results from our genome-wide SNP arrays to
screen for chromosome breakpoints and explore novel fusion genes in
several cancer cell lines.
Materials and methods
Cell lines, cell culture and
antibodies
Hep3B, HepG2, HuH6, HuH7, SK-Hep-1 and 293T cells
were obtained from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's
medium supplemented with 10% fetal bovine serum, 1% non-essential
amino acids, and 1% penicillin/streptomycin (Invitrogen, Carlsbad,
CA, USA). Primary anti-FNDC3B and anti-PRKCI antibodies were
purchased from Sigma-Aldrich (St. Louis, MO, USA) and BD
Biosciences (San Jose, CA, USA), respectively. HRP-conjugated
secondary antibodies were purchased from Millipore Corporation
(Billerica, MA, USA). The rabbit polyclonal antibody against
FNDC3B-PRKCI was generated at LTK BioLaboratories (Taoyuan, Taiwan)
by injecting a rabbit with a synthetic oligopeptide near the
FNDC3B-PRKCI fusion point (LLEWDEEPVMPM; FNDC3B, amino acids
413–418; and PRKCI, amino acids 198–203).
3′-Rapid amplification of cDNA ends (RACE) PCR. RNA
was extracted from Hep3B cells using TRIzol reagent (Invitrogen).
The 3′-end of the chimeric transcript was identified using a
3′-RACE System for rapid amplification of cDNA ends kit
(Invitrogen). RNA was reverse-transcribed into cDNA using the
adapter primer (GGCCACGCGTCGACTAGTACTTTTT TTTTTTTTTTTT). Then, the
product was PCR-amplified using a FNDC3B-specific primer
(TTCCCATGATGTCACCC AAT) and an abridged universal amplification
primer (GGCC ACGCGTCGACTAGTAC).
Analysis of copy number alterations and genomic PCR.
The copy number alterations were detected by Affymetrix GeneChip
Human Mapping 500K SNP Arrays (Affymetrix, Santa Clara, CA, USA)
and analysis by dChip software as previously described (9). In brief, the copy number (CN) for an
SNP probe in cancer cell lines was computed as follows:
CN = (SNP signal in Hep3B/mean signal of the
reference at SNP) × 2
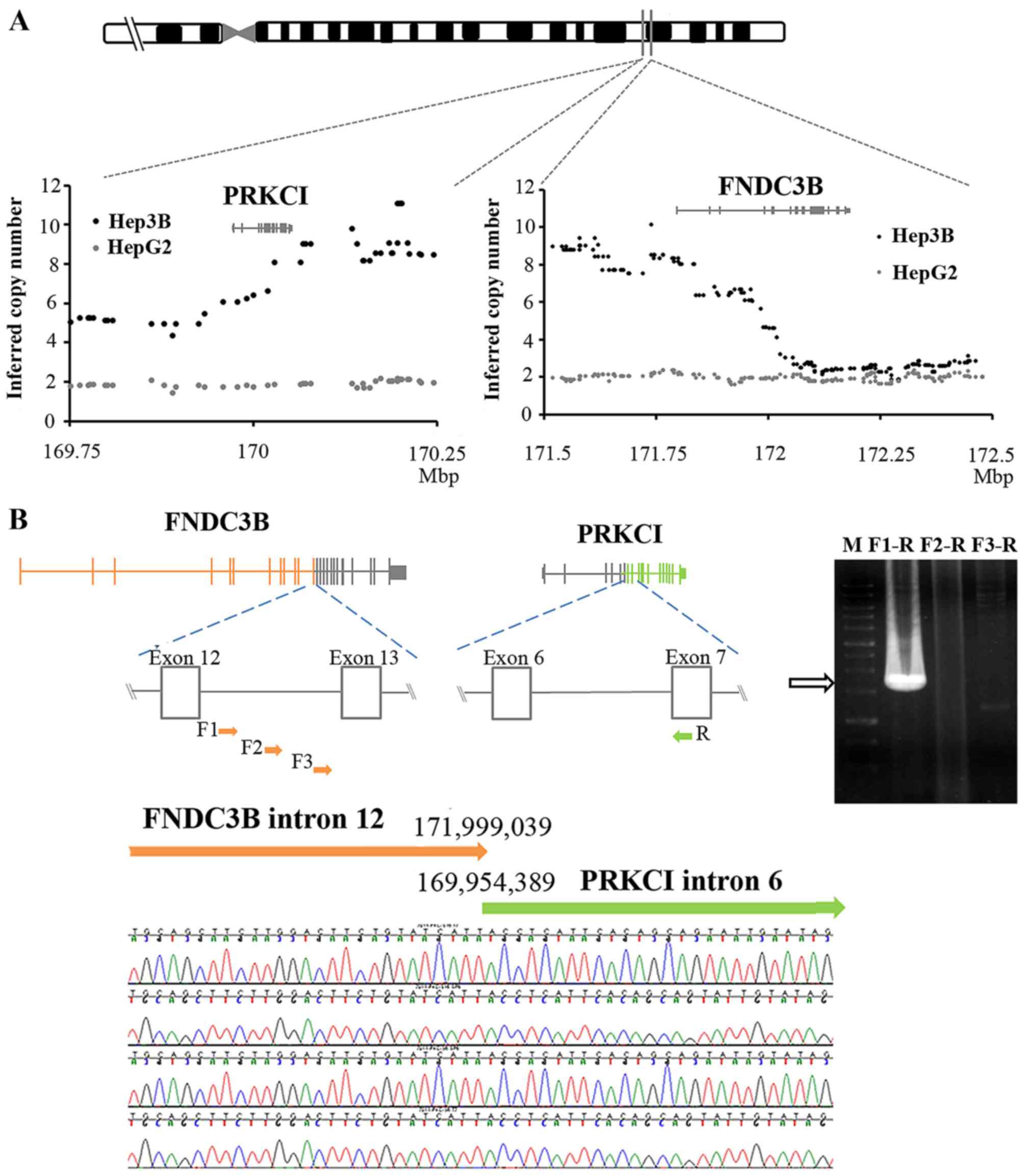
To search for 3q26.2 breakpoints, we designed 3
forward primers for FNDC3B intron 12 (F1, AGCTGGGAAGTTCAA
GGTCAAGGACTTGTATCTGG, F2, TAAGGGCAGGGAA CCCAAACAGACTGTTTATCTCC, and
F3, GATCAGGCT GGGCAGTTTGGGCCAATCTAAGT) to pair with the reverse
primer (R, ATGGATGACTGATCCATGGGCATCAC TGGT) for PRKCI exon
7. The PCR product was cloned and sequenced.
Cell migration, cell growth and
anchorage-independent growth assays. Full-length
FNDC3B-PRKCI was obtained by PCR from Hep3B cell cDNA, and
the product was ligated into a pCDNA3.0-HA vector (Invitrogen).
Cells for the wound-healing assay were seeded at a confluent
density and cultured overnight to permit growth into a monolayer.
The cultured cells were scratched with a p200 pipette tip, and the
wound was allowed to close for 20 h. The average decrease in the
area covered by the cells was calculated to evaluate the rate of
wound closure. Cell viability was determined by the cell
proliferation assay every 48 h for 6 days using the AlamarBlue
reagent (AbD Serotec, Kidlington, UK), and absorbance values at
wavelengths of 560 and 590 nm were evaluated to calculate the
growth curves. Approximately 10,000 cells were mixed in 0.25% top
agarose for the soft agar colony formation assay, and plated onto
0.5% bottom agarose in a culture medium in 60-mm dishes. All of the
experiments were conducted in triplicate. The dishes were incubated
at 37°C in a 5% CO2 incubator for 3 weeks, and the
medium was changed every 3 days. The colonies were visualized with
1% crystal violet (Sigma-Aldrich) staining and photographed under
light microscopy.
Results
To explore for fusion genes in cancer cells, we
established stringent criteria for defining HD and amplicon
breakpoints. First, the copy numbers for HDs and amplicon
breakpoints were higher or lower than its neighboring probe for a
copy number of at least 1.5, respectively. Then, the breakpoints
were located on a gene region. Overall, 80 breakpoints were
identified and located in 39 genes (Table I). In total, 27 out of the 39 genes
were identified to fuse with other genes by RNA-sequencing in
cancer patients from the The Cancer Genome Atlas (TCGA) and COMIC
fusion gene databases. These results suggest that most of the
breakpoints in cancer are localized in common fragile regions. For
example, Wang et al reported qPCR and fluorescence in
situ hybridization (FISH) assay results showing that
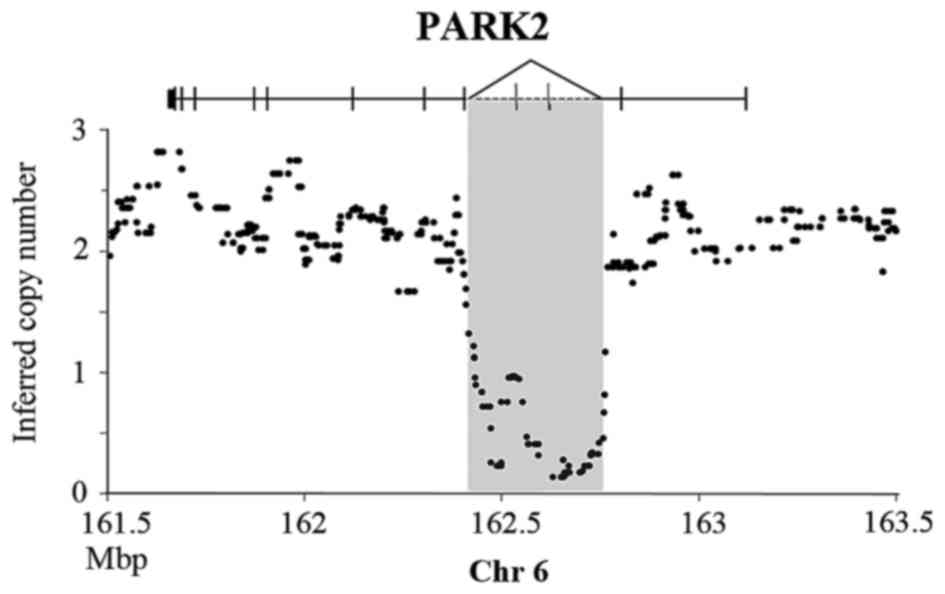
PARK2 lost exon 3 and 4 by the intra-chromosome
rearrangement in the PLC/PRF/5 cell line (10). Our data not only confirmed the
homozygous deletion in PLC/PRF/5, but also refined the breakpoints
in PARK2 intron 2 (162.47 Mb) and 4 (162.75 Mb) (Fig. 1).
 | Table I.Breakpoints observed in cancer cell
lines. |
Table I.
Breakpoints observed in cancer cell
lines.
| Cell line | Chr | 5′ Breakpoint | Gene | 3′ Breakpoint | Gene |
|---|
| Amplicons |
|
|
|
|
|
|
PLC/PRF/5 | 1 | 76795861 |
ST6GALNAC3 | 76881390 |
|
|
Hep3B | 3 | 171206621 | PRKCI | 173503700 | FNDC3B |
|
SNU387 | 3 | 7408728 | GRM7 | 7774941 | GRM7 |
|
PLC/PRF/5 | 5 | 53314739 | ARL15 | 53499505 |
|
|
PLC/PRF/5 | 5 | 138510925 | SIL1 | 138694271 |
|
|
Tong | 7 | 99315362 |
| 99842479 | ZCWPW1 |
|
Tong | 7 | 107532265 | LAMB4 | 107578077 |
|
|
Tong | 7 | 111630666 | DOCK4 | 112036838 |
|
|
Hep3B | 8 | 31790956 | NRG1 | 35099889 |
|
|
HA22T | 9 | 292324 | DOCKS | 416740 |
|
|
HA22T | 9 | 12689776 | TYRP1 | 13017530 | MPDZ |
|
HA22T | 10 | 12486578 | PRESER2 | 12533424 | UPF2 |
|
HA22T | 10 | 12626943 |
| 12761513 | CAMK1D |
|
Huh7 | 11 | 65636930 | PACS1 | 65754474 |
|
|
SNU387 | 11 | 65846608 | PACS1 | 68628370 |
|
|
Mahlavu | 16 | 76800885 | WWOX | 76844083 |
|
|
Hep3B | 17 | 41625323 |
| 41708649 | LRRC37A |
|
SK-Hep-1 | 17 | 41635580 |
| 41708649 | LRRC37A |
|
SNU449 | 17 | 19109505 |
| 19417180 | SLC47A1 |
|
HA22T | 19 | 40464311 |
| 41239370 | WDR62 |
|
Mahlavu | 19 | 48855794 |
| 51127600 | NOVA2 |
|
PLC/PRF/5 | 19 | 21336857 | ZNF708 | 22286718 |
|
|
Tong | 19 | 39827914 |
| 39895926 | ZNF599 |
|
HA59T | 20 | 29841434 | TPX2 | 30208930 | TPX2 |
|
Tong | 22 | 19458206 | PI4KA | 19520756 |
|
|
HepG2 | X | 10576540 | MID1 | 10685154 | MID1 |
|
Huh6 | X | 29019665 |
IL1RAPL1 | 29186133 |
IL1RAPL1 |
| Homozygous
deletions |
|
|
|
|
|
|
HA22T | 2 | 141590880 | LRP1B | 141738262 | LRP1B |
|
HA59T | 3 | 60520403 | FHIT | 60690206 | FHIT |
|
Hep3B | 3 | 53543957 | CACNA1D | 53646459 | CACNA1D |
|
Tong | 3 | 117643669 | LSAMP | 117718870 | LSAMP |
|
SNU398 | 4 | 93918392 | GRID2 | 94053969 | GRID2 |
|
PLC/PRF/5 | 6 | 162593039 | PARK2 | 162745092 | PARK2 |
|
HepG2 | 7 | 69045030 | AUTS2 | 69240841 | AUTS2 |
|
HepG2 | 7 | 78115615 | MAGI2 | 78249076 | MAGI2 |
|
HepG2 | 7 | 78581862 | MAGI2 | 78682833 | MAGI2 |
|
Mahlavu | 7 | 78153860 | MAGI2 | 78230785 | MAGI2 |
|
Tong | 8 | 3939796 | CSMD1 | 3947332 | CSMD1 |
|
Mahlavu | 9 | 9125223 | PTPRD | 9455190 | PTPRD |
|
SK-Hep-1 | 13 | 18983305 |
| 20439027 | LATS2 |
In the present study, we focused on fusion genes
produced by intra-chromosome rearrangement while inter-chromosome
rearrangement results in complex products with difficult to predict
fusion targets were not addressed. First, we selected the aberrant
region where both the 5′- and 3′-end breakpoints were found and
designed primers to check for potential fusion status using genomic
PCR. The mean distance in a 500K array is ~5.8 kb. Therefore, the
breakpoints were detectable by genomic PCR. Finally, a potential
fusion gene region was observed on chromosome 3q26 in Hep3B cells.
We hypothesized that the breakpoints were in PRKCI intron 7
and FNDC3B intron 12 after calculating the probe intensity
on 3q26 for the copy number variation (Fig. 2A). Then, we designed primers
targeting the intron of one of the genes to pair with a primer for
the exon of the other. Genomic PCR resulted in a 1.3-kb DNA
fragment using the forward primer in FNDC3B intron 12 and
the reverse primer for PRKCI exon 8 (Fig. 2B). The sequencing results suggested
that the breakpoints were located at 169.95 Mb (PRKCI) and
171.99 Mb (FNDC3B), respectively. According to the
orientation and localization of PRKCI and FNDC3B in
chromosome 3 and our findings, we hypothesized that
FNDC3B-PRKCI results from a 2-Mb segment duplication.
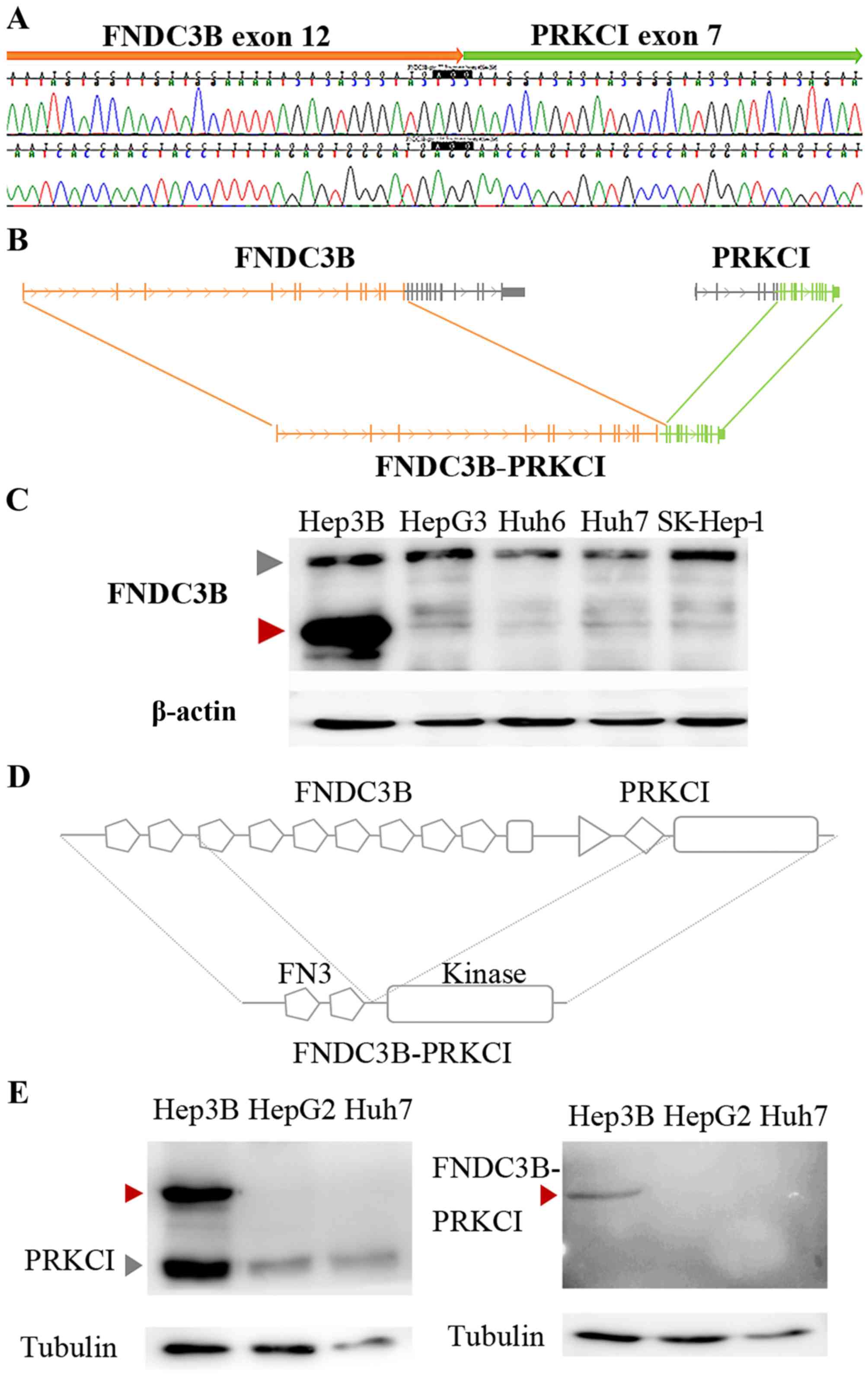
To establish our FNDC3B-PRKCI fusion model,
we performed 3′-RACE PCR for FNDC3B transcripts and detected
a PCR product smaller than normal FNDC3B. Sequencing
analyses revealed that the small transcripts were created by an
in-frame fusion of FNDC3B exons 1–12 with PRKCI exons
7–18 as predicted by our genomic DNA-sequencing results (Fig. 3A and B). Then, we performed western
blotting using an antibody that specifically recognizes the FNDC3B
N-terminal region. FNDC3B expression in Hep3B cells was not
different from other cell lines but an additional overexpressed
band was observed at ~90 kDa (Fig.
3C).
As FNDC3B-PRKCI was produced by in-frame
fusion, the chimeric protein contained two complete fibronectin
type III domains from FNDC3B and the serine-threonine kinase
domain from PRKCI (Fig. 3D).
Both FNDC3B- and PRKCI-specific antibodies recognized the
FNDC3B-PRKCI protein in Hep3B cells (Fig. 3C and E, left). A polyclonal antibody
that recognizes the peptide across the FNDC3B-PRKCI fusion
point was produced in rabbits to specifically detect the chimeric
protein. Western blotting revealed that the antibody specifically
detected the FNDC3B-PRKCI protein in Hep3B cells (Fig. 3E, right).
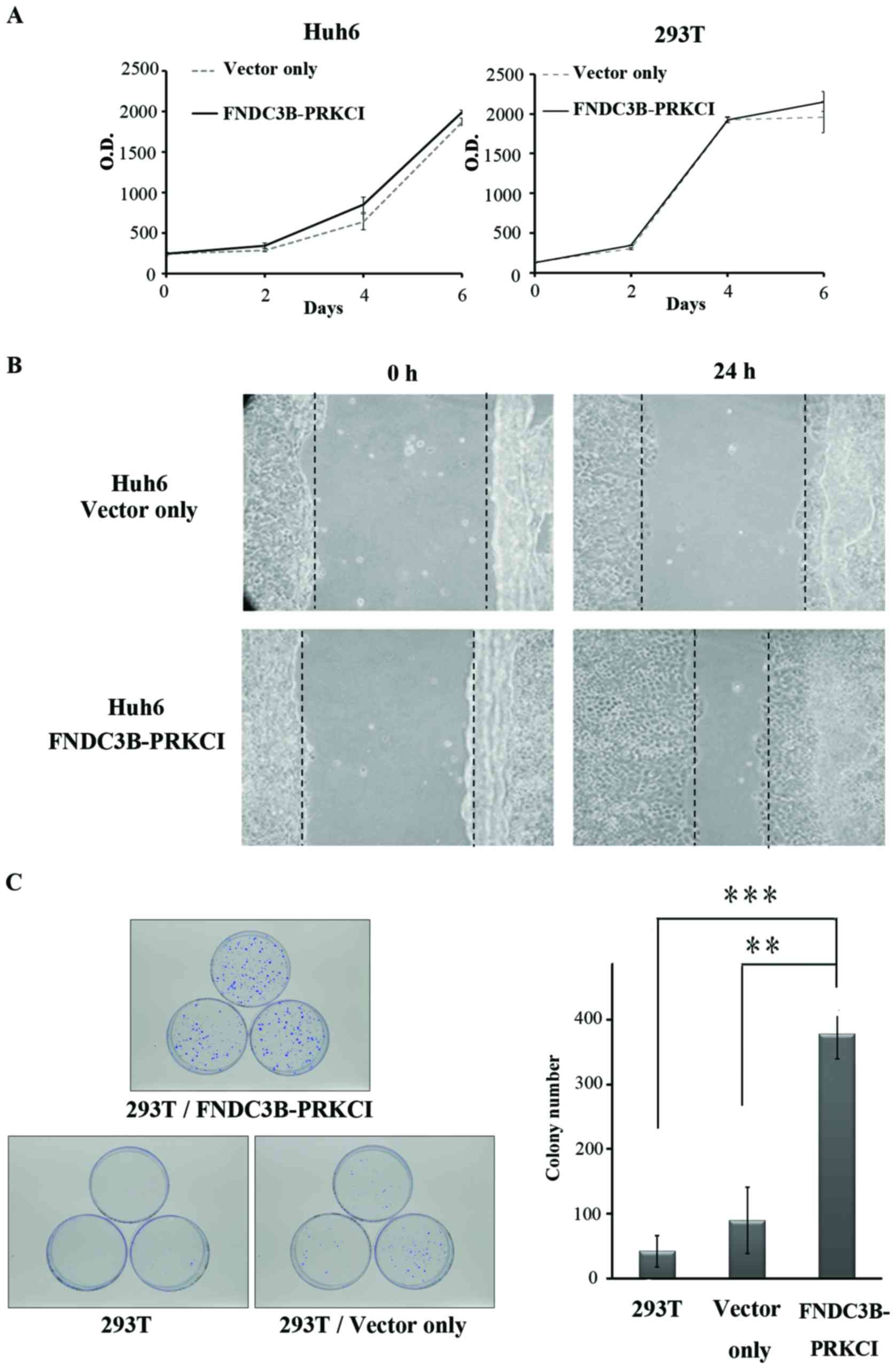
Huh6 cells were transfected with the fusion gene
cloned into an expression vector to explore the oncogenic
properties of FNDC3B-PRKCI. The 293T cell line was selected
to express the fusion protein since it was deficient in FNDC3B
(data not shown). FNDC3B-PRKCI did not exert any significant effect
on either Huh6 or 293T cells in regards to cell growth (Fig. 4A). However, overexpression of
FNDC3B-PRKCI significantly enhanced cell migration (Fig. 4B) and increased the number of
colonies as revealed in the anchorage-independent growth assay
(Fig. 4C). These results indicate
that FNDC3B-PRKCI is a potential oncogene in HCC.
Discussion
After predicting chromosome breakpoints and
performing genomic PCR and sequencing analyses, we suggest a
potential chromosome intra-rearrangement model at chromosome 3q26
in Hep3B cells. A chimeric transcript was observed by 3′-RACE
analysis and the FNDC3B-PRKCI in-frame fusion protein was detected
by western blotting. Finally, results of cell migration and colony
formation assays suggested that FNDC3B-PRKCI is a potential
oncogene.
Fusion genes created by joining two functional
fragments from different genes can provide a handy genetic window
for dissecting novel pathways involved in tumorigenesis. Moreover,
they can be used as diagnostic markers for cancer and targeted
therapy. However, it is difficult to explore fusion genes and
chromosome breakpoints in solid tumors. One major reason is that
solid tumors are commonly contaminated with normal or pre-malignant
tissues (3). For purity and
convenience, searching for fusion candidates in tumor cell lines
and confirming their presence in tumor samples using
high-throughput screening is a better solution (3). For example, recurrence breakpoints for
NRG1 were first found in breast and prostate cancer cell lines
(11). Then, they were discovered
in breast cancer samples at a follow-up screening using tissue
assays and paraffin-embedded tissue samples (12,13).
Gene fusion is caused by complex and small chromosomal
rearrangements such as duplications, deletions and insertions
(3). These small rearrangements
cannot be detected by traditional low resolution cytogenetic
methods, but are detectable by new high-throughput sequencing or
array platforms. Therefore, several new fusion genes in solid
tumors have recently been reported. For example, the first fusion
gene in HCC, ABCB11-LRP2, was detected by whole-genome sequencing
(14). We found genomic breakpoints
by applying high-resolution arrays and predicted a 2-Mb segment
duplication model in chromosome 3q26. Notably, the same genomic
breakpoints occurred in at least 3 HCC patients by analysis data
from TCGA HCC patients (data not shown).
FNDC3B is also named factor for adipocyte
differentiation 104 (FAD104) for it has been identified as a
regulator of differentiation in adipocytes and osteoblasts
(15,16). This gene is commonly overexpressed
in many types of cancer (17), but
its biological functions remain largely unknown. FNDC3B is mainly
composed of nine FNIII domains and one transmembrane domain. In
terms of function, most of the FNIII domains are commonly involved
in cell adhesion and growth signaling (17,18).
One study suggested that FNDC3B is involved in
epithelial-mesenchymal transition (EMT) (19), a cellular process in which
epithelial cells lose their polarized organization and acquire
mesenchymal characteristics. EMT is a critical process for the
conversion of early-stage tumors into invasive malignancies
(20,21). In our experiments, FNDC3B-PRKCI
enhanced cell migration and colony formation (Fig. 4B and C) suggesting that the first
two FNIII domains in FNDC3B are important for the EMT process.
Protein kinase C (PKC) is a family of
serine/threonine protein kinases. PRKCI belongs to an
atypical PKC subgroup in which catalytic activity is not dependent
on diacylglycerol, calcium or phosphatidylserine (22). Accumulating evidence indicates that
PRKCI is an oncogene and a prognostic marker in many human
types of cancer, such as lung, ovarian, prostate, gastric, breast
and liver cancer (23–28). PRKCI is required for
Ras-mediated transformation and it participates in multiple aspects
of the transformed phenotype including growth, invasion and
survival (29,30). For example, PRKCI-mediated
phosphorylation of IκK leads to activation of the canonical NF-κB
pathway and cell survival in prostate cancer (31). The presence of a complete
serine-threonine kinase domain in FNDC3B-PRKCI suggests that the
chimeric protein can transduce signals via phosphorylation.
However, further kinase activity and protein-protein interaction
studies are required to understand the detailed molecular mechanism
of FNDC3B-PRKCI in tumorigenesis.
In the present study, we combined genomic and
proteomic approaches to identify a novel fusion gene at the DNA,
RNA and protein levels. Functional-assay results suggest that
FNDC3B-PRKCI is a potential oncogene. In addition, a
specific antibody for FNDC3B-PRKCI was developed, which
could be beneficial for further patient screening.
Acknowledgements
The present study was funded by grants from the
Ministry of Science and Technology, Taiwan (NSC
101-2320-B-010-066-MY3, MOST 104-2314-B-532-005- and MOST
105-2319-B-010-001), and the National Yang-Ming University, Taiwan
(Ministry of Education, Aim for the Top University Plan). The
authors acknowledge the technical services provided by the
High-Throughput Genome Analysis Core Facility of VYM Genome
Research Center (VYMGC), National Yang-Ming University. The Core
Facility was supported by the National Core Facility Program for
Biotechnology (NCFPB), Ministry of Science and Technology.
Glossary
Abbreviations
Abbreviations:
|
HD
|
homozygous deletion
|
|
SNP
|
single nucleotide polymorphism
|
|
FNDC3B
|
fibronectin type III domain containing
3B
|
|
PRKCI
|
protein kinase C iota
|
|
HCC
|
hepatocellular carcinoma
|
|
RACE
|
rapid amplification of cDNA ends
|
References
|
1
|
Kaye FJ: Mutation-associated fusion cancer
genes in solid tumors. Mol Cancer Ther. 8:1399–1408. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prensner JR and Chinnaiyan AM: Oncogenic
gene fusions in epithelial carcinomas. Curr Opin Genet Dev.
19:82–91. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Edwards PA: Fusion genes and chromosome
translocations in the common epithelial cancers. J Pathol.
220:244–254. 2010.PubMed/NCBI
|
|
4
|
Licht JD: Acute promyelocytic leukemia -
weapons of mass differentiation. N Engl J Med. 360:928–930. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nambiar M, Kari V and Raghavan SC:
Chromosomal translocations in cancer. Biochim Biophys Acta.
1786:139–152. 2008.PubMed/NCBI
|
|
6
|
Heim S and Mitelman F: Molecular screening
for new fusion genes in cancer. Nat Genet. 40:685–686. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Howarth KD, Blood KA, Ng BL, Beavis JC,
Chua Y, Cooke SL, Raby S, Ichimura K, Collins VP, Carter NP, et al:
Array painting reveals a high frequency of balanced translocations
in breast cancer cell lines that break in cancer-relevant genes.
Oncogene. 27:3345–3359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maher CA, Kumar-Sinha C, Cao X,
Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N
and Chinnaiyan AM: Transcriptome sequencing to detect gene fusions
in cancer. Nature. 458:97–101. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen CF, Hsu EC, Lin KT, Tu PH, Chang HW,
Lin CH, Chen YJ, Gu DL, Lin CH, Wu JY, et al: Overlapping
high-resolution copy number alterations in cancer genomes
identified putative cancer genes in hepatocellular carcinoma.
Hepatology. 52:1690–1701. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang F, Denison S, Lai JP, Philips LA,
Montoya D, Kock N, Schüle B, Klein C, Shridhar V, Roberts LR, et
al: Parkin gene alterations in hepatocellular carcinoma. Genes
Chromosomes Cancer. 40:85–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adélaïde J, Huang HE, Murati A, Alsop AE,
Orsetti B, Mozziconacci MJ, Popovici C, Ginestier C, Letessier A,
Basset C, et al: A recurrent chromosome translocation breakpoint in
breast and pancreatic cancer cell lines targets the
neuregulin/NRG1 gene. Genes Chromosomes Cancer. 37:333–345.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang HE, Chin SF, Ginestier C, Bardou VJ,
Adélaïde J, Iyer NG, Garcia MJ, Pole JC, Callagy GM, Hewitt SM, et
al: A recurrent chromosome breakpoint in breast cancer at the
NRG1/neuregulin 1/heregulin gene. Cancer Res. 64:6840–6844.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Prentice LM, Shadeo A, Lestou VS, Miller
MA, deLeeuw RJ, Makretsov N, Turbin D, Brown LA, Macpherson N,
Yorida E, et al: NRG1 gene rearrangements in clinical breast
cancer: Identification of an adjacent novel amplicon associated
with poor prognosis. Oncogene. 24:7281–7289. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fernandez-Banet J, Lee NP, Chan KT, Gao H,
Liu X, Sung WK, Tan W, Fan ST, Poon RT, Li S, et al: Decoding
complex patterns of genomic rearrangement in hepatocellular
carcinoma. Genomics. 103:189–203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tominaga K, Kondo C, Johmura Y, Nishizuka
M and Imagawa M: The novel gene fad104, containing a
fibronectin type III domain, has a significant role in
adipogenesis. FEBS Lett. 577:49–54. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kishimoto K, Kato A, Osada S, Nishizuka M
and Imagawa M: Fad104, a positive regulator of adipogenesis,
negatively regulates osteoblast differentiation. Biochem Biophys
Res Commun. 397:187–191. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin F, Ren XD, Pan Z, Macri L, Zong WX,
Tonnesen MG, Rafailovich M, Bar-Sagi D and Clark RA: Fibronectin
growth factor-binding domains are required for fibroblast survival.
J Invest Dermatol. 131:84–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Obara M, Sakuma T and Fujikawa K: The
third type III module of human fibronectin mediates cell adhesion
and migration. J Biochem. 147:327–335. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai C, Rajaram M, Zhou X, Liu Q, Marchica
J, Li J and Powers RS: Activation of multiple cancer pathways and
tumor maintenance function of the 3q amplified oncogene
FNDC3B. Cell Cycle. 11:1773–1781. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pinzani M: Epithelial-mesenchymal
transition in chronic liver disease: Fibrogenesis or escape from
death? J Hepatol. 55:459–465. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Steinberg SF: Structural basis of protein
kinase C isoform function. Physiol Rev. 88:1341–1378. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Win HY and Acevedo-Duncan M: Role of
protein kinase C-iota in transformed non-malignant RWPE-1 cells and
androgen-independent prostate carcinoma DU-145 cells. Cell Prolif.
42:182–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takagawa R, Akimoto K, Ichikawa Y, Akiyama
H, Kojima Y, Ishiguro H, Inayama Y, Aoki I, Kunisaki C, Endo I, et
al: High expression of atypical protein kinase C lambda/iota in
gastric cancer as a prognostic factor for recurrence. Ann Surg
Oncol. 17:81–88. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kikuchi K, Soundararajan A, Zarzabal LA,
Weems CR, Nelon LD, Hampton ST, Michalek JE, Rubin BP, Fields AP
and Keller C: Protein kinase C iota as a therapeutic target in
alveolar rhabdomyosarcoma. Oncogene. 32:286–295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Justilien V, Walsh MP, Ali SA, Thompson
EA, Murray NR and Fields AP: The PRKCI and SOX2
oncogenes are coamplified and cooperate to activate Hedgehog
signaling in lung squamous cell carcinoma. Cancer Cell. 25:139–151.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Haverty PM, Hon LS, Kaminker JS, Chant J
and Zhang Z: High-resolution analysis of copy number alterations
and associated expression changes in ovarian tumors. BMC Med
Genomics. 2:212009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Du GS, Wang JM, Lu JX, Li Q, Ma CQ, Du JT
and Zou SQ: Expression of P-aPKC-iota, E-cadherin, and beta-catenin
related to invasion and metastasis in hepatocellular carcinoma. Ann
Surg Oncol. 16:1578–1586. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fields AP and Regala RP: Protein kinase C
iota: Ηuman oncogene, prognostic marker and therapeutic target.
Pharmacol Res. 55:487–497. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Murray NR, Kalari KR and Fields AP:
Protein kinase Cι expression and oncogenic signaling mechanisms in
cancer. J Cell Physiol. 226:879–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Win HY and Acevedo-Duncan M: Atypical
protein kinase C phosphorylates IKKalphabeta in transformed
non-malignant and malignant prostate cell survival. Cancer Lett.
270:302–311. 2008. View Article : Google Scholar : PubMed/NCBI
|