Introduction
Excessive ROS production leads to oxidative stress,
which is known to contribute to the pathogenesis of several
diseases including inflammation (1), diabetes and obesity (2) and to play a role in carcinogenesis
(3,4). In physiological conditions, ROS
production is involved in many cell functions such as signalling
(5), and growth (4). Excluding the mitochondria, cellular
ROS production involves two other systems (6). The first is NADPH oxidases (NOX), the
main driver of cellular ROS production in physiological (7) and pathological processes (8). The NOX family counts seven NOX
isoenzymes, i.e. NOX1 to 5 plus dual oxidases (DUOX)1 and 2, some
of which are expressed in cancer cells (9–11). The
second ROS-producing system in cells are the NO synthases (NOS)
(12,13). Three NOS isoenzymes are
differentiated on tissue expression patterns (12,14,15).
NO production plays a role in cell signalling (14), vascular relaxation (12), and immunity (15). However, the inducible isoform of NOS
(iNOS) appears to play a role in metastasis, especially in breast
cancer (16). NOS and NOX products
can interact, and this mechanism is a powerful regulatory modulator
of their activity (17).
Leptin has been widely studied as one of many
different adipokines that are modulated during obesity (18). Leptin is upregulated in obesity
where it is involved in critical steps of cell homeostasis from
cell growth (19) to metabolism
(20) and energy production
(21). A crosslink between ROS
production and leptin signalling has been reported in several
tissues including endothelium (22)
and ovaries (23), and could be one
of the signaling pathways activated through the leptin receptor
OB-R in the presence of leptin (24). However, there is surprisingly little
data on how leptin affects oxidative stress, especially in cancer
(25). Several studies have
investigated the impact of leptin on different steps in breast
carcinogenesis (26) from cell
proliferation (27,28), inflammatory response (29) and modulation of the cell
microenvironment (30) to cell
signalling pathways (27,31), but few of them have focused on
intracellular ROS production (32,33).
ROS production is involved in the pathogenesis of
both obesity (2) and cancer
(3) while leptin plays a well-known
role in breast carcinogenesis (34). Yet very few studies have assessed
the ability of adipokines to modulate ROS production (35) via NOX and NOS enzymes in cancer
cells.
Here we attempted to determine whether leptin,
mimicking normal and obesity situations, modulates the activity and
gene expression of NOS and NOX enzymes and whether ROS-producing
enzymes are healthy-state or metastatic neoplastic-dependent.
Regarding literature, plasma leptin concentrations were defined
around 10 to 30 ng/ml and 50 to 150 ng/ml respectively for a lean
and an obese adult woman (36).
Thus, we chose leptin doses at 10 ng/ml for physiological and 100
ng/ml for obese conditions, which are also relevant to tissue
concentrations (30). We used
healthy (HMEC) and neoplastic (MCF-7 and MDA-MB-231) mammary
epithelial cells that express the leptin receptor (Ob-R) (37) and are sensitive to leptin signalling
(28) to assess and characterize
cellular ROS production under leptin via different fluorescent
probes (38). We then determined
whether this ROS production could be due to NOX or NOS enzymes.
Materials and methods
Cell culture
Healthy human mammary epithelial cells (HMEC)
obtained from 55-year-old Caucasian women (Lonza, Basel,
Switzerland) were grown in complete MEBM medium supplemented with
hydrocortisone (0.5 µg/m1), epithelial growth factor (10 ng/ml),
insulin (5 µg/ml), gentamicin (50 µg/ml)/amphotericin-B (50 ng/ml)
and bovine pituitary extract (0.4%) as recommended by the
manufacturer (Lonza). MCF-7 and MDA-MB-231 neoplastic human mammary
epithelial cells obtained from 69- and 51-year-old Caucasian women,
respectively (ATCC, Molsheim, France), were grown in RPMI-1640
medium (Biowest, Nuaillé, France) containing 10% fetal calf serum,
L-glutamine (2 mM), penicillin (50 IU/ml) and streptomycin (50
µg/ml) (Sigma-Aldrich, Saint-Quentin-Fallavier, France). Culture
was carried out at 37°C in a humidified atmosphere with 5%
CO2 until passage 15. For the experiments, the mammary
epithelial cells were cultured at a density of 22,500
cells/cm2.
Treatment with leptin
Mammary epithelial cells were synchronized in
serum-free medium for 24 h before initiation of leptin treatment.
Cells (HMEC, MCF7 and MDA-MB-231) were grown for 0–2 h in their
media either with or without recombinant human leptin (R&D
Systems, Abingdon, UK) at physiological (10 ng/ml) or obese (100
ng/ml) concentrations. Cells were harvested after trypsinization.
Total cell lysates were obtained by two successive thawing-freezing
cycles in Tris HCl 25 mM buffer pH 7.4 containing Tween-20 0.1%
(Sigma-Aldrich), with 15-sec periods in an ultra-sound bath, and
then stored at −80°C until analysis.
RNA isolation and reverse
transcription
After treatment with leptin, total RNA was isolated
from the epithelial cells by TRIzol® reagent
(Invitrogen, Saint Aubin, France) according to the manufacturer's
protocol, and quantified using a NanoDrop spectrophotometer
(NanoDrop®2000, Thermo Scientific, Waltham, MA, USA).
Reverse transcription was performed in a thermocycler
(Mastercycler® gradient; Eppendorf, Montesson, France)
on 1 µg of total RNA for each condition using a high-capacity cDNA
reverse transcription kit (Applied Biosystems, Saint Aubin, France)
with random hexamer pdN6 primers.
Quantitative real-time PCR
(q-PCR)
q-PCR was performed using SYBR®Green
reagents according to the manufacturer's instructions on a StepOne
system (Applied Biosystems). Each condition was assayed in
triplicate. Relative quantification was obtained by the comparative
Cq method, based on the formula 2−∆∆Cq (39). Expression levels were normalized to
the housekeeping gene (β-actin) for each time point and expressed
as fold-change from the basal level corresponding to untreated
cells at time 0. Sequences and fragment sizes of the human-specific
primers used are reported in Table
I.
 | Table I.Summary of PCR primers. |
Table I.
Summary of PCR primers.
| Gene name | Accession no. | Primer
sequences | Amplicon length
(bp) |
|---|
| NOX 1 | AJ_438989 | F:
5′-TCG-ACC-ACC-AAT-ATT-CAC-CA-3′ | 225 |
|
|
| R:
5′-TGG-CCT-TGT-CAA-AGT-TTA-AT-3′ |
| NOX 2 | NM_000397 | F:
5′-AGA-GTT-CGA-AGA-CAA-CTG-GA-3′ | 233 |
|
|
| R:
5′-CCT-CCT-TCA-GGG-TTC-TTT-AT-3′ |
|
| NOX 3 | NM_015718 | F:
5′-CAA-ACA-CAA-CCA-CTG-AAT-TG-3′ | 231 |
|
|
| R:
5′-TGT-TGT-GCA-GAG-AGA-GAC-TG-3′ |
|
| NOX 4 | NM_016931 | F:
5′-CTT-TTG-GAA-GTC-CAT-TTG-AG-3′ | 231 |
|
|
| R:
5′-ATC-AAG-CGG-CCC-CCT-TTT-TTC-AC-3′ |
|
| NOX 5 | NT_010194 | F:
5′-CTC-ATT-CTC-ACA-CTC-CTC-GAC-AGC-3′ | 238 |
|
|
| R:
5′-GTC-TGT-TCT-CTT-GCC-AAA-AC-3′ |
|
| DUOX1 | NP_059130 | F:
5′-GCA-GGA-CAT-CAA-CCC-TGC-ACT-CTC-3′ | 348 |
|
|
| R:
5′-CTG-CCA-TCT-ACC-ACA-CGG-ATC-TGC-3′ |
|
| DUOX2 | NP_054799 | F:
5′-GAT-GGT-GAC-CGC-TAC-TGG-TT-3′ | 303 |
|
|
| R:
5′-GCC-ACC-ACT-CCA-GAG-AGA-AG-3′ |
|
| iNOS | NP_000616 | F:
5′-AGC-ATG-TAC-CCT-CGG-TTC-TG-3′ | 250 |
|
|
| R:
5′-GGG-GAT-CTG-AAT-GTG-CTG-TT-3′ |
|
| eNOS | NP_000594 | F:
5′-GAA-GAG-GAA-GGA-GTC-CAG-TAA-CA-3′ | 438 |
|
|
| R:
5′-GGA-CTT-GCT-GCT-TTG-CAG-GTT-TTC-3′ |
|
| β-actin | NM_001101 | F:
5′-TCG-TGC-GTG-ACA-TTA-AGG-AG-3′ | 262 |
|
|
| R:
5′-AGC-ACT-GTG-TTG-GCG-TAC-AG-3′ |
|
| 18S | NR_003286 | F:
5′-GTC-TGT-GAT-GCC-CTT-AGA-3′ | 177 |
|
|
| R:
5′-AGC-TTA-TGA-CCC-GCA-CTT-AC-3′ |
|
Reactive oxygen species (ROS)
production
ROS production was measured by fluorescence using a
microplate reader (Fluoroscan Ascent Microplate
Fluorometer®, Thermo Scientific) for extracellular
production and by flow cytometry for intracellular production (FC
500 MPL, Beckman Coulter, Villepinte, France). The probes used
(FluoProbes®, Interchim, Montluçon, France) are given in
Table II. Total cellular ROS
production was determined by dichlorofluorescein (DCF). Cytosolic
superoxide anion (O2•−) was assayed using
dihydroethidine (DHE). Mitochondrial ROS production was assayed
using dihydrorhodamine (DHR). Nitric oxide and peroxynitrite
production was assayed using diaminofluorescein (DAF). Probe stock
solutions (20 mM solution in DMSO) were stored under nitrogen at
−80°C until analysis. Probes were used at 2 µM final concentration
in cell media. Probe oxidation was measured at a wavelength of 488
nm for excitation and 520 nm for emission. Auto-oxidation of probe
was subtracted. Results were normalized to the basal fluorescence
signal without leptin and expressed in relative fluorescence units
(RFU).
| Table II.Fluorescence probes. |
Table II.
Fluorescence probes.
|
|
| Fluorescent
probes |
|---|
|
|
|
|
|---|
| Species
detected | Symbol | DCF | DHE | DHR | DAF | Fluo-3 |
|---|
| Cytosolic
superoxide anion |
O2•− | X | X |
|
|
|
| Mitochondrial
superoxide anion |
O2•− | X |
| X |
|
|
| Hydrogen
peroxide |
H2O2 | X |
| X |
|
|
| Hydroxyl
radical | HO· | X |
|
|
|
|
| Nitric oxide | NO· | X |
|
| X |
|
| Peroxynitrite |
ONOO− | X |
|
| X |
|
| Calcium |
Ca2+ |
|
|
|
| X |
For extracellular production analysis, synchronized
cells were incubated for 30 min in the dark at 37°C in their media
with the appropriate probe (2 µM). After adding leptin (10 or 100
ng/ml) or medium (for control condition), cells were assayed for
fluorescence every 10 min over a 120-min window in the microplate
reader.
For intracellular production analysis, synchronized
cells were incubated in presence of leptin (10 or 100 ng/ml) or
medium (for control condition). After trypsinization, the cell
suspension loaded with the appropriate probe (2 µM) was incubated
for 30 min in the dark at 37°C then, submitted to fluorescent flow
cytometry analysis on a log scale for 10,000 events (cell
counts).
Intracellular calcium influx
To assess the calcium-dependent signalling pathway
in cell ROS production, calcium influx was measured by fluorescence
on the microplate reader using the Fluo-3 calcium probe (Interchim)
at 2 µM as previously described (40). The effects of calcium channel
blocking were measured in the presence of verapamil (20 µM)
(Sigma-Aldrich). After synchronization, the cells were incubated
for 30 min in the dark at 37°C in the appropriate growth medium
with verapamil before addition of Fluo-3 or dichlorofluorescein.
After adding leptin (10 or 100 ng/ml) or medium (for control
condition) with or without verapamil, the cells were assayed for
fluorescence every 10 min over a 120-min window. Probe oxidation
was measured at a wavelength of 488 nm for excitation and 520 nm
for emission. Results were expressed in relative fluorescence units
(RFU).
NOX catalytic activity
NOX catalytic activity was measured by fluorescence
using a microplate reader (Fluoroscan Ascent Microplate
Fluorometer, Thermo Scientific) in presence of dichlorofluorescein
(DCF) (Interchim).
Briefly, for experiments on the microplate reader:
cells were incubated in the dark at 37°C in the reagent buffer (100
mM TrisHCl, 2 mM MgCl2, 0.5% Tween-20, 100 mM NADPH, 10
µM DCF, pH 7.4) (Sigma-Aldrich) in the presence or not of leptin at
10 or 100 ng/ml. Plates were assayed for fluorescence every 60 sec
over a 10-min window. Stimulation of the fluorescent signal was
obtained by the addition of calcium chloride (2 mM) in the reagent
buffer. NOX inhibition was assayed by the addition of
diphenyliodonium (DPI) (40 µM) to inhibit FAD enzymes or apocynin
(ApoC) (40 µM) to inhibit NOX 1, 2 and 4 (Sigma-Aldrich), as
previously described (41). Results
were expressed in relative catalytic units normalized to the
protein content of the cell.
NOX5 silencing
To confirm the involvement of NOX5 in ROS
production, its expression was inhibited via siRNA. We used
21-nucleotide double-stranded siRNAs (5 nM) (Qiagen HP genome wide
siRNA databank, forward 5′-GCC CUA UUU GAC UCC GAU ATT-3′, reverse
5′-UAU CGG AGU CAA AUA GGG CAA-3′) targeting the NOX5 mRNA
sequence. Non-silencing RNA (5 nM) was used as negative siRNA
control (scrambled siRNA).
The cells were siRNA-transfected as per the
manufacturer's protocol (Qiagen, Courtaboeuf, France). Briefly,
22,500/cm2 cells were cultured in 6-well plates. After
incubation with siRNA for 1 h, the wells were completed with the
serum-free medium for 24 h. Synchronized cells were assayed for ROS
production by fluorescence using a microplate reader for 120 min as
described above, either with or without 10 or 100 ng/ml recombinant
human leptin. Cells were then harvested and quantitative RT-PCR was
performed to compare NOX5 mRNA silencing in the presence and
absence of siRNA.
Statistical analysis
Each experiment was performed in triplicate and the
average value was treated as a single data point. Statistical
analyses were performed using GraphPad Prism5 (GraphPad Software,
Inc., La Jolla, CA, USA). Data are expressed as means ± standard
deviation. Values for fluorescence measurements are expressed as a
ratio of the control at baseline. Between-group comparisons were
performed by one-way ANOVA followed by a Bonferroni multiple
comparisons test. Level of significance was set at 0.05.
Significance is indicated by different subscript letters or flagged
as p<0.05, p<0.01 and p<0.001.
Results
Basal expression of ROS-producing
enzymes and basal ROS production
ROS-producing enzymes of mammary epithelial cells
were characterized in our culture cell conditions without leptin by
q-PCR using previously-described primers (Table I) (42,43).
Of the different ROS-producing systems studied, i.e. NADPH oxidase
(NOX) isoenzymes 1, 2, 4 and 5, dual oxidase (DUOX) isoenzymes 1
and 2, and the endothelial and inducible nitric oxide synthase
isoforms (eNOS and iNOS), only NADPH oxidase 5 (NOX5) was expressed
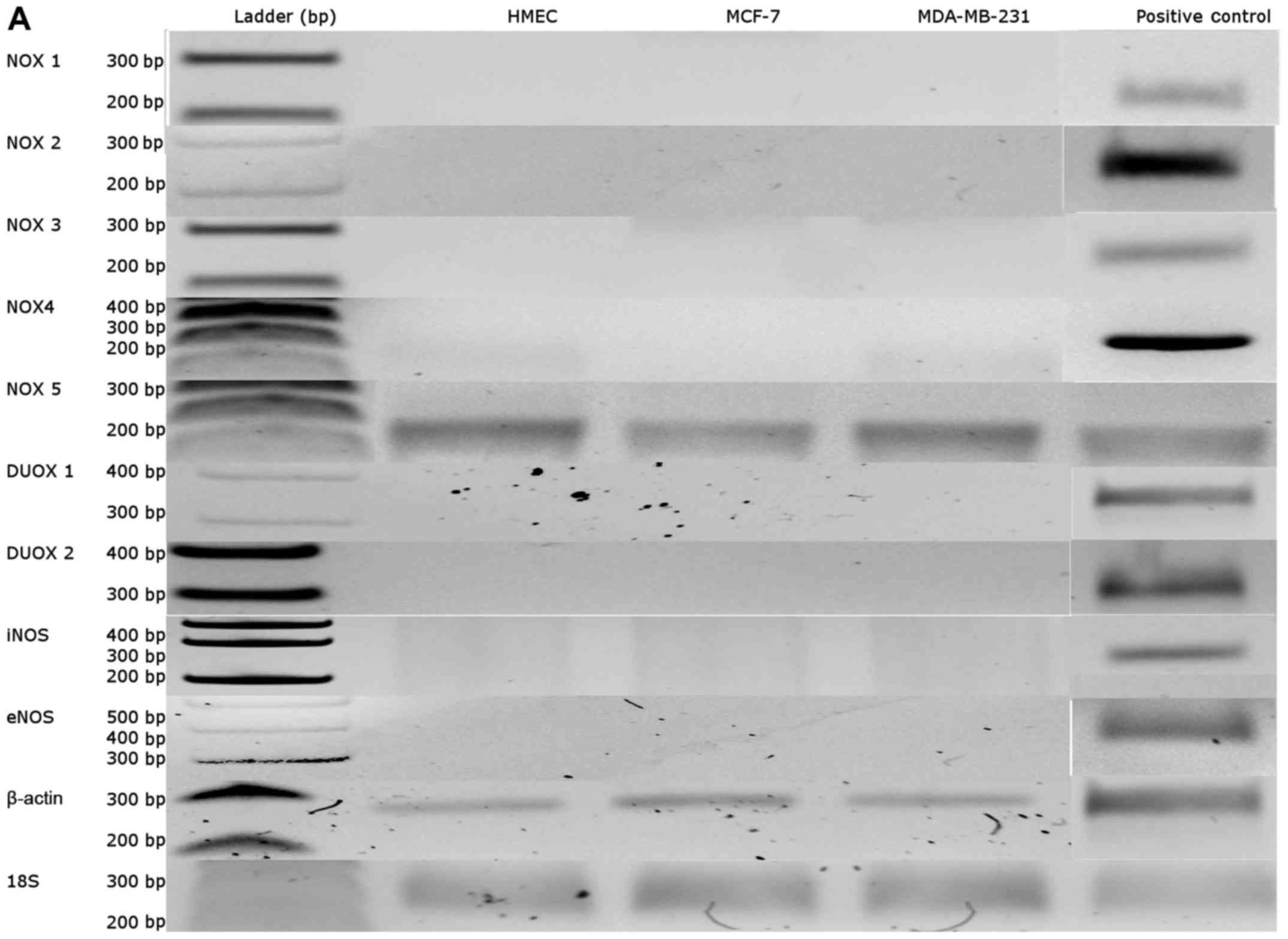
in our cell models (Fig. 1A).
NOX5 mRNA expression was 6-fold higher in neoplastic cells
than in healthy cells (p<0.05, Fig.
1B).
Relative catalytic activity assayed by fluorescence
in presence of NADPH and calcium showed similar profiles but was
higher in neoplastic cells than HMEC (p<0.05, Fig. 1C). This observation was related to
the difference in NOX5 expression. However, the basal
cytosolic superoxide anion production was higher in neoplastic
cells than in healthy cells (p<0.05, Fig. 1D). Basal total cellular ROS
production (without leptin) assayed with DCF was similar in the
HMEC and neoplastic cell models (Fig.
1E).
Leptin-induced ROS production in human
mammary epithelial cell lines
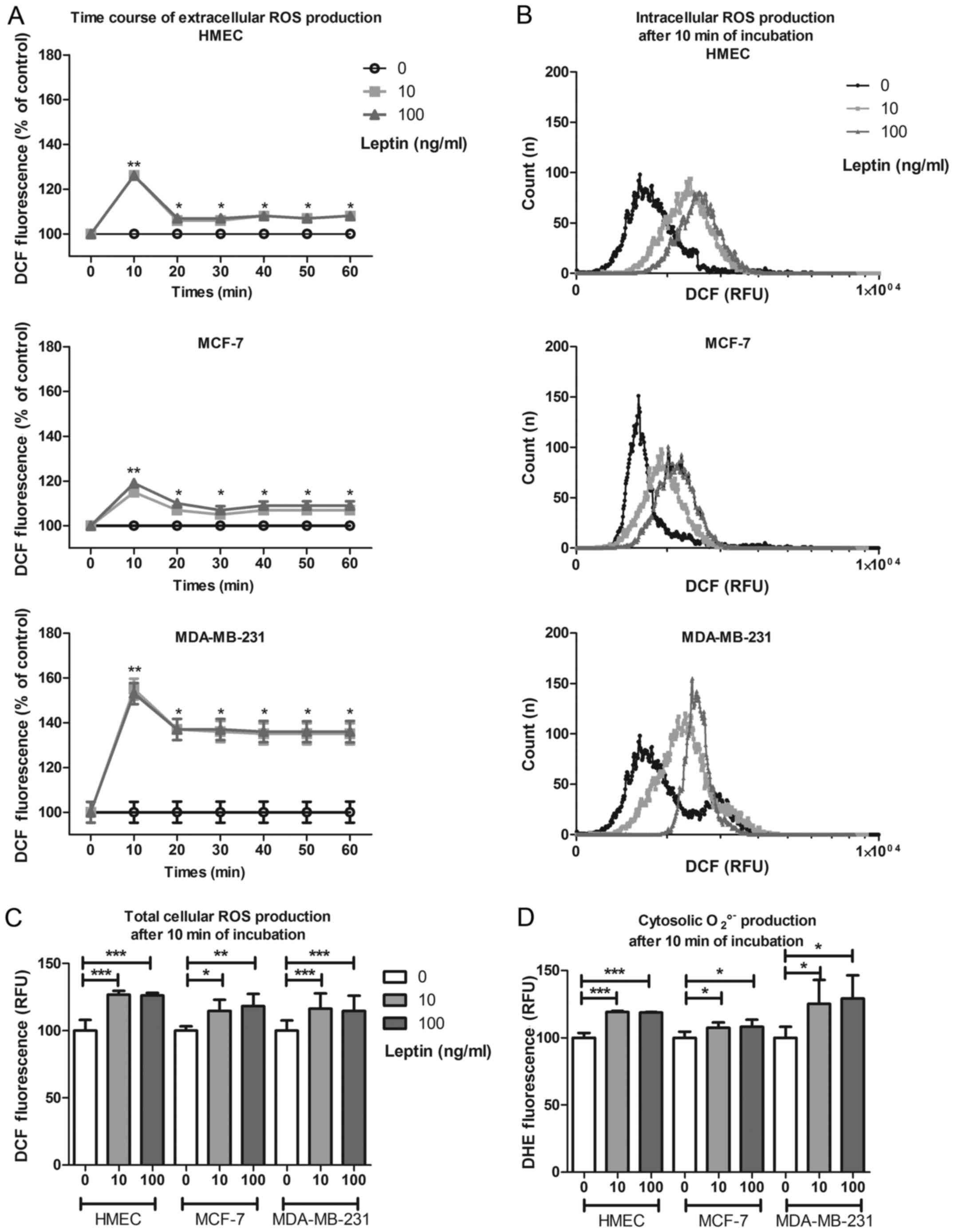
The time-course of total cellular ROS production
(DCF) in the culture medium in the presence of leptin showed an
increase at 10 min, irrespective of cell model or leptin dose,
(126±4%, 115±2% and 155±19% of basal production for HMEC, MCF-7 and
MDA-MB-231, respectively; p<0.05, Fig. 2A). This short-term increase was
followed by mid-term production for at least 1 h at 108% for HMEC
and MCF-7 and at 135% for MDA-MB-231. This same increase in ROS
production was confirmed by flow cytometry in each cell type
(Fig. 2B). In presence of leptin,
and whatever the cell model, total cellular ROS production
significantly increased at 10 min (p<0.05, Fig. 2C) without any difference between the
two concentrations used. Cytosolic superoxide anion production
assayed by DHE increased at 10 min in the presence of leptin (119,
108 and 125% of basal production for HMEC, MCF-7 and MDA-MB-231,
respectively; p<0.05, Fig.
2D).
NOX isoenzyme involvement evaluated by
inhibition with DPI and ApoC
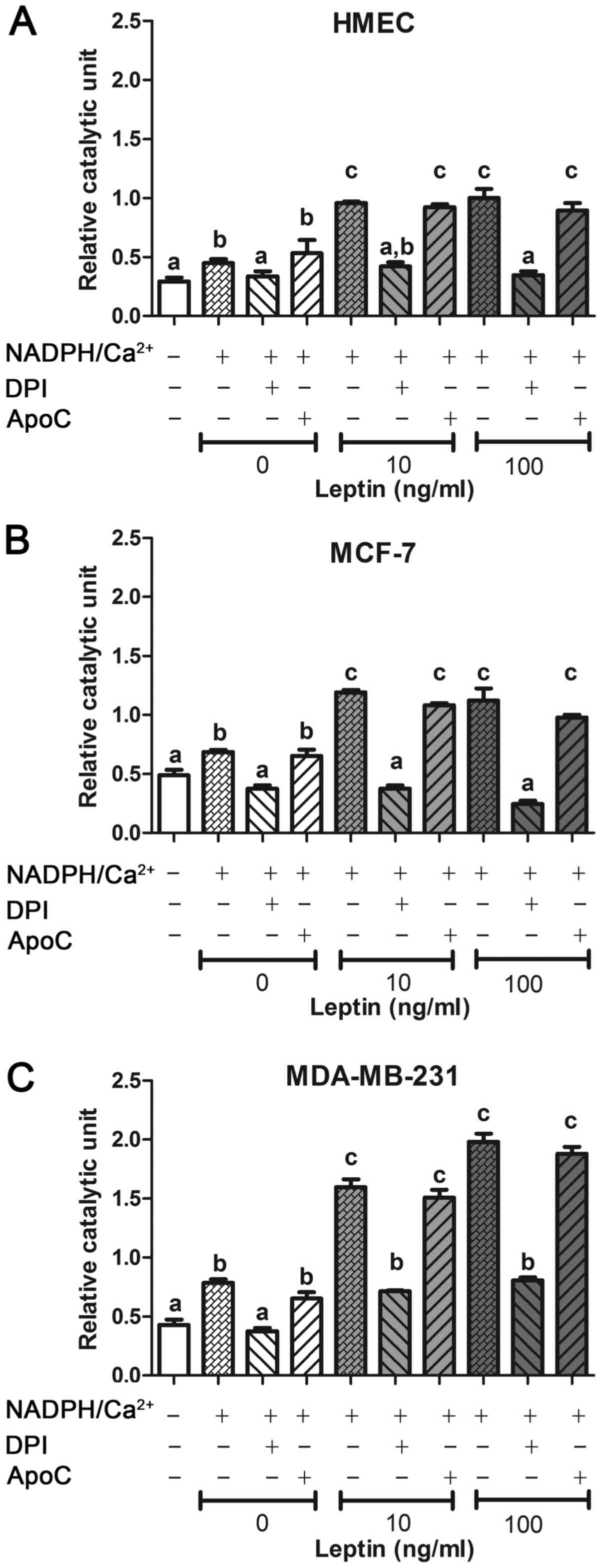
The putative implication of a NOX enzyme was assayed
on cells in the presence of two well-known NOX inhibitors: DPI and
ApoC (41). For each cell line,
after incubation with leptin, DCF fluorescence signal increased in
the presence of NADPH and calcium and was greater in neoplastic
cells than in HMEC (p<0.05, Fig.
3). In presence of NOX inhibitor DPI (which is efficient on all
the isoenzymes as a FAD inhibitor), the fluorescence signal was
totally inhibited only with DPI and was not different from the
blank signal (Fig. 3). In the
presence of NOX inhibitor ApoC (which is efficient on NOX1, NOX2
and NOX4) at 40 µM, relative catalytic activity showed no change in
each cell line. DPI, but not ApoC, was able to inhibit this ROS
production stimulated by NADPH and calcium. Taken together, these
results suggest that the ROS-producing system was a FAD enzyme,
which needs NADPH and calcium at the exclusion of NOX1, NOX2 or
NOX4. NOX5 should be the isoenzyme present in these cells.
NOX5 involvement in ROS production
under leptin was confirmed using siRNA
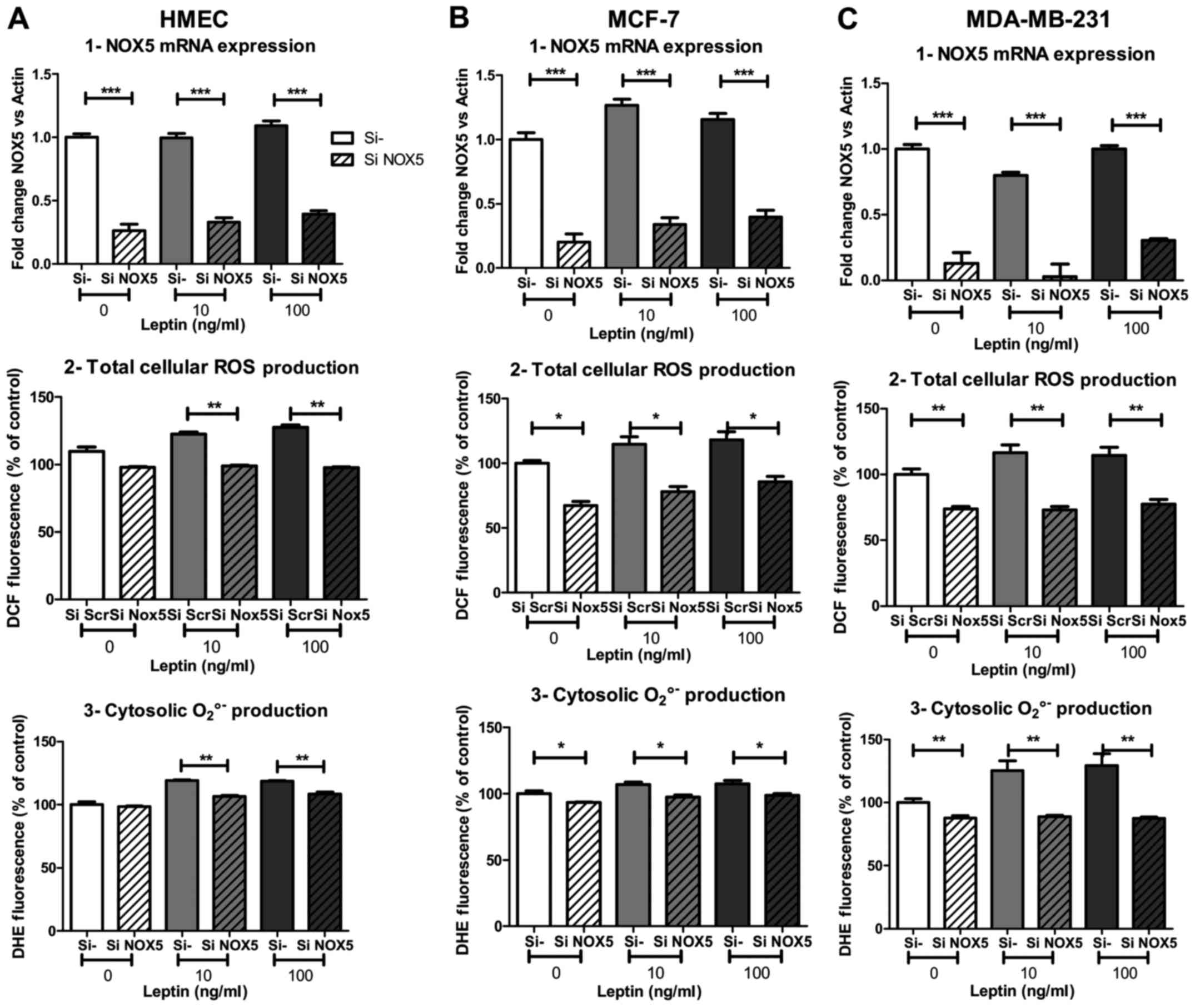
To better understand and confirm the involvement of
NOX5 in ROS production, we used siRNA against NOX5 (siNOX5)
to inhibit its expression. Twenty-four hours of NOX5 silencing
potently inhibited NOX5 mRNA expression under basal
conditions (−74, −80 and −87% of mRNA expression in HMEC, MCF-7 and
MDA-MB-231, respectively; p<0.001, Fig. 4A1, B1 and C1) and at both leptin
concentrations. Surprisingly, despite the marked inhibition of mRNA
expression, total cellular ROS production under basal conditions
was not affected by the silencing in HMEC compared to neoplastic
cells (−33 and −26% of inhibition in MCF-7 and MDA-MB-231,
respectively; p<0.05 and p<0.01 Fig. 4A2, B2 and C2). Basal cytosolic
superoxide anion production also seemed to be independent of NOX5,
as only 25% of basal signal was inhibited in neoplastic cells
(p<0.05 and p<0.01, respectively for MCF-7 and MDA-MB-231)
and no change was observed in HMEC (Fig. 4A3, B3 and C3).
However, after NOX5 silencing, ROS production failed
to increase whatever the leptin concentration and whatever the
probe used for ROS detection. Total cellular ROS production under
DCF decreased −22, −32 and −37% in HMEC, MCF-7 and MDA-MB-231,
respectively, with 10 ng/ml of leptin (p<0.01, p<0.05,
p<0.01, respectively; Fig. 4A2, B2
and C2). In the same experimental conditions, cytosolic
superoxide anion production under DHE decreased −10, −8 and −29% in
HMEC, MCF-7 and MDA-MB-231, respectively, with 10 ng/ml of leptin
(p<0.01, p<0.05, p<0.01, respectively; Fig. 4A3, B3 and C3). In the three cell
models, the increase in ROS production in the presence of leptin
was completely suppressed by NOX5 siRNA treatment.
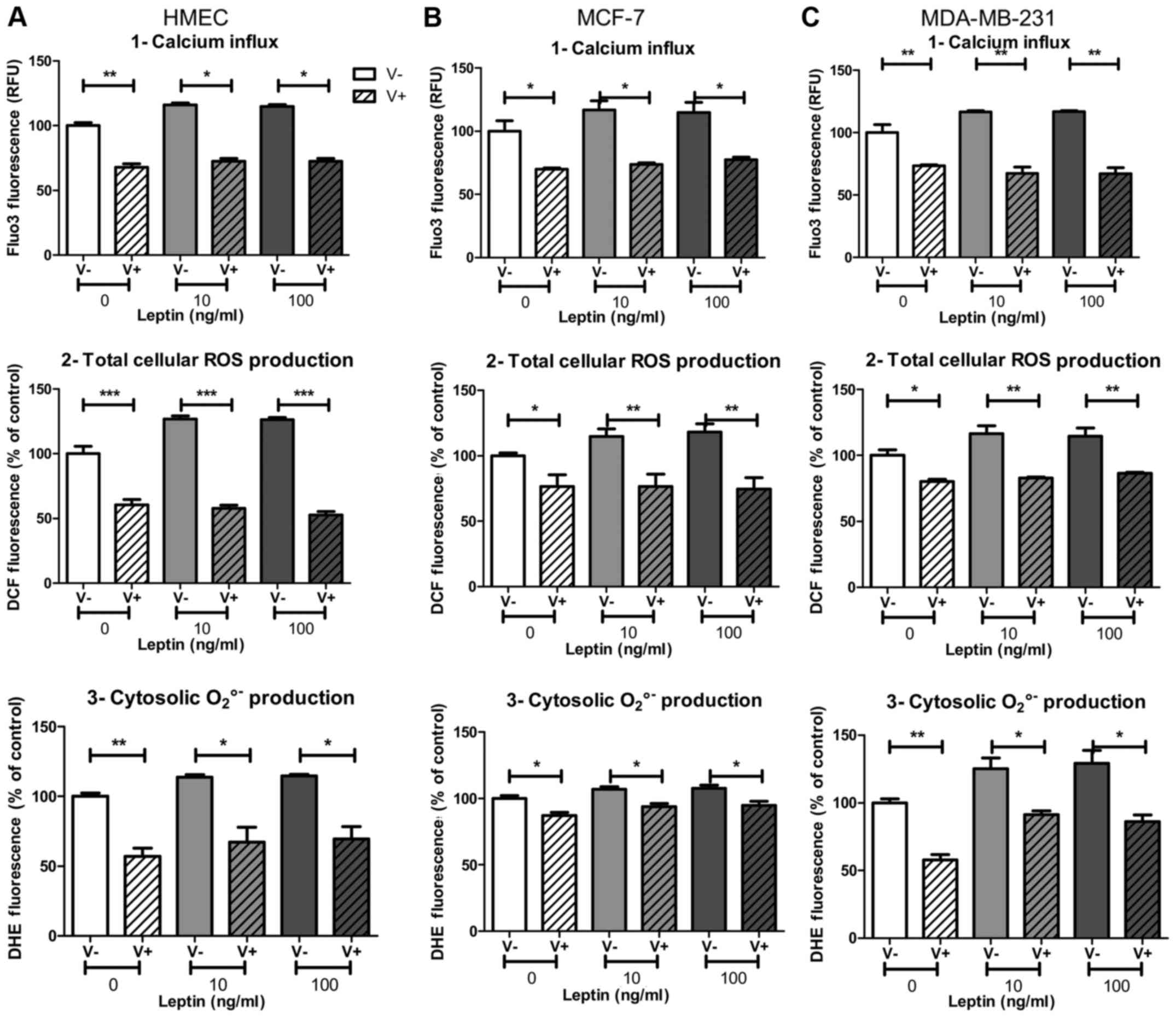
NOX5 activation by leptin was calcium
flux-dependent
Calcium influx, which is a potent NOX5 activator
(11), could be partly blocked by
the calcium channel blocker verapamil. Whatever the cell model,
leptin at both concentrations induced an increase in fluorescence
of the calcium influx probe Fluo-3 (+19, +23 and +20% in HMEC,
MCF-7 and MDA-MB-231, respectively; p<0.05, Fig. 5A1, B1 and C1) that was totally
inhibited with verapamil. All the cell models were sensitive to
verapamil, which is able to reduce calcium flux.
In the presence of verapamil, ROS production
decreased in both basal and leptin conditions. However, basal ROS
production seemed to be largely independent of calcium influx,
since at least 45% of ROS production remained in the presence of
the inhibitor verapamil. In the presence of leptin, verapamil
decreased the signal measured for both total cellular ROS
production and cytosolic superoxide anion production. In HMEC and
MCF-7 cells, verapamil treatment decreased total ROS production and
cytosolic superoxide anion production down to same level as basal
conditions (p<0.01, Fig. 5A2, B2
and p<0.05, Fig. 5A3, B3,
respectively). In MDA-MB-231 cells, verapamil treatment totally
suppressed total cellular ROS production under leptin to the same
level as basal conditions (p<0.01, Fig. 5C2), whereas cytosolic superoxide
anion production remained higher than without leptin (p<0.05,
Fig. 5C3).
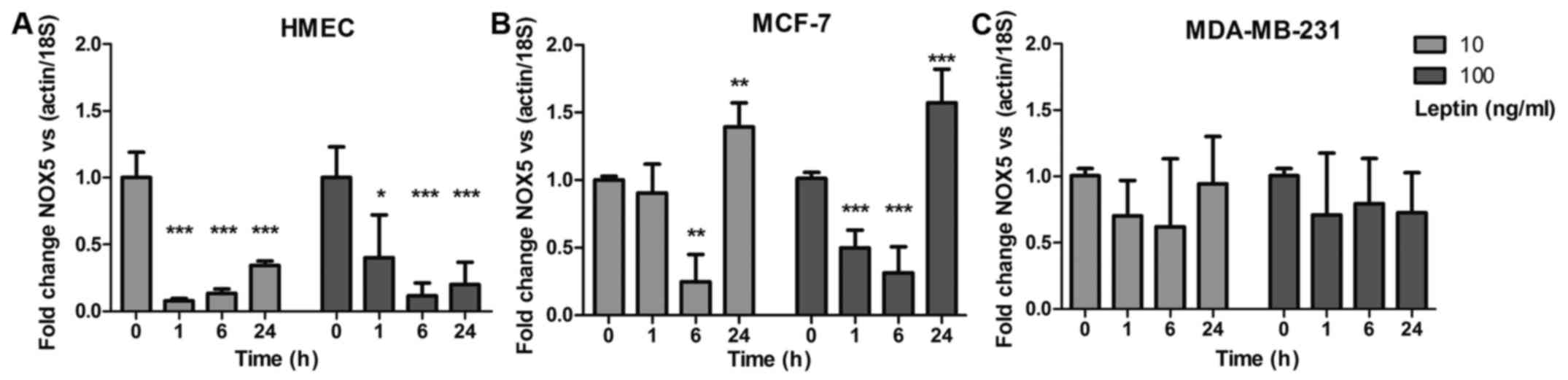
Leptin downregulates NOX5 mRNA
expression
Gene expression of the ROS production enzymes was
determined at 1, 6 and 24 h by q-PCR after addition of 10 or 100
ng/ml of leptin. Only NOX5 mRNA expression showed a
downregulation in the three cell models. In healthy HMEC,
NOX5 expression was downregulated at 1, 6 and 24 h
(p<0.001) whatever the leptin concentration, whereas neoplastic
cells showed differences (Fig. 6A).
In MCF-7 cells, NOX5 expression was weakly downregulated at
6 h (p<0.01) then increased at 24 h with both leptin
concentrations (p<0.01; Fig.
6B). In MDA-MB-231 cells, whatever the leptin concentration,
NOX5 expression showed no significant downregulation
(Fig. 6C). None of the other
NOX isoforms nor the NOS isoforms showed any
detectable expression at any point in the 24-h experiment (data not
shown).
Discussion
Leptin was able to modulate long-term ROS production
after 24 h in mammary epithelial cells by modifying the
mitochondrial function (33).
However, mammary epithelial cells contain several NOX isoenzymes
(10,44) and there is very little data on
whether leptin can modulate them. We thus focused our study on
short-term ROS production and the potent role of NOX enzymes in the
first hour of leptin stimulation at two concentrations (10 ng/ml
mimicking normal body mass index (BMI), 100 ng/ml mimicking
obesity) on three mammary cell lines. These cell lines represented
the major types of mammary epithelial cells i.e. HMEC for healthy
cells, MCF-7 for estrogen sensitive neoplastic cells and MDA-MB-231
for metastatic triple-negative cells (31). Whatever the cell line, leptin
briefly increased ROS production, which remained significantly
higher than basal level for more than 1 h. This study demonstrates
for the first time that leptin is involved in short-term ROS
production in epithelial mammary cells. The results are in
agreement with previous work linking leptin to oxidative stress in
other cell types such as endothelial cells, hepatic epithelial
cells, cardiac myofibroblasts and ovarian cells (22,23,25,35).
However, leptin did not present a difference of effect despite our
hypothesis on hyperleptinemia due to obesity. One of leptin
regulation mechanism is the internalization of the complex
leptin/receptor (26). In that
case, cells became insensitive to leptin signal until the recycling
of the receptor. As we used synchronized cells during a short time
period, this mechanism could be pre-eminent in our in vitro
experiments and could explain the lack of difference between leptin
doses.
The NOX isoenzyme family is one of the most potent
generators of ROS in cells (45).
These enzymes are clearly involved in many pathological situations
(46), especially carcinogenesis
(47). Until recently, there was
only scant data on the NOX isoenzyme family and the ROS production
system in mammary epithelial cells (9), despite the important role of NOX
isoforms as cellular signalling pathways (7). In our conditions, only NOX5
mRNA presented significant expression levels in the three cell
lines. NOX5 has been studied less than other NOX isoenzymes
(11,47) and its biological functions remain
unclear, especially in cancer (48,49).
These data confirm the expression profile previously found in
mammary tumor cell lines such as MCF-7 where NOX5 is the major NOX
isoform expressed (9) and in breast
tumors where NOX5 isoenzyme expression is positive in approximately
60% of tumors assessed (47).
However, at variance with previous papers, the other NOX isoforms
such as NOX2 (10), NOX3, or NOX4
(44), which are less expressed in
breast cancer cells than NOX5, never presented significant mRNA
expression during our short-time-window (less than 24 h)
conditions.
In order to confirm the implication of a NOX enzyme
in ROS production, we first assayed two pharmacological NOX
inhibitors (50): DPI, efficient on
all NOX isoenzymes as well as other FAD enzymes and ApoC which
interacts only with NOX1, 2 and 4 complexes (51). In our experiment, DPI but not ApoC
was able to inhibit the short-term ROS production induced by
leptin. These results argue for a FAD-dependent ROS production with
NOX1, 2 and 4 apparently ruled out. Taken together, our findings
suggest that NOX5 is likely the isoenzyme involved in this
short-term response.
Secondly, we used siRNA against NOX5 to confirm its
potent role. After 24 h of siNOX5 treatment, basal total cellular
ROS production in absence of leptin partly decreased in the
neoplastic cell lines but not in HMEC cells despite a significant
decrease in NOX5 mRNA expression. Cytosolic superoxide anion
production remained unchanged whatever the cell lines used. These
results suggest that other potential ROS sources, such as
mitochondrial oxidases (33,44) or
the respiratory chain (52), are
involved in basal ROS production in these cells, as DCF was able to
detect ROS coming from mitochondria. It could also be hypothesized
that NOX5 protein turnover was slower than the decrease in mRNA
expression, especially in HMEC cells, which presented slower
growth. However, in presence of leptin, siNOX5 treatment abolished
short-term ROS production whatever the cell line, both in terms of
total cellular ROS and cytosolic superoxide anion. These results
confirm that NOX5 is involved in part of the short-term
leptin-induced ROS production.
The NOX5 complex features two domains for calcium
binding which acts as the main regulator of its activity (11). In a final step of our experiment, we
focused on the effect of calcium flux on short-term ROS production.
In the presence of leptin, and as previously described (53,54),
we showed a calcium influx in the presence of Fluo-3 probe.
Verapamil partly inhibited the calcium influx in the three cell
lines, as Fluo-3 fluorescence only decreased by 30%. In the
presence of verapamil and leptin, total ROS production and
cytosolic superoxide anion production decreased to under basal
levels in both healthy HMEC and neoplastic MCF-7 cells. In
MDA-MB-231 cells, only total cellular ROS production under leptin
was suppressed down to basal levels by verapamil, whereas cytosolic
superoxide anion production remained higher than in the basal
condition without leptin. These data confirm that a
calcium-dependent pathway is involved in short-term ROS production
under leptin (53), which
reinforces the idea that NOX5 plays a potent role in HMEC and MCF-7
cells but suggests another source of superoxide anion in MDA-MB-231
cells. However, as no other NOX isoenzyme was expressed in this
short-time window, the identity of this other source remains
unclear.
As the main long-term regulator of NOX5 is its mRNA
expression level (49), we analyzed
NOX5 mRNA expression after leptin stimulation in the three
cell lines which showed different responses. In HMEC cells,
NOX5 mRNA expression decreased from 1 to 24 h at both
concentrations of leptin, whereas the neoplastic cells did not show
this downregulation pattern. After short decreases at 1 and 6 h, in
MCF-7 cells, NOX5 expression increased higher than the basal
level at 24 h. In MDA-MB-231 cells, NOX5 expression was not
modified during the 24 h time-window whatever the leptin
concentration. In healthy cells, a feedback loop seems to exist
when it is altered in MCF-7 cells, resulting in a higher mRNA
expression at 24 h. The metastasis of MDA-MB-231 cells lose this
feedback loop for NOX5 expression, that could be related to
their more aggressive phenotype and their constitutive oxidative
stress (55). The downregulation of
NOX5 expression under leptin appears to be less effective in
neoplastic cells and could be linked to the adverse effect of
obesity in breast cancer risk (26,29).
Herein, for the first time, leptin is demonstrated
to induce short-term ROS production in both healthy and neoplastic
human mammary epithelial cells for the two doses used (physio and
obesity). This short-time ROS production under leptin appeared to
be partly due to NOX5 as analysis of the mRNA expression levels of
NOX-family members (FAD-dependent enzymes) showed a unique and
significant NOX5 expression in our conditions. Moreover,
calcium influx inhibition by verapamil and FAD-dependent enzyme
activity inhibition by DPI prevented ROS production that reinforce
the NOX5 implication hypothesis. The main difference between the
three mammary epithelial cell lines concerns the feedback loop of
NOX5 mRNA expression which is less effective in neoplastic than in
healthy cells. This lack of regulation in the neoplastic cells
could be associated to the in-tumor major re-oxidative stress and
related to the neoplastic processes in carcinogenesis (55). Taken together, our results point to
the role for leptin in ROS production via NOX5 isoenzyme in mammary
epithelial cells. This leptin ROS stimulation could be a potent
actor in neoplastic cell signalling and could contribute to the
increased risk of breast cancer associated to obesity (32,48,56).
Acknowledgements
This work was supported by a PhD fellowship (Sinda
Mahbouli) from the Conseil Régional d'Auvergne and the European
research fund (FEDER). We thank Jacques Nunès for his valuable
advice and expert knowledge on cell signaling and cancer cell
biochemistry.
Glossary
Abbreviations
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
NOX
|
NADPH oxidase
|
|
DUOX
|
dual oxidases
|
|
HMEC
|
human mammary epithelial cells
|
|
NOS
|
NO synthases
|
|
q-PCR
|
quantitative real-time PCR
|
|
DCF
|
dichlorofluorescein
|
|
RFU
|
relative fluorescence unit
|
|
RLU
|
relative luminescence units
|
|
siRNA
|
small interfering RNA
|
|
DPI
|
diphenyliodonium
|
|
ApoC
|
apocynin
|
|
BMI
|
body mass index
|
References
|
1
|
Basu S: Bioactive eicosanoids: Role of
prostaglandin F(2α) and F2-isoprostanes in inflammation
and oxidative stress related pathology. Mol Cells. 30:383–391.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fernández-Sánchez A, Madrigal-Santillán E,
Bautista M, Esquivel-Soto J, Morales-González A, Esquivel-Chirino
C, Durante-Montiel I, Sánchez-Rivera G, Valadez-Vega C and
Morales-González JA: Inflammation, oxidative stress, and obesity.
Int J Mol Sci. 12:3117–3132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Valko M, Rhodes CJ, Moncol J, Izakovic M
and Mazur M: Free radicals, metals and antioxidants in oxidative
stress-induced cancer. Chem Biol Interact. 160:1–40. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xia C, Meng Q, Liu L-Z, Rojanasakul Y,
Wang X-R and Jiang B-H: Reactive oxygen species regulate
angiogenesis and tumor growth through vascular endothelial growth
factor. Cancer Res. 67:10823–10830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li J, Stouffs M, Serrander L, Banfi B,
Bettiol E, Charnay Y, Steger K, Krause KH and Jaconi ME: The NADPH
oxidase NOX4 drives cardiac differentiation: Role in regulating
cardiac transcription factors and MAP kinase activation. Mol Biol
Cell. 17:3978–3988. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kovac S, Angelova PR, Holmström KM, Zhang
Y, Dinkova-Kostova AT and Abramov AY: Nrf2 regulates ROS production
by mitochondria and NADPH oxidase. Biochim Biophys Acta.
1850:794–801. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang F, Zhang Y and Dusting GJ: NADPH
oxidase-mediated redox signaling: Roles in cellular stress
response, stress tolerance, and tissue repair. Pharmacol Rev.
63:218–242. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meitzler JL, Antony S, Wu Y, Juhasz A, Liu
H, Jiang G, Lu J, Roy K and Doroshow JH: NADPH oxidases: A
perspective on reactive oxygen species production in tumor biology.
Antioxid Redox Signal. 20:2873–2889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Juhasz A, Ge Y, Markel S, Chiu A,
Matsumoto L, van Balgooy J, Roy K and Doroshow JH: Expression of
NADPH oxidase homologues and accessory genes in human cancer cell
lines, tumours and adjacent normal tissues. Free Radic Res.
43:523–532. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rao Malla R, Raghu H and Rao JS:
Regulation of NADPH oxidase (Nox2) by lipid rafts in breast
carcinoma cells. Int J Oncol. 37:1483–1493. 2010.PubMed/NCBI
|
|
11
|
Bedard K, Jaquet V and Krause K-H: NOX5:
From basic biology to signaling and disease. Free Radic Biol Med.
52:725–734. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ziche M, Morbidelli L, Choudhuri R, Zhang
HT, Donnini S, Granger HJ and Bicknell R: Nitric oxide synthase
lies downstream from vascular endothelial growth factor-induced but
not basic fibroblast growth factor-induced angiogenesis. J Clin
Invest. 99:2625–2634. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mattila JT and Thomas AC: Nitric oxide
synthase: Non-canonical expression patterns. Front Immunol.
5:4782014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lander HM, Jacovina AT, Davis RJ and
Tauras JM: Differential activation of mitogen-activated protein
kinases by nitric oxide-related species. J Biol Chem.
271:19705–19709. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alblas J, Honing H, de Lavalette CR, Brown
MH, Dijkstra CD and van den Berg TK: Signal regulatory protein
alpha ligation induces macrophage nitric oxide production through
JAK/STAT- and phosphatidylinositol 3-kinase/Rac1/NAPDH
oxidase/H2O2-dependent pathways. Mol Cell
Biol. 25:7181–7192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ranganathan S, Krishnan A and
Sivasithambaram ND: Significance of twist and iNOS expression in
human breast carcinoma. Mol Cell Biochem. 412:41–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Delledonne M, Zeier J, Marocco A and Lamb
C: Signal interactions between nitric oxide and reactive oxygen
intermediates in the plant hypersensitive disease resistance
response. Proc Natl Acad Sci USA. 98:pp. 13454–13459. 2001;
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sáinz N, Barrenetxe J, Moreno-Aliaga MJ
and Martínez JA: Leptin resistance and diet-induced obesity:
Central and peripheral actions of leptin. Metabolism. 64:35–46.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Garonna E, Botham KM, Birdsey GM, Randi
AM, Gonzalez-Perez RR and Wheeler-Jones CPD: Vascular endothelial
growth factor receptor-2 couples cyclo-oxygenase-2 with
pro-angiogenic actions of leptin on human endothelial cells. PLoS
One. 6:e188232011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ehrhardt RA, Foskolos A, Giesy SL,
Wesolowski SR, Krumm CS, Butler WR, Quirk SM, Waldron MR and
Boisclair YR: Increased plasma leptin attenuates adaptive
metabolism in early lactating dairy cows. J Endocrinol.
229:145–157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park HK and Ahima RS: Physiology of
leptin: energy homeostasis, neuroendocrine function and metabolism.
Metabolism. 64:24–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bouloumie A, Marumo T, Lafontan M and
Busse R: Leptin induces oxidative stress in human endothelial
cells. FASEB J. 13:1231–1238. 1999.PubMed/NCBI
|
|
23
|
Bilbao MG, Di Yorio MP, Galarza RA, Varone
CL and Faletti AG: Regulation of the ovarian oxidative status by
leptin during the ovulatory process in rats. Reproduction.
149:357–366. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bourgeais J, Gouilleux-Gruart V and
Gouilleux F: Oxidative metabolism in cancer: A STAT affair?
JAK-STAT. 2:e257642013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dattaroy D, Pourhoseini S, Das S, Alhasson
F, Seth RK, Nagarkatti M, Michelotti GA, Diehl AM and Chatterjee S:
Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via
leptin-mediated NADPH oxidase in experimental and human
nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver
Physiol. 308:G298–G312. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nalabolu MR, Palasamudram K and Jamil K:
Adiponectin and leptin molecular actions and clinical significance
in breast cancer. Int J Hematol Oncol Stem Cell Res. 8:31–40.
2014.PubMed/NCBI
|
|
27
|
Naviglio S, Di Gesto D, Illiano F, Chiosi
E, Giordano A, Illiano G and Spina A: Leptin potentiates
antiproliferative action of cAMP elevation via protein kinase A
down-regulation in breast cancer cells. J Cell Physiol.
225:801–809. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dubois V, Jardé T, Delort L, Billard H,
Bernard-Gallon D, Berger E, Geloen A, Vasson MP and Caldefie-Chezet
F: Leptin induces a proliferative response in breast cancer cells
but not in normal breast cells. Nutr Cancer. 66:645–655. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vona-Davis L and Rose DP: The
obesity-inflammation-eicosanoid axis in breast cancer. J Mammary
Gland Biol Neoplasia. 18:291–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Andò S and Catalano S: The multifactorial
role of leptin in driving the breast cancer microenvironment. Nat
Rev Endocrinol. 8:263–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guo S, Liu M, Wang G, Torroella-Kouri M
and Gonzalez-Perez RR: Oncogenic role and therapeutic target of
leptin signaling in breast cancer and cancer stem cells. Biochim
Biophys Acta. 1825:207–222. 2012.PubMed/NCBI
|
|
32
|
Badid N, Ahmed FZB, Merzouk H, Belbraouet
S, Mokhtari N, Merzouk SA, Benhabib R, Hamzaoui D and Narce M:
Oxidant/antioxidant status, lipids and hormonal profile in
overweight women with breast cancer. Pathol Oncol Res. 16:159–167.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Blanquer-Rosselló MM, Santandreu FM,
Oliver J, Roca P and Valle A: Leptin modulates mitochondrial
function, dynamics and biogenesis in MCF-7 cells. J Cell Biochem.
116:2039–2048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schmidt S, Monk JM, Robinson LE and
Mourtzakis M: The integrative role of leptin, oestrogen and the
insulin family in obesity-associated breast cancer: Potential
effects of exercise. Obes Rev. 16:473–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Martínez-Martínez E, Jurado-López R,
Valero-Muñoz M, Bartolomé MV, Ballesteros S, Luaces M, Briones AM,
López-Andrés N, Miana M and Cachofeiro V: Leptin induces cardiac
fibrosis through galectin-3, mTOR and oxidative stress: Potential
role in obesity. J Hypertens. 32:1104–1114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Grossmann ME, Ray A, Nkhata KJ, Malakhov
DA, Rogozina OP, Dogan S and Cleary MP: Obesity and breast cancer:
Status of leptin and adiponectin in pathological processes. Cancer
Metastasis Rev. 29:641–653. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jardé T, Caldefie-Chézet F,
Goncalves-Mendes N, Mishellany F, Buechler C, Penault-Llorca F and
Vasson MP: Involvement of adiponectin and leptin in breast cancer:
Clinical and in vitro studies. Endocr Relat Cancer. 16:1197–1210.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Woolley JF, Stanicka J and Cotter TG:
Recent advances in reactive oxygen species measurement in
biological systems. Trends Biochem Sci. 38:556–565. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schefe JH, Lehmann KE, Buschmann IR, Unger
T and Funke-Kaiser H: Quantitative real-time RT-PCR data analysis:
Current concepts and the novel ‘gene expression's CT difference’
formula. J Mol Med (Berl). 84:901–910. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Minta A, Kao JP and Tsien RY: Fluorescent
indicators for cytosolic calcium based on rhodamine and fluorescein
chromophores. J Biol Chem. 264:8171–8178. 1989.PubMed/NCBI
|
|
41
|
Rossary A, Arab K, Goudable J and Steghens
JP: Fatty acids regulate NOX activity. Ann Biol Clin (Paris).
65:33–40. 2007.(In French). PubMed/NCBI
|
|
42
|
Rossary A, Arab K and Steghens J-P:
Polyunsaturated fatty acids modulate NOX 4 anion superoxide
production in human fibroblasts. Biochem J. 406:77–83. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lamas B, Vergnaud-Gauduchon J,
Goncalves-Mendes N, Perche O, Rossary A, Vasson M-P and Farges M-C:
Altered functions of natural killer cells in response to L-Arginine
availability. Cell Immunol. 280:182–190. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Graham KA, Kulawiec M, Owens KM, Li X,
Desouki MM, Chandra D and Singh KK: NADPH oxidase 4 is an
oncoprotein localized to mitochondria. Cancer Biol Ther.
10:223–231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li J-M and Shah AM: ROS generation by
nonphagocytic NADPH oxidase: Potential relevance in diabetic
nephropathy. J Am Soc Nephrol. 14 Suppl 3:S221–S226. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cave AC, Brewer AC, Narayanapanicker A,
Ray R, Grieve DJ, Walker S and Shah AM: NADPH oxidases in
cardiovascular health and disease. Antioxid Redox Signal.
8:691–728. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Antony S, Wu Y, Hewitt SM, Anver MR,
Butcher D, Jiang G, Meitzler JL, Liu H, Juhasz A, Lu J, et al:
Characterization of NADPH oxidase 5 expression in human tumors and
tumor cell lines with a novel mouse monoclonal antibody. Free Radic
Biol Med. 65:497–508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dho SH, Kim JY, Kwon E-S, Lim JC, Park SS
and Kwon K-S: NOX5-L can stimulate proliferation and apoptosis
depending on its levels and cellular context, determining cancer
cell susceptibility to cisplatin. Oncotarget. 6:39235–39246. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen F, Wang Y, Barman S and Fulton DJR:
Enzymatic regulation and functional relevance of NOX5. Curr Pharm
Des. 21:5999–6008. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Aldieri E, Riganti C, Polimeni M, Gazzano
E, Lussiana C, Campia I and Ghigo D: Classical inhibitors of NOX
NAD(P)H oxidases are not specific. Curr Drug Metab. 9:686–696.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Petrônio MS, Zeraik ML, Fonseca LM and
Ximenes VF: Apocynin: Chemical and biophysical properties of a
NADPH oxidase inhibitor. Molecules. 18:2821–2839. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ameri K, Jahangiri A, Rajah AM, Tormos KV,
Nagarajan R, Pekmezci M, Nguyen V, Wheeler ML, Murphy MP, Sanders
TA, et al: HIGD1A regulates oxygen consumption, ROS production, and
AMPK activity during glucose deprivation to modulate cell survival
and tumor growth. Cell Rep. 10:891–899. 2015. View Article : Google Scholar
|
|
53
|
Martín-Romero C and Sánchez-Margalet V:
Human leptin activates PI3K and MAPK pathways in human peripheral
blood mononuclear cells: Possible role of Sam68. Cell Immunol.
212:83–91. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Trachootham D, Lu W, Ogasawara MA, Nilsa
RD and Huang P: Redox regulation of cell survival. Antioxid Redox
Signal. 10:1343–1374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nourazarian AR, Kangari P and Salmaninejad
A: Roles of oxidative stress in the development and progression of
breast cancer. Asian Pac J Cancer Prev. 15:4745–4751. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen F, Yu Y, Haigh S, Johnson J, Lucas R,
Stepp DW and Fulton DJR: Regulation of NADPH oxidase 5 by protein
kinase C isoforms. PLoS One. 9:e884052014. View Article : Google Scholar : PubMed/NCBI
|