Introduction
Cancer cachexia is a multifactorial metabolic
syndrome characterized by muscle wasting (with or without fat
wasting) and systemic inflammation (1). It occurs in nearly 85% of terminal
cancer patients, and is responsible for ~20% of all cancer-related
deaths (2). Muscle atrophy is the
major physiological effect of cancer cachexia. It is induced by
multiple mechanisms, including an imbalance between muscle protein
synthesis and degradation (3,4),
myocyte apoptosis (5–7), and muscle regeneration dysfunction
(8). Muscle regeneration by
satellite cells is the main means of repairing muscle damage.
Muscle damage, particularly to the sarcolemma, is an important
feature of cancer cachexic muscle atrophy. In animals and humans
with cancer cachexic, various tumor factors impair not only the
sarcolemma, but also the ability to regenerate muscle tissue.
Satellite cells are undifferentiated mononuclear
myogenic cells (9). In resting
adult muscles, satellite cells are quiescent in a reversible G0
state. When muscle fibers are damaged, satellite cells become
activated and proliferate to produce muscle precursor cells,
commonly referred to as myoblasts (10). The myoblasts then fuse into existing
myofibers in need of repair (11).
Tumor factors can inhibit myoblasts from fusing into myofibers and
thereby induce myoblast apoptosis, which impairs muscle
regeneration (8,12).
The most common mechanism of apoptosis in normal and
diseased tissue involves the mitochondrial pathway (13). Apoptotic stimuli converge at
mitochondria and cause mitochondrial outer membrane
permeabilization (MOMP) (14). The
Bcl-2 family of proteins plays a critical role in apoptosis by
regulating mitochondrial integrity (15). The Bcl-2 protein family contains
both pro- and anti-apoptotic members, and the Bax protein is a
pro-apoptotic member. During apoptosis, cytosolic Bax translocates
to pores in the mitochondrial outer membrane (MOM), where it
impairs mitochondrial integrity, induces the loss of mitochondrial
membrane potential (Δψm), and causes the release of
cytochrome c (CYTC) into the cytosol. The released CYTC then
triggers the caspase-3/poly (ADP-ribose) polymerase (PARP)
proteolytic cascade, which induces apoptosis. Bid is another
pro-apoptotic member of the Bc1-2 protein family. JNK activates
caspase-8 and thereby induces Bid cleavage (16). The resulting truncated tBid protein
then binds to Bax, and initiates the activation of Bax (15). As an anti-apoptotic member of the
Bc1-2 family, the Bc1-2 protein binds with Bax on the MOM, and then
inactivates Bax to protect mitochondrial integrity.
Ghrelin is a multifunctional circulating hormone
that consists of 28 amino acids and exists in two different forms:
acylated ghrelin (AG) and unacylated ghrelin (UnAG), respectively.
Both forms originate from the same precursor, and the only
structural difference between them is an octanoylated Ser3 found in
AG. Both AG and UnAG are predominantly synthesized in stomach cells
and then secreted into blood serum (17,18).
Ghrelin receptors are widely expressed in the central nervous
system, intestines, pancreas, liver, adipose tissue, skeletal and
cardiac muscle, and play important roles in numerous biological
functions, including appetite regulation, gastric motility,
pancreatic, cardiovascular and immune function, and muscle
metabolism in both humans and animals (18–20).
Both AG and UnAG can act directly on myoblasts to promote their
differentiation and fusion, although the identity of their
receptor(s) remains unknown (21).
Accumulating evidence suggests that AG and UnAG inhibit apoptosis
via the mitogen-activated protein kinase (MAPK) and
phosphatidylinositol-3-kinase (PI3K)/Akt pathways (22–24).
Moreover, AG/UnAG also regulate Bcl-2 family proteins and inhibit
apoptosis by preventing mitochondrial dysfunction (25,26).
It has never been investigated whether AG or UnAG
may inhibit myoblast apoptosis induced by tumor factors. In the
present study, we used a Transwell-plate system to develop a novel
myoblast-carcinoma cell coculture model. This model allows
myoblasts and carcinoma cells to grow in the same culture medium
and establish intercellular communications without the need for
cell-to-cell contact. We then examined whether this type of culture
environment induced myoblast apoptosis. We also investigated
whether AG or UnAG inhibited myoblast apoptosis, and if so, the
possible mechanisms involved.
Materials and methods
Cell culture
Mouse C2C12 myoblasts and CT26 colon carcinoma cells
were obtained from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and maintained, respectively, in Dulbecco's
modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA, USA) that
was supplemented with 10% fetal bovine serum (FBS) (Gibco,
Auckland, New Zealand) and 1% penicillin-streptomycin (Invitrogen).
The cells were cultured in an atmosphere of 5% CO2 at
37°C.
To establish the coculture model, C2C12 myoblasts
were seeded into the lower wells of a 6-well Transwell clear plate
(3450; Corning, Corning, NY, USA) at a density of 20,000
cells/cm2; CT26 cells were seeded (20,000
cells/cm2) into the upper inserts (0.4-µm pore polyester
membrane) of another 6-well plate (#3516; Corning). After 24 h of
culture, the upper inserts were placed into the lower wells that
contained myoblasts. The base of each insert contained a membrane
with 0.4-µm pores that allowed the movement of secreted factors,
and thus permitted paracrine interactions to occur between the two
different cell types. Next, the medium in both the lower wells and
upper inserts was changed to fresh medium with or without AG/UnAG
(1465/2951, 100 nM; Tocris Bioscience (Ellisville, MO, USA), and
the cells were cocultured for 24 h. After 24 h of coculture, the
medium was collected for analysis by ELISA, and the myoblasts were
harvested for analysis by RT-qPCR, western blotting, flow cytometry
or the
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazole-carbocyanine
iodide (JC-1) staining assay.
The coculture combinations consisted of sham
myoblasts (without CT26 cells in the insert) and without ghrelin
(NC group); myoblasts with CT26 cells but without ghrelin (CO
group); myoblasts with CT26 cells and acylated ghrelin (AG group);
myoblasts with CT26 cells and unacylated ghrelin (UnAG group).
Flow cytometry analysis
Both adherent and floating myoblasts were collected
and washed twice with PBS. An Annexin V/PI Staining kit (C1062;
Beyotime, Shanghai, China) was used according to the manufacturer's
instructions to quantitatively assess the apoptosis status of the
myoblasts. A Mitochondrial Membrane Potential Assay kit with JC-1
(C2006; Beyotime) was used according to the manufacturer's
instructions to assess the mitochondrial membrane potential of
myoblasts. Fluorescence intensity was detected with an Accuri C6
flow cytometer (BD Biosciences, San Diego, CA, USA). A minimum of
1×105 cells were recorded in each sample, and results
were analyzed using FlowJo software (version 7.6.2, FlowJo; LLC,
Ashland, OR, USA).
JC-1 staining assay
Myoblasts were washed twice with PBS, and then
incubated with JC-1 dye (C2006; Beyotime) according to the
manufacturer's instructions. Images of stained cells were acquired
with an Olympus IX71 fluorescence microscope (Olympus Corporation,
Tokyo, Japan). JC-1 dye can exist in two different states:
aggregates and monomers. When excited at 488 nm, monomers emit at
530 nm (green) and aggregates emit at 590 nm (red). Red emissions
signify healthy mitochondria, as healthy mitochondria are
polarized, and the JC-1 taken up by such mitochondria forms
aggregates. JC-1 does not accumulate in depolarized mitochondria,
but rather remains in the cytoplasm as monomers.
Western blot analysis
Myoblasts were washed twice with ice-cold PBS, and
then scraped into a 4°C cell lysis buffer (#9803; Cell Signaling
Technology, Danvers, MA, USA) that was supplemented with a protease
inhibitor cocktail (05892970001; Roche Diagnostics GmbH, Mannheim,
Germany). Protein concentrations were measured using a BCA kit
(BCA1; Sigma-Aldrich, St. Louis, MO, USA). Mitochondrial proteins
were isolated using a Mitochondria Isolation kit for Cultured Cells
(ab110170; Abcam, Cambridge, UK). Aliquots of total protein (20
µg/lane) were separated by electrophoresis on a 4–20% SDS-PAGE gel,
and the separated proteins bands were transferred onto PVDF
membranes. The membranes were blocked with 5% skim milk, and then
incubated overnight at 4°C with a primary antibody; after which,
they were incubated with a HRP-conjugated secondary antibody
(ab97051, 1:2,000; Abcam). The immunostained proteins were
visualized with enhanced chemiluminescence reagents (GE2301;
GenView Scientific, Arcade, NY, USA). Images of the membranes were
recorded with a ChemiDoc XRS+ System, and analyzed using Quantity
One software (version 4.6.6) (both from Bio-Rad, Hercules, CA,
USA). The following antibodies were used as primary antibodies:
Abcam: anti-caspase-3 (ab184787); anti-PARP (ab191217); anti-Bax
(ab32503); anti-Bcl-2 (ab182858); anti-Bcl-2 (phospho-S70,
ab138406); anti-Bid (ab63541); anti-Bid cleavage site (ab10640);
anti-cytochrome c (ab133504); anti-COX IV (ab202554);
anti-p38 (ab170099); anti-p38 (phospho-T180 + Y182, ab195049);
anti-JNK1 + JNK2 + JNK3 (ab179461); anti-JNK1 + JNK2 + JNK3
(phospho-Y185 + Y185 + Y223, ab76572); anti-ERK1 + ERK2 (ab184699);
anti-Erk1 (phospho-T202 + Y204) + Erk2 (phospho-T185 + Y187)
(ab76299); anti-Akt (ab179463); anti-Akt (phospho-S473, ab81283);
anti-GSK3 β (ab32391); anti-GSK3 β (phospho-S9, ab131097);
anti-GAPDH (ab181602).
Real-time quantitative RT-PCR
analysis
Total RNA was extracted from myoblasts with TRIzol
reagent (15596026; Invitrogen) in accordance with the
manufacturer's instructions. Reverse transcription was performed
with SuperScript II reverse transcriptase (18064022; Invitrogen).
The resulting cDNA for specific transcripts was used for real-time
quantitative PCR (RT-qPCR) performed with PowerUp SYBR-Green Master
Mix (A25742; Life Technologies, Carlsbad, CA, USA) and a 7500
Real-Time PCR System (Applied Biosystems, Foster City, CA, USA).
Gene expression data was normalized to that of a housekeeper gene
(GAPDH). Relative gene expression levels were obtained using the
2−ΔΔCt method. The following RT-qPCR primer sequences
were used: Bcl-2, 5′-GTGGTGGAGGAACTCTTCAG-3′ and
5′-GTTCCACAAAGGCATCCCAG-3′; GAPDH, 5′-ATGACAATGAATACGGCTACAGCAA-3′
and 5′-GCAGCGAACTTTATTGATGGTATT-3′.
ELISA
A Mouse TNF alpha ELISA kit (ab46105), Mouse IL-1
beta ELISA kit (ab100704) (both from Abcam), and Mouse IFN-gamma
Quantikine ELISA kit (MIF00; R&D Systems; Minneapolis, MN, USA)
were used to measure TNF-α, IL-1β and IFN-γ concentrations,
respectively, in samples of cell culture medium according to the
manufacturer's instructions. The assay plates were read using a
SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA,
USA).
Statistical analysis
Each experiment was repeated at least three times,
and all data were analyzed using IBM SPSS Statistics for Windows,
version 19.0 (IBM Corp., Armonk, NY, USA). Results are shown as the
mean ± SD. Statistical comparisons between groups were analyzed
using one-way ANOVA followed by the Tukey's test when equal
variances were assumed. When equal variances were not assumed,
Dunnett's T3 test was applied. Two-sided P-values <0.05 were
considered statistically significant.
Results
AG and UnAG attenuate
coculture-induced apoptosis in myoblasts
Fig. 1A and B show
results of the flow cytometric assays used to assess the apoptotic
status of myoblasts. The coculture significantly increased the
numbers of myoblasts undergoing apoptosis (P<0.001), and both AG
and UnAG attenuated those increases (P<0.001). Moreover, as
shown in Fig. 1C-E, a western blot
analysis revealed that the coculture was associated with an
increased level of cleaved caspase-3 protein and a decreased level
of pro-caspase-3 protein, which increased the cleaved
caspase-3/pro-caspase-3 ratio in myoblasts. Moreover, similar
changes were observed in the cleaved-PARP/PARP ratio, and AG and
UnAG also attenuated these changes.
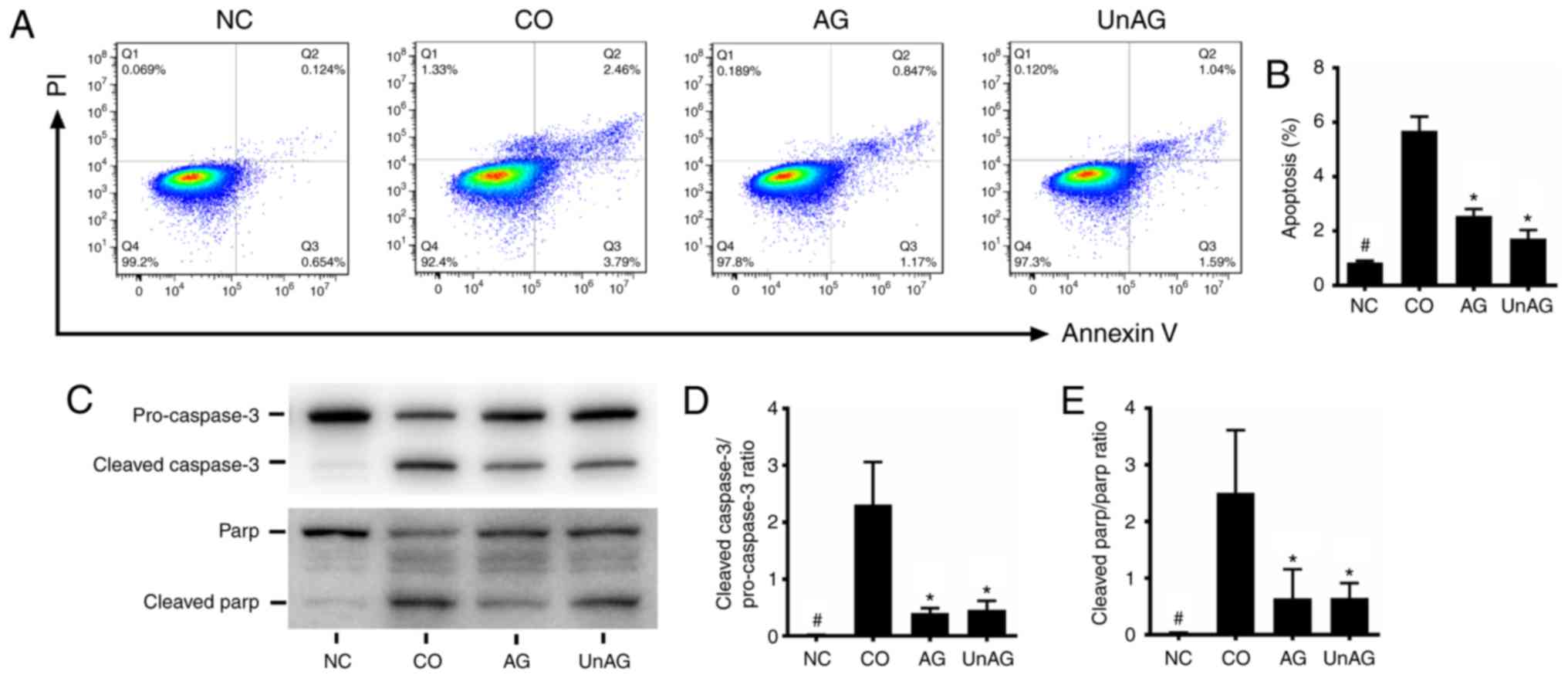 | Figure 1.Effect of AG and UnAG on
coculture-induced apoptosis in myoblasts. (A) Twenty-four hours
after coculture, apoptosis of myoblasts was detected by flow
cytometry. The signals from apoptotic myoblasts are localized in
the Q2 and Q3 quadrants of the resulting pseudocolor graph. (B)
Statistical graph of apoptosis in the different groups. Significant
differences were detected between CO and NC groups
(#P<0.001), between CO and AG/UnAG groups
(*P<0.001), by one-way ANOVA followed by Tukeys test, F=126.284,
P<0.0001. (C) Western blotting of cleaved caspase-3,
pro-caspase-3, cleaved-PARP, PARP and GAPDH in myoblasts. (D)
Quantification of cleaved caspase-3/pro-caspase-3 ratio.
Significant differences were detected between CO and NC groups
(#P<0.001), between CO and AG/UnAG groups (*P=0.001,
P=0.002; respectively), by one-way ANOVA followed by Tukeys test,
F=21.587, P<0.0001. (E) Quantification of cleaved-PARP/PARP
ratio. Significant differences were detected between CO and NC
groups (#P=0.005), between CO and AG/UnAG groups
(*P=0.025, P=0.027; respectively), by one-way ANOVA followed by
Tukeys test, F=8.927, P=0.0006. Data are represented as mean ± SD.
The coculture combinations consisted of sham myoblasts (without
CT26 cells in the insert) and without ghrelin (NC group); myoblasts
with CT26 cells but without ghrelin (CO group); myoblasts with CT26
cells and acylated ghrelin (AG group); myoblasts with CT26 cells
and unacylated ghrelin (UnAG group). |
AG and UnAG ameliorate the
coculture-induced mitochondrial dysfunction in myoblasts
The JC-1 stain flow cytometric assay was used to
assess the Δψm of mitochondria in the myoblasts. As
shown in Fig. 2A-C, the coculture
significantly decreased the membrane potential of mitochondria in
the myoblasts (P<0.01), and both AG and UnAG ameliorated these
changes (P<0.01). Moreover, western blot analysis revealed that
coculture-induced impairment of mitochondria resulted in the
release of CYTC from the mitochondria into the cytosol, and both AG
and UnAG attenuated the effect (Fig.
2D-F).
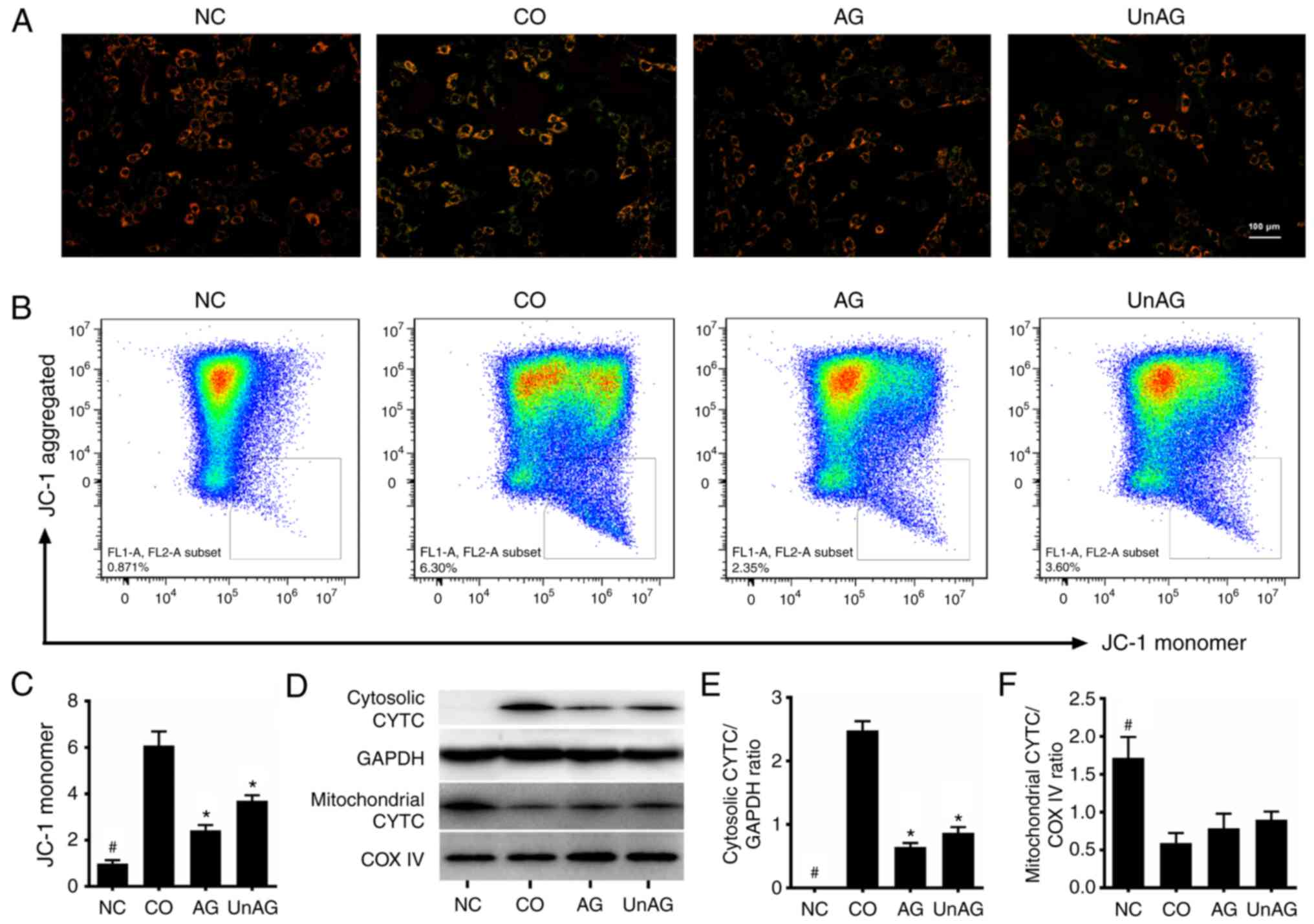 | Figure 2.Effect of AG and UnAG on
coculture-induced mitochondrial dysfunction in myoblasts. (A)
Detection of mitochondrial membrane potential by JC-1 staining
(×100). Scale bar represents 100 µm. (B) Flow cytometry analysis of
JC-1-stained myoblasts. (C) Statistical graph of JC-1 monomer in
the different groups (n=4). Significant differences were detected
between CO and NC groups (#P=0.001), between CO and
AG/UnAG groups (*P=0.009, P=0.002; respectively), by one-way ANOVA
followed by Dunnett's T3 test, F=149.332, P<0.0001. (D) Western
blotting of cytosolic cytochrome c (CYTC), mitochondrial CYTC,
GAPDH and COX IV in myoblasts. (E) Quantification of cytosolic CYTC
was normalized to GAPDH. Significant differences were detected
between CO and NC groups (#P<0.001), between CO and
AG/UnAG groups (*P<0.001), by one-way ANOVA followed by Tukeys
test, F=286.821, P<0.001. (F) Quantification of mitochondrial
CYTC was normalized to COX IV. Significant differences were
detected between CO and NC groups (#P<0.001), by
one-way ANOVA followed by Tukeys test, F=20.972, P<0.001. Data
are represented as mean ± SD. CYTC, cytochrome c. The coculture
combinations consisted of sham myoblasts (without CT26 cells in the
insert) and without ghrelin (NC group); myoblasts with CT26 cells
but without ghrelin (CO group); myoblasts with CT26 cells and
acylated ghrelin (AG group); myoblasts with CT26 cells and
unacylated ghrelin (UnAG group). |
Action of Bcl-2 family proteins in
myoblasts
We performed western blotting and RT-qPCR assays to
assess the role of Bcl-2 family proteins. As shown in Fig. 3A, B, F and I, the coculture
decreased the levels of Bcl-2 protein in mitochondria, and both AG
and UnAG ameliorated these changes. However, the levels of Bc1-2
mRNA and cytosolic protein were not affected by either coculture or
AG/UnAG administration. Regarding Bax, the coculture decreased its
cytosolic protein levels and increased its mitochondrial protein
levels (Fig. 3A, C and G), and thus
increased the Bax/Bcl-2 ratio in mitochondria Fig. 3H). Moreover, the coculture also
increased the levels of tBid protein and reduced the levels of Bid
protein in the cytosol (Fig. 3A, D and
E), and both AG and UnAG attenuated those changes.
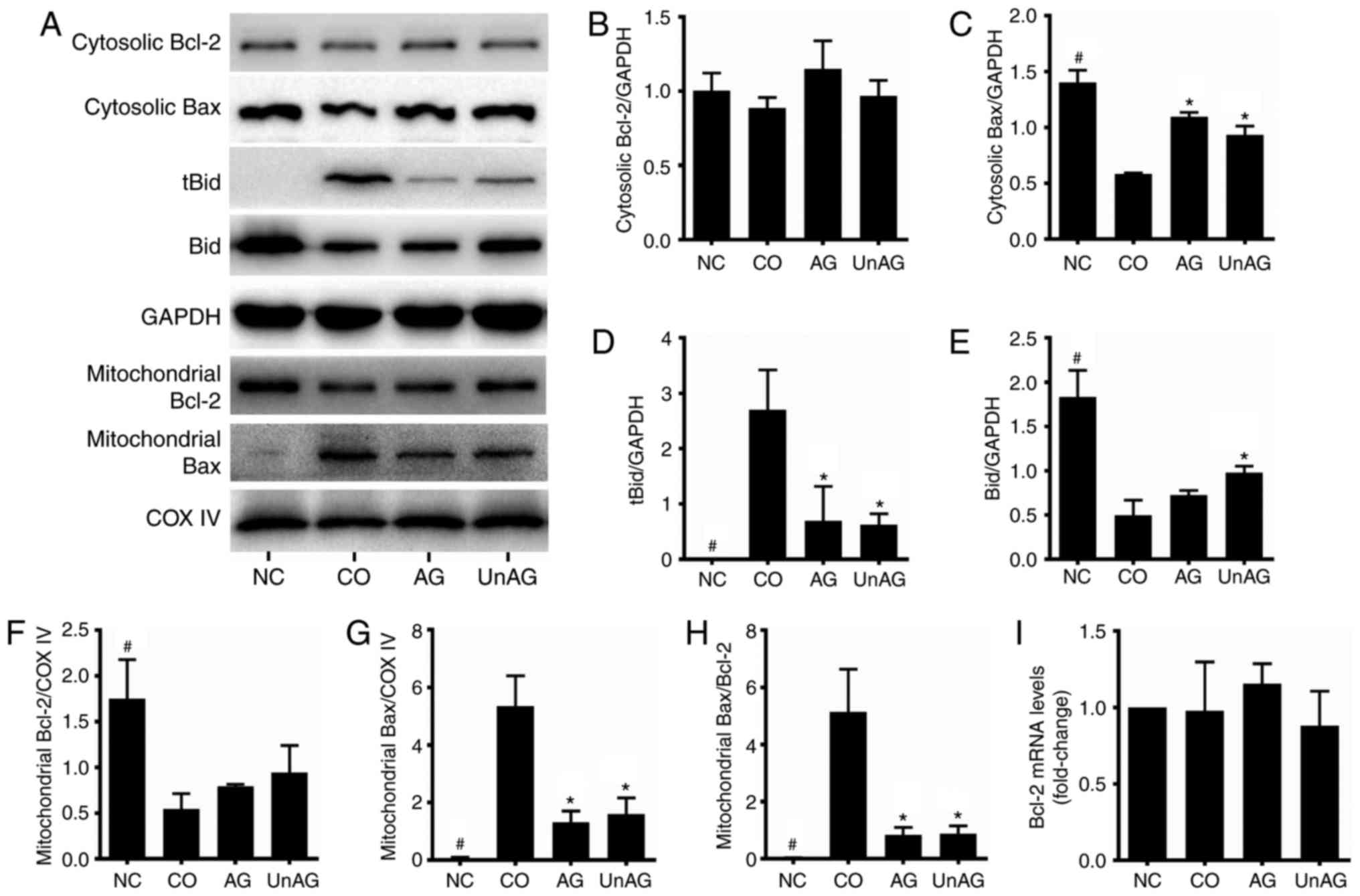 | Figure 3.Regulation of Bcl-2 family proteins
by coculture and AG/UnAG administration in myoblasts. (A) Western
blotting of protein levels. (B) Quantification of cytosolic Bcl-2
was normalized to GAPDH. No significant differences were detected
between 4 groups, by one-way ANOVA followed by Tukeys test. (C)
Quantification of cytosolic Bax was normalized to GAPDH.
Significant differences were detected between CO and NC groups
(#P<0.001), between CO and AG/UnAG groups
(*P<0.001, P=0.01; respectively), by one-way ANOVA followed by
Tukeys test, F=71.961, P<0.001. (D) Quantification of tBid was
normalized to GAPDH. Significant differences were detected between
CO and NC groups (#P=0.001), between CO and AG/UnAG
groups (*P=0.006, P=0.005; respectively), by one-way ANOVA followed
by Tukeys test, F=14.980, P=0.001. (E) Quantification of Bid was
normalized to GAPDH. Significant differences were detected between
CO and NC groups (#P<0.001), between CO and UnAG
groups (*P=0.01), by one-way ANOVA followed by Tukeys test,
F=62.589, P<0.001. (F) Quantification of mitochondrial Bcl-2 was
normalized to COX IV. Significant differences were detected between
CO and NC groups (#P=0.003), by one-way ANOVA followed
by Tukeys test, F=10.816, P=0.003. (G) Quantification of
mitochondrial Bax was normalized to COX IV. Significant differences
were detected between CO and NC groups (#P<0.001),
between CO and AG/UnAG groups (*P<0.001), by one-way ANOVA
followed by Tukeys test, F=40.226, P<0.001. (H) Quantification
of mitochondrial Bax/Bcl-2 ratio. Significant differences were
detected between CO and NC groups (#P<0.001), between
CO and AG/UnAG groups (*P=0.001), by one-way ANOVA followed by
Tukeys test, F=27.339, P<0.001. (I) The levels of Bcl-2 mRNA in
myoblasts. mRNA levels were normalized to GAPDH. No significant
differences were detected between 4 groups, by one-way ANOVA
followed by Tukeys test. Data are represented as mean ± SD. The
coculture combinations consisted of sham myoblasts (without CT26
cells in the insert) and without ghrelin (NC group); myoblasts with
CT26 cells but without ghrelin (CO group); myoblasts with CT26
cells and acylated ghrelin (AG group); myoblasts with CT26 cells
and unacylated ghrelin (UnAG group). |
MAPK pathway activity in
myoblasts
We performed western blot assays to assess MAPK
pathway activity. As shown in Fig.
4A-C, the coculture significantly increased the levels of p-JNK
and JNK proteins in the myoblasts (P<0.01), and both AG and UnAG
ameliorated these changes. Moreover, the p-JNK/JNK ratio was also
increased by coculture, and that increase was ameliorated by UnAG
(Fig. 4D). No significant
difference in the levels of p-P38, P38, p-ERK and ERK proteins was
observed among the four different groups (Fig. 4E and F).
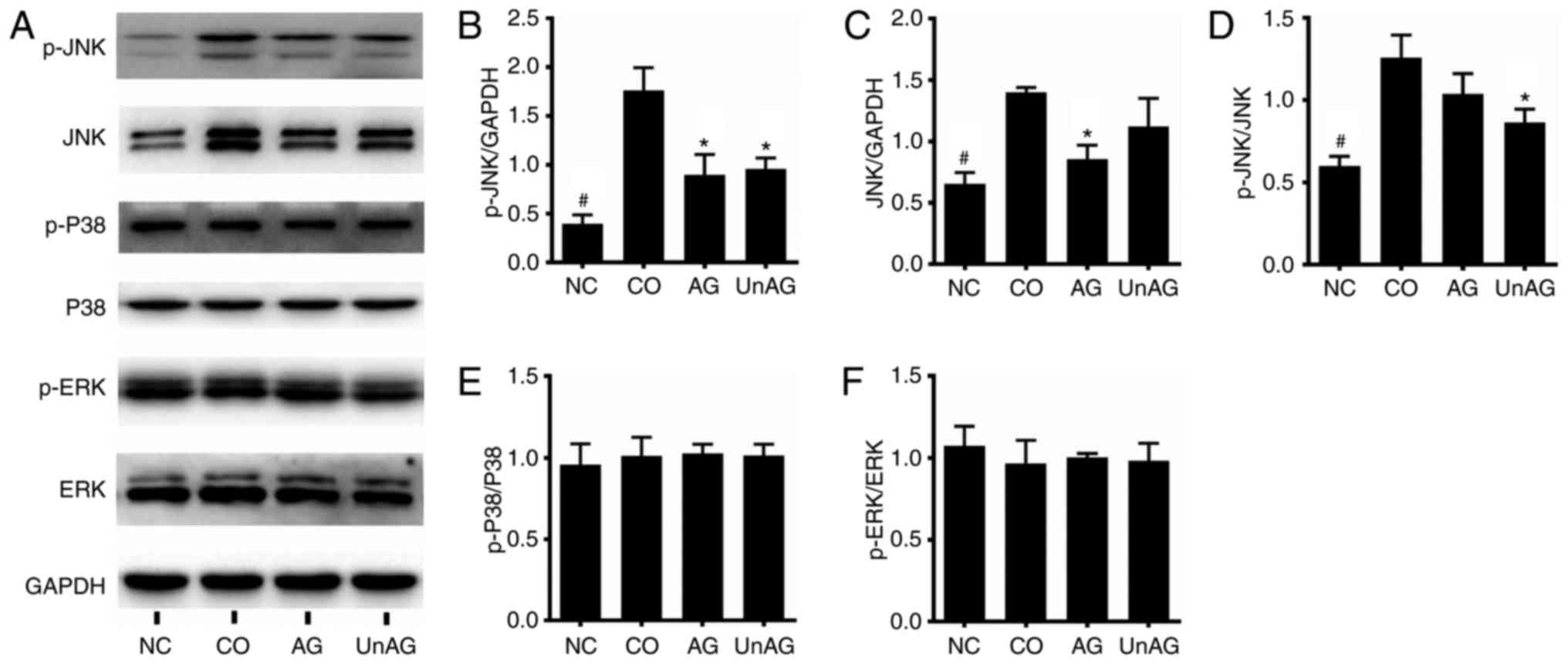 | Figure 4.Regulation of the MAPK pathway by
coculture and AG/UnAG administration in myoblasts. (A) Western
blotting of protein levels. (B) Quantification of p-JNK was
normalized to GAPDH. Significant differences were detected between
CO and NC groups (#P<0.001), between CO and AG/UnAG
groups (*P=0.001, P=0.002; respectively), by one-way ANOVA followed
by Tukeys test, F=32.370, P<0.001. (C) Quantification of JNK was
normalized to GAPDH. Significant differences were detected between
CO and NC groups (#P=0.001), between CO and AG groups
(*P=0.005), by one-way ANOVA followed by Tukeys test, F=17.031,
P=0.001. (D) Quantification of p-JNK/JNK ratio. Significant
differences were detected between CO and NC groups
(#P=0.001), between CO and UnAG groups (*P=0.013), by
one-way ANOVA followed by Tukeys test, F=16.480, P=0.001. (E)
Quantification of p-P38 was normalized to P38. No significant
differences were detected between 4 groups, by one-way ANOVA
followed by Tukeys test. (F) Quantification of p-ERK was normalized
to ERK. No significant differences were detected between 4 groups,
by one-way ANOVA followed by Tukeys test. Data are represented as
mean ± SD. The coculture combinations consisted of sham myoblasts
(without CT26 cells in the insert) and without ghrelin (NC group);
myoblasts with CT26 cells but without ghrelin (CO group); myoblasts
with CT26 cells and acylated ghrelin (AG group); myoblasts with
CT26 cells and unacylated ghrelin (UnAG group). |
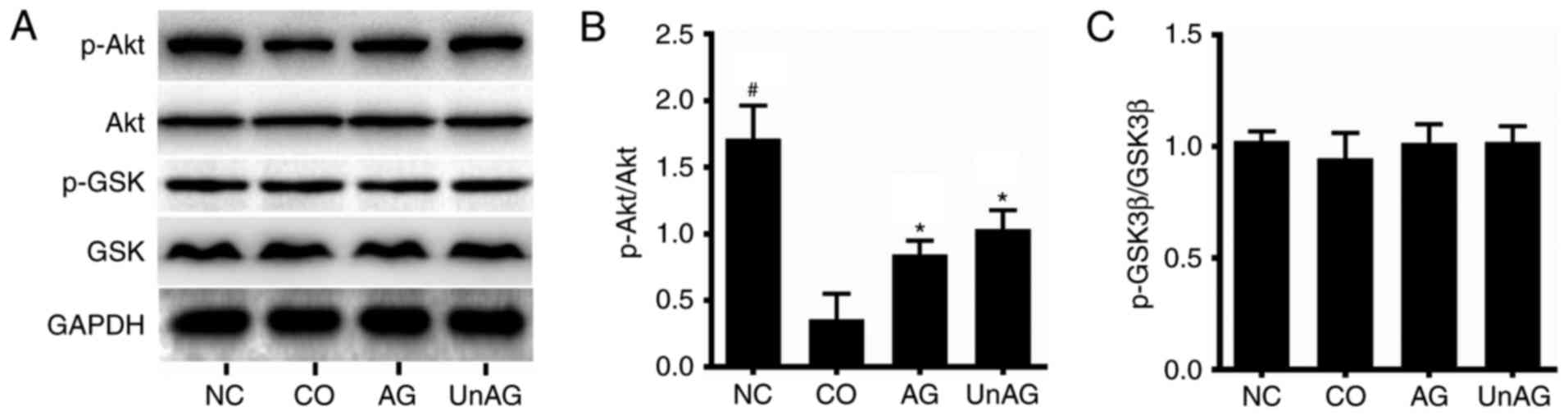
Activity of the PI3K/Akt/glycogen
synthase kinase 3β (GSK3β) pathway in myoblasts
Western blot analysis was performed to assess
PI3K/Akt/GSK3β pathway activity. As shown in Fig. 5A and B, the coculture was associated
with a decreased level of p-AkT protein, while Akt protein levels
were unaffected. This change resulted in decreased p-AkT protein
levels and p-Akt/Akt ratios in myoblasts; once again however, both
AG and UnAG ameliorated these changes. No significant difference in
the levels of p-GSK3β and GSK3β proteins was observed in the four
different groups (Fig. 5C).
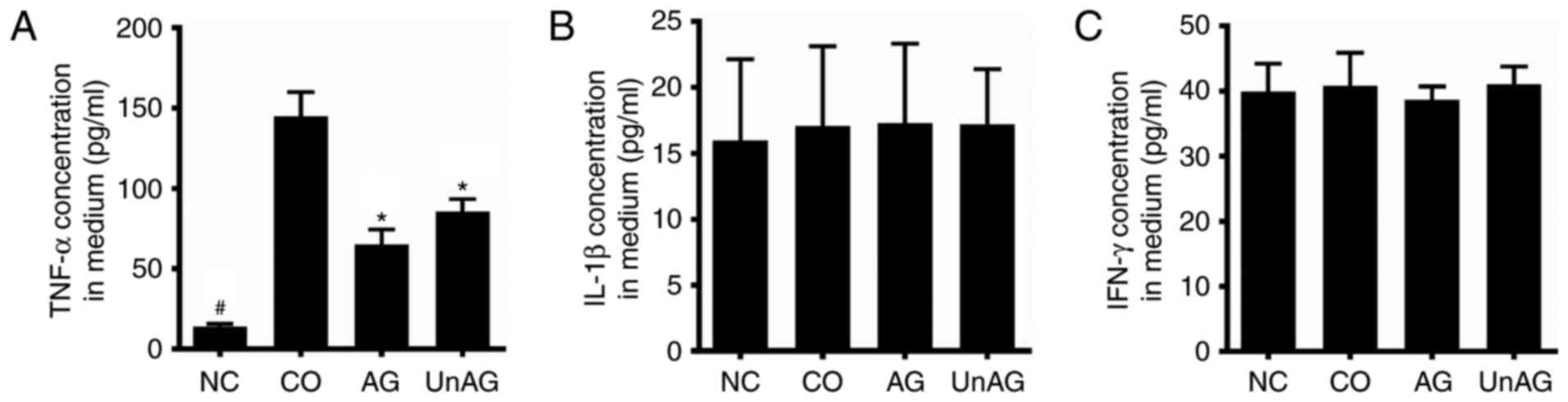
Concentrations of pro-inflammatory
cytokines in the coculture medium
ELISA assays were performed to assess the
concentrations of TNF-α, IL-1β, and IFN-γ in samples of coculture
medium. As shown in Fig. 6A, the
coculture increased the mean TNF-α concentration in medium by
~11-fold, and both AG and UnAG ameliorated this affect. However,
there was no statistically significant difference in the IL-1β and
IFN-γ concentrations in samples of culture medium from the four
different groups (Fig. 6B and
C).
Discussion
Cancer cachexia is a life-threatening syndrome
associated with myofiber damage. Tumor factors can induce myoblast
apoptosis, and thereby impair muscle regeneration. Ghrelin is a
multifunctional hormone with an anti-apoptosis effect (19,20).
In the present study, we demonstrated that either AG or UnAG could
ameliorate the increase in myoblast apoptosis caused by coculture,
indicating that AG and UnAG could maintain the regeneration
capability of muscle tissue, and thereby inhibit muscle
atrophy.
A mitochondrial-centered control pathway is the most
common mechanism of apoptosis (13), and changes in Δψm can
serve as markers of mitochondrial function (27). In this scenario, apoptotic signals
converge at mitochondrial membranes, where they cause MOMP and the
loss of Δψm, which lead to the release of toxic proteins
(such as CYTC) from the mitochondria into cytosol (28). Next, the cytosolic CYTC works in
conjunction with APAF1 to form an apoptosome, which triggers the
caspase-3/PARP proteolytic cascade; which in turn, activates the
downstream pathway to induce apoptotic cellular dismantling and
clearance (29,30). Our results showed that both AG and
UnAG prevented the loss of Δψm induced by the coculture.
Moreover, AG and UnAG also inhibited the activation of caspase-3
and PARP. These findings indicate that AG and UnAG protect
myoblasts from apoptosis by inhibiting coculture-induced
mitochondrial damage.
The Bcl-2 family of proteins plays a critical role
in regulating mitochondrial integrity. In healthy cells, Bax
proteins reside mainly in the cytosol (31,32).
Apoptotic stimuli activate Bax proteins and target them to the MOM.
The Bax protein molecules then form dimers and larger oligomers
with each other and create pores directly in the MOM that result in
cytochrome c release and apoptotic cell death (33). The Bcl-2 protein is exclusively
membrane-bound and attaches to various organelles, including
mitochondria. Bcl-2 can bind to the active form of Bax and inhibit
its activity to protect mitochondrial integrity. The Bax/Bcl-2
ratio in mitochondria determines how mitochondria response to
apoptotic stimuli (34). A previous
study demonstrated that tumor factors increase the Bax/Bcl-2
protein ratio in skeletal muscle tissue (6). Similar to that finding, our results
showed that the coculture increased the levels of Bax protein and
decreased the levels of Bcl-2 protein in mitochondria. These
changes increased the Bax/Bcl-2 ratio in mitochondria and induced
apoptosis. The coculture also decreased the Bax protein levels in
the cytosol, which indicated that Bax became targeted to
mitochondria during the coculture. We found that both AG and UnAG
ameliorated these events to protect mitochondria, which is
consistent with results from previous studies (23,24,26).
Bcl-2 has a long biological half-life, and its expression does not
significantly change during apoptosis (35). Consistent with that finding, we did
not detect any significant differences in the levels of Bcl-2 mRNA
and cytoplasmic protein in the four different groups.
Bax primarily resides in the cytosol and must be
activated by tBid to help target it towards the MOM (36). Consistent with that finding, our
results showed that the coculture increased the levels of tBid and
decreased the levels of Bid in the cytosol, suggesting that the
coculture had activated Bid to induce the activation of Bax. Both
AG and UnAG inhibited the activation of Bid to prevent
apoptosis.
Previous studies demonstrated that apoptotic stimuli
activate MAPK proteins to regulate the activity of Bcl-2 family
proteins (16,34,37,38).
Both JNK and P38 can phosphorylate Bcl-2 to inhibit its
anti-apoptotic activity. JNK can also activate Bid via activation
of caspase-8. AG has been reported to inhibit both JNK and P38
activity, and thereby regulate the activity of Bcl-2 family
proteins (24). Moreover, AG and
UnAG can also activate ERK to inhibit apoptosis (22,23).
In the present study, we found that the coculture increased the
levels of p-JNK and JNK proteins, and also the p-JNK/JNK ratio in
myoblasts, which indicated the activation of JNK. We also found
that both AG and UnAG suppressed these increases. These data
suggest that AG and UnAG can ameliorate the increase in JNK
activity caused by coculture. However, we did not detect any
difference in the levels of p-P38, P38, p-ERK and ERK proteins in
the four different groups. Taken together, our results indicate
that AG and UnAG suppressed JNK activity, and thereby attenuated
the coculture-induced apoptosis of myoblasts.
Activated Akt promotes the survival of various cell
types and prevents apoptosis induced by various stimuli (39–41).
Both AG and UnAG inhibit apoptosis via activation of Akt (22,23).
Consistent with those findings, our results showed that AG and UnAG
ameliorated the decreased Akt activity in myoblasts (as shown by a
decreased p-Akt/Akt ratio) induced by the coculture. Akt has been
reported to inhibit apoptosis via phosphorylation of its downstream
target, GSK3β (23,40). Phosphorylation of GSK3β inhibits its
pro-apoptosis activity. However, in the present study, the levels
of p-GSK3β and GSK3β were not affected by either the coculture or
AG/UnAG administration. These discrepancies are likely related to
differences in the strain of the cells, the apoptotic stimuli, the
dose of ghrelin administered, and the duration of exposure. Akt
also inhibits the cleavage of Bid, which suppresses the production
of tBid to prevent the activation of Bax. Thus, Akt prevents cell
apoptosis by inhibiting Bax activity (39). Akt may act through this mechanism to
inhibit coculture-induced apoptosis in myoblasts. Taken together,
these data indicate that both AG and UnAG suppressed
coculture-induced myoblast apoptosis via activation of Akt.
Increased levels of serum pro-inflammatory cytokines
secreted by either immune cells or tumors are commonly seen in
cancer cachexic animals (2,42,43).
Several pro-inflammatory cytokines are known to stimulate cell
apoptosis via activation of JNK and inhibition of Akt (38,44).
In the present study, we found that coculture increased the TNF-α
concentrations in samples of culture medium. Moreover, both AG and
UnAG attenuated those increases, indicating that AG and UnAG
inhibited TNF-α secretion, and thus impaired TNF-α-induced
apoptosis in myoblasts.
Cancer cachexia causes ~20% of all cancer-related
deaths (2), and its pathogenesis is
not completely understood. Although some tumor-bearing animal
models have been developed to study cancer cachexia (45,46),
to the best of our knowledge, cell coculture models have never been
used to simulate cancer cachexic muscle apoptosis. Our cell
coculture model uses a Transwell system to grow two types of cells
in the same culture medium, and allows intercellular communications
to occur via cellular secretions. This model has been used to
investigate cell-cell interactions between multiple cell types,
such as between adipocytes and skeletal muscle fibers (47), and osteoblasts and mesenchymal
stromal cells (48). In the present
study, coculture of C2C12 myoblasts with CT26 colon carcinoma cells
increased the TNF-α concentrations in samples of culture medium and
induced apoptosis in myoblasts, indicating that these two types of
cells had interacted with each other via secreted factors. The
muscle apoptosis associated with cancer cachexia results from
tumor-host interactions mediated by pro-inflammatory cytokines.
Moreover, increased TNF-α concentrations are often detected in
tumor-bearing animals and cancer cachexic patients (2,42,43).
Such findings suggest that our coculture model can, at least in
part, simulate cancer-induced muscle satellite cell apoptosis in
vitro. Since many other tumor factors are also involved in
cancer cachexia, additional studies are needed to investigate
whether these factors are involved in our coculture model.
To the best of our knowledge, we demonstrated for
the first time that AG and UnAG inhibit cancer-induced apoptosis in
myoblasts. Previous studies have shown that both AG and UnAG can
directly act on skeletal muscles, which contain numerous
high-affinity binding sites (21,49,50).
However, the identity of the AG/UnAG receptor remains unknown, and
requires further investigation.
In conclusion, cancer cachexia is a devastating
syndrome for cancer patients, and elucidating the mechanisms
involved in such cachexia should enable the development of new
treatment agents that can improve patient survival and quality of
life. We demonstrated that coculture of C2C12 myoblasts with CT26
colon carcinoma cells increased the TNF-α concentrations in culture
medium. Additionally, the coculture activated JNK and suppressed
Akt activity to regulate the activity of Bcl-2 family proteins and
impair mitochondrial integrity. This impairment led to myoblast
apoptosis. AG and UnAG inhibited all of these effects and protected
cocultured myoblasts against apoptosis. Based on these results, we
proposed that our cell coculture model can simulate cancer-induced
myoblast apoptosis, and represents a new approach for investigating
cancer cachexic myoblast apoptosis in vitro. We also
speculate that AG and UnAG can maintain the regeneration capability
of muscle tissue, and thereby attenuate cancer-induced muscle
atrophy by inhibiting myoblast apoptosis. Thus, the findings
described in the present study may contribute to development of an
AG/UnAG-based treatment for cancer cachexia.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81272465).
Glossary
Abbreviations
Abbreviations:
|
AG
|
acylated ghrelin
|
|
UnAG
|
unacylated ghrelin
|
|
TNF-α
|
tumor necrosis factor-α
|
|
JNK
|
c-Jun N-terminal kinase
|
|
CYTC
|
cytochrome c
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
MOMP
|
mitochondrial outer membrane
permeabilization
|
|
MOM
|
mitochondrial outer membrane
|
|
MAPK
|
mitogen-activated protein kinase
|
|
PI3K
|
phosphatidylinositol-3-kinase
|
|
Akt
|
protein kinase B
|
|
IL-1β
|
interleukin-1β
|
|
IFN-γ
|
interferon-γ
|
References
|
1
|
Fearon K, Strasser F, Anker SD, Bosaeus I,
Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N,
Mantovani G, et al: Definition and classification of cancer
cachexia: An international consensus. Lancet Oncol. 12:489–495.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Donohoe CL, Ryan AM and Reynolds JV:
Cancer cachexia: Mechanisms and clinical implications.
Gastroenterol Res Pract. 2011:6014342011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Glass DJ: Signaling pathways perturbing
muscle mass. Curr Opin Clin Nutr Metab Care. 13:225–229. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schiaffino S, Dyar KA, Ciciliot S, Blaauw
B and Sandri M: Mechanisms regulating skeletal muscle growth and
atrophy. FEBS J. 280:4294–4314. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Busquets S, Figueras MT, Fuster G,
Almendro V, Moore-Carrasco R, Ametller E, Argilés JM and
López-Soriano FJ: Anticachectic effects of formoterol: A drug for
potential treatment of muscle wasting. Cancer Res. 64:6725–6731.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ishiko O, Sumi T, Yoshida H, Hyun Y and
Ogita S: Expression of apoptosis regulatory proteins in the
skeletal muscle of tumor-bearing rabbits compared with
diet-restricted rabbits. Int J Mol Med. 8:279–283. 2001.PubMed/NCBI
|
|
7
|
Belizário JE, Lorite MJ and Tisdale MJ:
Cleavage of caspases-1, −3, −6, −8 and −9 substrates by proteases
in skeletal muscles from mice undergoing cancer cachexia. Br J
Cancer. 84:1135–1140. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He WA, Berardi E, Cardillo VM, Acharyya S,
Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P,
et al: NF-κB-mediated Pax7 dysregulation in the muscle
microenvironment promotes cancer cachexia. J Clin Invest.
123:4821–4835. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bossola M, Marzetti E, Rosa F and Pacelli
F: Skeletal muscle regeneration in cancer cachexia. Clin Exp
Pharmacol Physiol. 43:522–527. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chargé SB and Rudnicki MA: Cellular and
molecular regulation of muscle regeneration. Physiol Rev.
84:209–238. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Talbert EE and Guttridge DC: Impaired
regeneration: A role for the muscle microenvironment in cancer
cachexia. Semin Cell Dev Biol. 54:82–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He WA, Calore F, Londhe P, Canella A,
Guttridge DC and Croce CM: Microvesicles containing miRNAs promote
muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci
USA. 111:pp. 4525–4529. 2014; View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hotchkiss RS, Strasser A, McDunn JE and
Swanson PE: Cell death. N Engl J Med. 361:1570–1583. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Green DR: Apoptotic pathways: Ten minutes
to dead. Cell. 121:671–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moldoveanu T, Follis AV, Kriwacki RW and
Green DR: Many players in BCL-2 family affairs. Trends Biochem Sci.
39:101–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deng Y, Ren X, Yang L, Lin Y and Wu X: A
JNK-dependent pathway is required for TNFalpha-induced apoptosis.
Cell. 115:61–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gnanapavan S, Kola B, Bustin SA, Morris
DG, McGee P, Fairclough P, Bhattacharya S, Carpenter R, Grossman AB
and Korbonits M: The tissue distribution of the mRNA of ghrelin and
subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol
Metab. 87:29882002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Molfino A, Gioia G and Muscaritoli M: The
hunger hormone ghrelin in cachexia. Expert Opin Biol Ther.
13:465–468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chopin L, Walpole C, Seim I, Cunningham P,
Murray R, Whiteside E, Josh P and Herington A: Ghrelin and cancer.
Mol Cell Endocrinol. 340:65–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guillory B, Splenser A and Garcia J: The
role of ghrelin in anorexia-cachexia syndromes. Vitam Horm.
92:61–106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Filigheddu N, Gnocchi VF, Coscia M,
Cappelli M, Porporato PE, Taulli R, Traini S, Baldanzi G, Chianale
F, Cutrupi S, et al: Ghrelin and des-acyl ghrelin promote
differentiation and fusion of C2C12 skeletal muscle cells. Mol Biol
Cell. 18:986–994. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baldanzi G, Filigheddu N, Cutrupi S,
Catapano F, Bonissoni S, Fubini A, Malan D, Baj G, Granata R,
Broglio F, et al: Ghrelin and des-acyl ghrelin inhibit cell death
in cardiomyocytes and endothelial cells through ERK1/2 and PI
3-kinase/AKT. J Cell Biol. 159:1029–1037. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chung H, Seo S, Moon M and Park S:
Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3 beta
and ERK1/2 pathways mediate protective effects of acylated and
unacylated ghrelin against oxygen-glucose deprivation-induced
apoptosis in primary rat cortical neuronal cells. J Endocrinol.
198:511–521. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mao Y, Wang J, Yu F, Li Z, Li H, Guo C and
Fan X: Ghrelin protects against palmitic acid or
lipopolysaccharide-induced hepatocyte apoptosis through inhibition
of MAPKs/iNOS and restoration of Akt/eNOS pathways. Biomed
Pharmacother. 84:305–313. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu J, Xu H, Shen X and Jiang H: Ghrelin
protects MES23.5 cells against rotenone via inhibiting
mitochondrial dysfunction and apoptosis. Neuropeptides. 56:69–74.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Q, Huang WD, Lv XY and Yang YM:
Ghrelin protects H9c2 cells from hydrogen peroxide-induced
apoptosis through NF-κB and mitochondria-mediated signaling. Eur J
Pharmacol. 654:142–149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Galluzzi L, Vitale I, Abrams JM, Alnemri
ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry
WS, Fulda S, et al: Molecular definitions of cell death
subroutines: Recommendations of the nomenclature committee on cell
death 2012. Cell Death Differ. 19:107–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zou H, Henzel WJ, Liu X, Lutschg A and
Wang X: Apaf-1, a human protein homologous to C. elegans CED-4,
participates in cytochrome c-dependent activation of caspase-3.
Cell. 90:405–413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hsu YT, Wolter KG and Youle RJ:
Cytosol-to-membrane redistribution of Bax and Bcl-XL
during apoptosis. Proc Natl Acad Sci USA. 94:pp. 3668–3672. 1997;
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wolter KG, Hsu YT, Smith CL, Nechushtan A,
Xi XG and Youle RJ: Movement of Bax from the cytosol to
mitochondria during apoptosis. J Cell Biol. 139:1281–1292. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gross A, Jockel J, Wei MC and Korsmeyer
SJ: Enforced dimerization of BAX results in its translocation,
mitochondrial dysfunction and apoptosis. EMBO J. 17:3878–3885.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamamoto K, Ichijo H and Korsmeyer SJ:
BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal
protein kinase pathway normally activated at G2/M. Mol
Cell Biol. 19:8469–8478. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Willimott S and Wagner SD:
Post-transcriptional and post-translational regulation of Bcl2.
Biochem Soc Trans. 38:1571–1575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Walensky LD: Direct BAKtivation. Nat
Struct Mol Biol. 20:536–538. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De Chiara G, Marcocci ME, Torcia M,
Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G,
Armirotti A, Amodei S, et al: Bcl-2 Phosphorylation by p38 MAPK:
Identification of target sites and biologic consequences. J Biol
Chem. 281:21353–21361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kyriakis JM and Avruch J: Mammalian
MAP004B signal transduction pathways activated by stress and
inflammation: A 10-year update. Physiol Rev. 92:689–737. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yamaguchi H and Wang HG: The protein
kinase PKB/Akt regulates cell survival and apoptosis by inhibiting
Bax conformational change. Oncogene. 20:7779–7786. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu Z, Dai J, Liao Y and Wang T: Sox9
protects against human lung fibroblast cell apoptosis induced by
LPS through activation of the AKT/GSK3β pathway. Biochemistry.
82:606–612. 2017.PubMed/NCBI
|
|
41
|
Li S, Chen JW, Xie X, Tian J, Deng C, Wang
J, Gan HN and Li F: Autophagy inhibitor regulates apoptosis and
proliferation of synovial fibroblasts through the inhibition of
PI3K/AKT pathway in collagen-induced arthritis rat model. Am J
Transl Res. 9:2065–2076. 2017.PubMed/NCBI
|
|
42
|
Fearon KC, Glass DJ and Guttridge DC:
Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell
Metab. 16:153–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Argiles JM, Busquets S, Stemmler B and
López-Soriano FJ: Cancer cachexia: Understanding the molecular
basis. Nat Rev Cancer. 14:754–762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Granata R, Settanni F, Biancone L, Trovato
L, Nano R, Bertuzzi F, Destefanis S, Annunziata M, Martinetti M,
Catapano F, et al: Acylated and unacylated ghrelin promote
proliferation and inhibit apoptosis of pancreatic beta-cells and
human islets: Involvement of 3′,5′-cyclic adenosine
monophosphate/protein kinase a, extracellular signal-regulated
kinase 1/2, and phosphatidyl inositol 3-Kinase/Akt signaling.
Endocrinology. 148:512–529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tsubouchi H, Yanagi S, Miura A, Matsumoto
N, Kangawa K and Nakazato M: Ghrelin relieves cancer cachexia
associated with the development of lung adenocarcinoma in mice. Eur
J Pharmacol. 743:1–10. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Penna F, Costamagna D, Pin F, Camperi A,
Fanzani A, Chiarpotto EM, Cavallini G, Bonelli G, Baccino FM and
Costelli P: Autophagic degradation contributes to muscle wasting in
cancer cachexia. Am J Pathol. 182:1367–1378. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wohlers LM, Powers BL, Chin ER and
Spangenburg EE: Using a novel coculture model to dissect the role
of intramuscular lipid load on skeletal muscle insulin
responsiveness under reduced estrogen conditions. Am J Physiol
Endocrinol Metab. 304:E1199–E1212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Voss JO, Loebel C, Bara JJ, Fussinger MA,
Duttenhoefer F, Alini M and Stoddart MJ: Effect of short-term
stimulation with interleukin-1β and differentiation medium on human
mesenchymal stromal cell paracrine activity in coculture with
osteoblasts. Biomed Res Int. 2015:7142302015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sheriff S, Kadeer N, Joshi R, Friend LA,
James JH and Balasubramaniam A: Des-acyl ghrelin exhibits
pro-anabolic and anti-catabolic effects on C2C12 myotubes exposed
to cytokines and reduces burn-induced muscle proteolysis in rats.
Mol Cell Endocrinol. 351:286–295. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Porporato PE, Filigheddu N, Reano S,
Ferrara M, Angelino E, Gnocchi VF, Prodam F, Ronchi G, Fagoonee S,
Fornaro M, et al: Acylated and unacylated ghrelin impair skeletal
muscle atrophy in mice. J Clin Invest. 123:611–622. 2013.PubMed/NCBI
|