Introduction
Melanoma incidence has markedly risen, and although
targeted therapy with B-Raf proto-оncogene, serine/threonine kinase
(BRAF) or mitogen-activated protein kinase kinase (MEK) inhibitors
is effective for patients with BRAF-mutated melanoma (1), alkylating agents such as temozolomide
(TMZ) remain a common therapy for BRAF wild-type patients (2). However, melanomas quickly acquire drug
resistance, and recurrence of metastases is observed in almost all
cases. To overcome such resistance, numerous studies have focused
on identifying the mechanisms involved (3–5).
Several studies have demonstrated that autophagy, a
lysosome-dependent degradation process in which cellular organelles
are absorbed and degrade for recycling within the cell, may play a
significant role in limiting the efficacy of chemotherapy. It is
essential that cells undergo autophagy to maintain their vitality
and integrity under starvation conditions, infections, and some
diseases, such as neurodegenerative diseases, cancers and aging
(6,7). Cytoprotective autophagy is often
upregulated under anticancer therapy and concurrently with cell
death pathways, leading to adaption to therapeutic stress and
recurrence. Based on studies that autophagy inhibition can increase
the antitumor efficacy of therapies that induce autophagy (8–10),
numerous clinical trials have been launched (8,11). It
has been demonstrated that high levels of autophagy before
treatment predict invasiveness, poor response to cytotoxic
chemotherapy, and shortened survival in metastatic melanoma
(10,12). Elevated levels of autophagy in
primary tumors have also been correlated with fast proliferation
and progression (13). Several
anticancer drugs, including TMZ and vemurafenib, are known to
induce cytoprotective autophagy in cancer cells (9,14).
The endoplasmic reticulum (ER) is a critical
cellular organelle for quality control of secretory proteins.
Stress ER is the phenomenon of a functional overload of the protein
secretion apparatus by misfolded protein chains. For normal protein
maturation, a finely tuned correspondence between the biosynthetic
load and the functional capacity of the ER is necessary.
Disturbances to this balance results in overloading the ER, leading
to misfolding and, ultimately, accumulation in the ER of inactive
or chemically aggressive proteins (15). Despite the fact that melanoma cells
are adapted to a high level of stress, suppression of adaptation
mechanisms is a new direction for developing a therapeutic strategy
(16).
ER stress leads to activation of two protein
degradation pathways, the ubiquitin-proteasome via ER-assisted
degradation and lysosome-mediated protein degradation via autophagy
(17). The unfolded protein
response (UPR) is a complex of closely interconnected signal
branches, united by a common trigger mechanism. This mechanism is
represented by a triad of transmembrane proteins (PERK, IRE1 and
ATF6), each of which, under normal physiological conditions, is
inactivated by chaperone 78 kDa glucose-regulated protein 78
(GRP78), also referred to as BiP (18).
It has been demonstrated that induction of GRP78, a
major target of UPR, leads to general translation arrest,
upregulation of chaperones and folding enzymes, and degradation of
misfolded proteins (19). Thus,
GRP78 represents a prosurvival arm of the UPR (20). GRP78 maintains ER integrity and
assists in autophagosome formation independent of Beclin
1-dependent autophagy. The knockdown of GRP78 causes suppression of
the autophagy caused by ER stress (21) and silencing GRP78-dependent
autophagy enhances the cytotoxic effects of TMZ on glioma cells
(22). In addition, GRP78 induces
activation of AMPK and TSC2, which leads to inhibition of mTOR and
simultaneous knockdown of GRP78, and Beclin 1 synergistically
restores antiestrogen sensitivity in resistant cells (23). Recently, Cerezo et al
reported that the HA15 compound (thiazole benzenesulfonamides)
displayed anti-melanoma activity in vitro and in
vivo, both on BRAF resistant and sensitive melanomas, by
targeting GRP78, leading to melanoma cell death by concomitant
induction of autophagic and apoptotic mechanisms (24). UPR and autophagy occur at the same
time and participate in pathological processes, including
chemoresistance of tumors (25).
In the present study, we aimed to investigate the
role of GRP78-dependent autophagy in inducing sensitivity of
melanoma cells to TMZ-treatment regimes.
Materials and methods
Cell lines
Metastatic melanoma cell lines Mel MTP, Mel Z and
Mel IL were derived from patients under treatment at N.N. Blokhin
National Medical Scientific Center for Oncology (26,27).
Cell lines were cultured in RPMI-1640 (Gibco, Paisley, UK)
supplemented with 10% fetal bovine serum (FBS) (HyClone; GE
Healthcare Life Sciences, Logan, UT, USA), 2 mM L-glutamine
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), 10 U/ml penicillin
(Sigma-Aldrich; Merck KGaA), and 0.1 mg/ml streptomycin
(Sigma-Aldrich; Merck KGaA) at 37°C under a 5% CO2
humidified atmosphere.
Transfection
Small interfering (si) RNAs targeting GRP78 were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA)
and a control siRNA (siCTRL, sequence, ATAGAGCGATCACATACAGCC) was
constructed by Syntol (Moscow, Russia). Melanoma cells
(2×105 cells/well) were seeded onto 6-cm Petri dishes
(Nunc, Roskilde, Denmark) and transfected with 10 nM GRP78 or
siCTRL using the Lipofectamine RNAiMAX reagent (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's protocol. Twenty-four hours after transfection, the
cells were treated with 100 µM TMZ (Sigma-Aldrich; Merck KGaA) and
20 µM chloroquine (CQ) (Sigma-Aldrich; Merck KGaA) for 24 or 48 h,
and cell viability and the percentages of autophagy and apoptosis
were determined.
Cell proliferation assay
Melanoma cell lines Mel MTP, Mel Z and Mel IL were
plated (8×103 cells/well) into 96-well plates (Nunc).
After 24 h, TMZ (100 µM) alone or combined with CQ (20 µM) was
added, and the cells were incubated for 48 h. Control cells were
treated with an equal amount of dimethyl sulfoxide (DMSO).
Cytotoxicity was assessed by incubating cells with 20 µl of
3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT)
reagent (Sigma-Aldrich; Merck KGaA) for 4 h, and measuring the
absorbance at 540 nm with a microplate analyzer (Multiscan FC;
Invitrogen; Thermo Fisher Scientific, Inc.) in triplicate.
Colony formation assay
Melanoma cells (2×105) were seeded in
6-well plates. Medium containing 100 µM TMZ, and 20 µM CQ or an
equal amount of DMSO as a vehicle control was added to the
appropriate wells and cells were incubated for 24 h. After
incubation, cells were reseeded on new 6-well plates
(2×103 cells/well) in triplicate and cultivated for 12
days, and the medium was changed every 3–4 days. At the end of the
experiment, colonies were fixed in 1% formalin, stained with 0.5%
crystal violet, and counted using ImageJ software [National
Institutes of Health (NIH) Bethesda, MD, USA]. Three independent
experiments were carried out.
Immunoblotting
Cells were lysed with lysis buffer containing 20 mM
Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA,
1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM
β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml
leupeptin, 1 mM phenylmethanesulfonyl fluoride (PMSF), 10 µl/ml
inhibition cocktail, and 100 µM DTT for 40 min at 4°С. The cells
were then centrifuged at 13,500 × g for 15 min at 4°С. The total
protein content was analyzed using a Quant-IT protein assay kit
according to manufacturer's protocol (Invitrogen; Thermo Fisher
Scientific, Inc.) on a Quibit 2.0 fluorometer (Invitrogen; Thermo
Fisher Scientific, Inc.). An equal amount of protein (40–60 µg)
from each group was separated using 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then
transferred to nitrocellulose membranes. Membranes were incubated
with 5% dry milk (Applichem, Darmstadt, Germany) for 90 min, and
then incubated with Beclin 1 (1:1,000; cat. no. 3738; Cell
Signaling Technology, Inc., Danvers, MA, USA), LC3B (1:500; cat.
no. NB100-2220B; Novus Biologicals, Cambridge, UK), р62/SQSTM1
(1:1,000; cat. no. 5114; Cell Signaling Technology, Inc.),
caspase-7 (1:100; cat. no. sc-56063; Santa Cruz Biotechnology,
Inc.), cleaved PARP (1:1000, cat. no. 44698G; Invitrogen; Thermo
Fisher Scientific, Inc.), GRP78 (1:1,000; cat. no. 3183; Cell
Signaling Technology, Inc.) and β-actin (1:4,000; cat. no. A5441;
Sigma-Aldrich; Merck KGaA) antibodies at 4°C overnight. The
membranes were washed in Tris-buffered saline, containing Tween-20
and incubated with appropriate horseradish peroxidase-conjugated
secondary anti-mouse antibodies (1:10,000; cat. no. NA991VS) and
anti-rabbit (1:10,000; cat. no. NA934VS; both from GE Healthcare,
Chicago, IL, USA) for 90 min at room temperature. Immunoreactive
proteins were detected using enhanced chemiluminescence reagent
Clarity ECL (Bio-Rad Laboratories GmbH, Munich, Germany). The
density of bands was determined on a ChemiDoc Touch Imaging System
(Bio-Rad Laboratories GmbH) and quantified using ImageJ software
(NIH).
Cell cycle analysis
After 24 h of treatment with 100 µM TMZ and 20 µM
CQ, the cells were washed with PBS; the cell pellets were
resuspended in 500 µl of 50 µg/ml solution of propidium iodide (PI)
in buffer (BD Biosciences, Franklin Lakes, NJ, USA) and incubated
in the dark at room temperature for 15 min. The PI fluorescence was
assessed on a NovoCyte 2000R flow cytometer (ACEA Biosciences, San
Diego, CA, USA) and the cell cycle distribution was analyzed using
ModFit 3.2 software (Verity Software House, Topsham, ME, USA).
Apoptosis
Apoptosis was determined by caspase-7 activity
within the cells 24 h after drug treatment. Cells were treated with
100 µM TMZ alone or in combination with 20 µM CQ. Control cells
were treated with DMSO. After treatment, cells were trypsinized,
centrifuged, permeabilized in 200 µl 0.1% Triton X-100-citrate
buffer, and incubated for 30 min at 4°C with mouse anti-caspase-7
antibodies (1:100; Santa Cruz Biotechnology, Inc.). After
incubation, the cells were washed and incubated with anti-mouse
antibody AlexaFluor® 488 (1:2,000; cat. no. A11001; Life
Technologies; Thermo Fisher Scientific, Inc.), washed and fixed in
1% formalin, and followed by analysis on a NovoCyte 2000R flow
cytometer (ACEA Biosciences) using NovoExpress v.1.2.4 software.
Results are presented as the percent increases relative to the
control.
Quantitation of autophagy
Melanoma cells were seeded (3×105
cells/well) in 24-well plates (BD Falcon). Twenty-four hours later,
the cells were transfected with 30 viral particles/cell using
Premo® Autophagy Tandem Sensor RFP-GFP-LC3B and Premo
Autophagy Sensor RFP-p62, as described in the manufacturer's manual
(Life Technologies; Thermo Fisher Scientific, Inc.) and incubated
overnight. The next day, the cells were treated with TMZ and CQ or
equal amounts of DMSO as a vehicle control and further incubated
for 24 h. Imaging was performed using IN Cell Analyzer 6000 and In
Cell Investigator software (GE Healthcare Life Sciences).
Quantitative real-time PCR
Total RNA was extracted from cells using TRIzol
reagent (Sigma-Aldrich; Merck KGaA), as previously described
(28). For cDNA synthesis, RNA (250
ng) was reverse-transcribed in a final volume of 20 µl using
iScript™ Select cDNA Synthesis kit according to manufacturer's
instructions (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
No-reverse transcriptase controls were performed by omitting the
addition of the reverse transcriptase enzyme, and no-template
controls were performed by the addition of nuclease-free water. A
relative quantitation of Beclin 1 mRNA expression normalized to two
endogenous reference genes (β-actin and GAPDH) was performed using
a Bio-Rad CFX96 Real-Time System (Bio-Rad, Laboratories, Inc.) and
iTaq® Universal SYBR®-Green SuperMix (Bio-Ra,
Laboratories, Inc.). Primers are listed in Table I. The PCR reaction mixture (final
volume, 10 µl) contained 5 µl of 2X SuperMix, 5 pmol of GADPH,
β-actin and Beclin 1 and 2 µl (50 ng) of cDNA. The thermocycling
conditions were: 5 min at 95°C, followed by 39 cycles of 5 sec at
95°C, 30 sec at 60°C and 30 sec at 72°C. At the end of the 39 PCR
cycles, melting curve analysis was performed by continuously
recording the fluorescence during progressive heating up to 95°C
with a ramp rate of 0.5°C/sec. All samples were analyzed in
duplicate wells of a 96-well plate. The results of real-time RT-PCR
were represented by the parameter ∆∆Cq (29).
 | Table I.Primer sequencing. |
Table I.
Primer sequencing.
| Gene | Sense | bp |
|---|
| GAPDH | F:
5′-GGGGAGCCAAAAGGGTCATCATCT-3′ | 212 |
|
| R:
5′-GACGCCTGCTTCACCACCTTCTTG-3′ |
|
| β-actin | F:
5′-GTGGGGCGCCCCAGGCACCA-3′ | 201 |
|
| R:
5′-CTCCTTAATGTCACGCACGATTTC-3′ |
|
| Beclin
1 | F:
5′-GAGTTTCAAGATCCTGGACCGTGTCA-3′ | 282 |
|
| R:
5′-CTGTTGGCACTTTCTGTGGACATCA-3′ |
|
Statistical analysis
Each treatment condition was set up in triplicate,
and each experiment was repeated three times independently. Data
are expressed as the mean ± standard deviation (SD), and the
concentration-response curves were produced using the GraphPad
Prism v.5.0 software (GraphPad, Software, Inc., La Jolla, CA, USA).
Statistical analysis was carried out using the Student's t-test. A
P-value of <0.05 was considered to indicate a statistically
significant difference.
Results
Enhanced cytotoxic effect of TMZ under
inhibition of ER stress-induced autophagy depends on the initial
autophagy level in melanoma cells
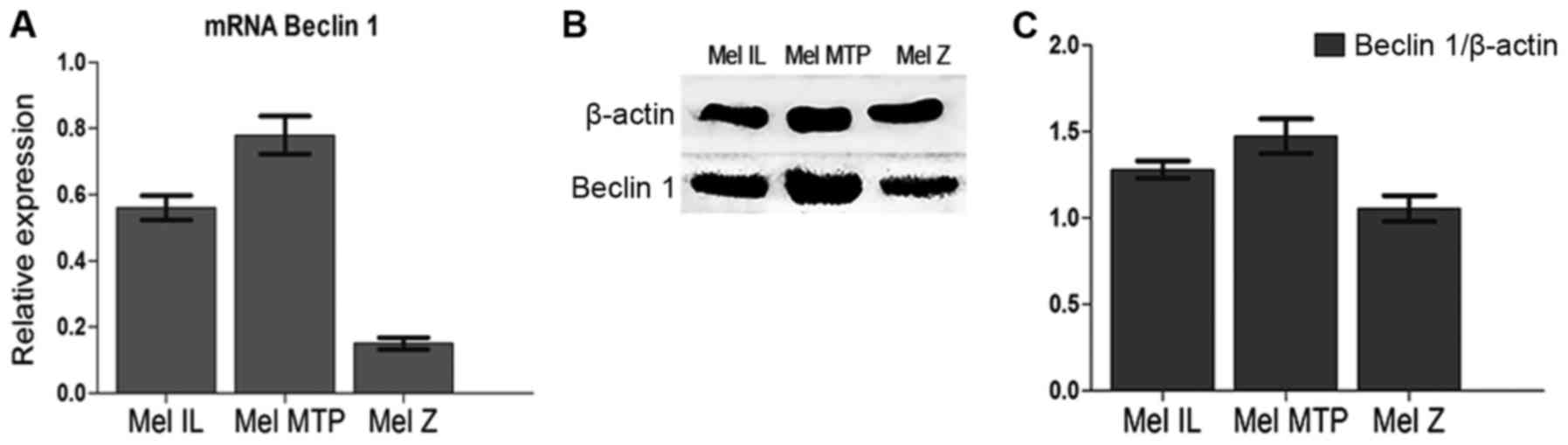
All experiments were carried out on three melanoma
cell lines with different basal autophagy levels [Mel Z (low basal
autophagy), Mel IL (medium basal autophagy), and Mel MTP (high
basal autophagy], which were investigated by Beclin 1 mRNA
expression (qRT-PCR) (Fig. 1A) and
Beclin 1 protein expression (western blotting) (Fig. 1B and C).
Previously, we demonstrated that the autophagy
inhibitor chloroquine (CQ) enhances the cytotoxic effect of TMZ on
both BRAF-mutated and wild-type melanoma cell lines (30).
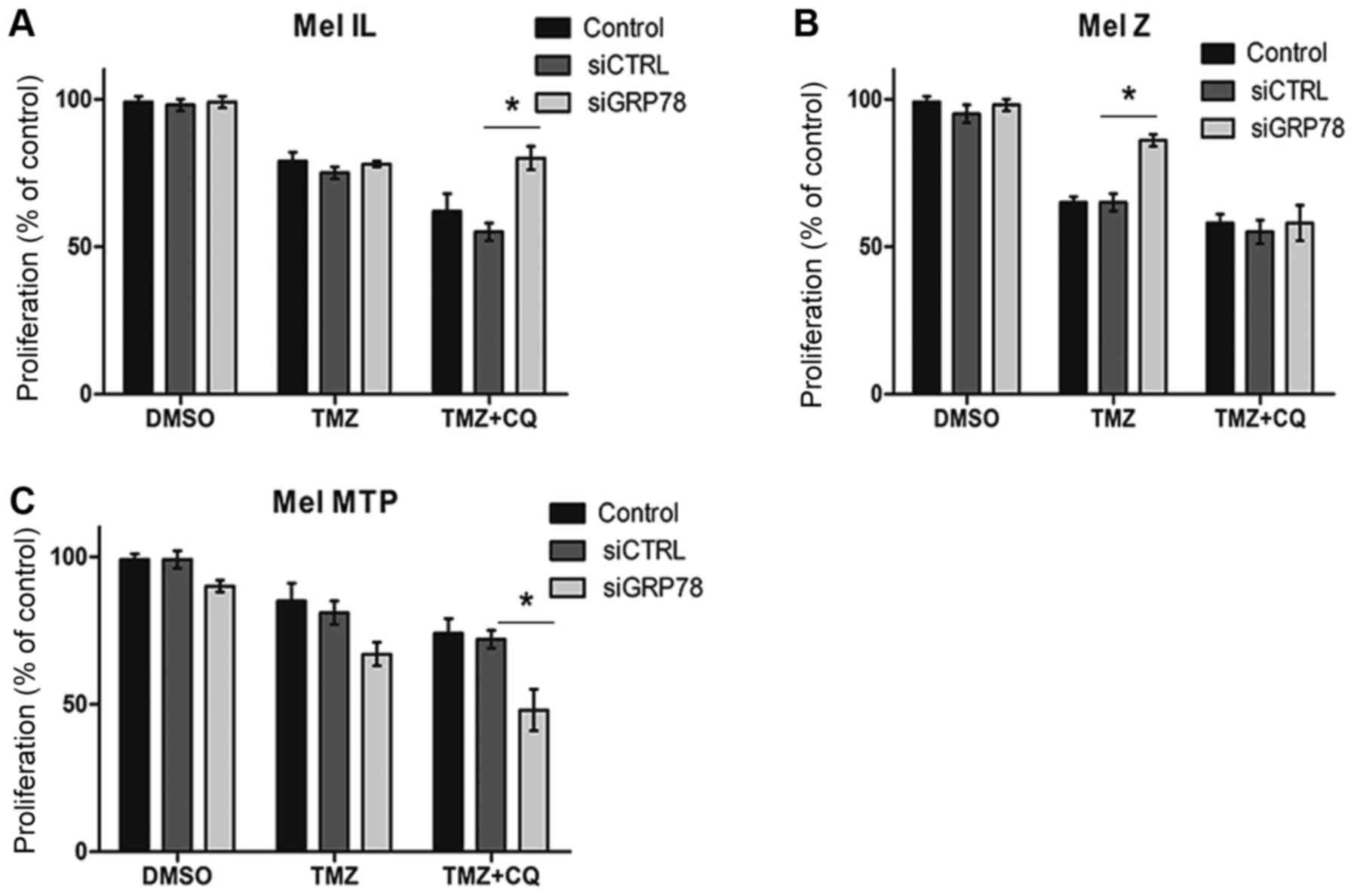
First, we determined the cytotoxic effect of TMZ on
the melanoma cell lines with knockdown of GRP78 by MTT assay. We
initially transfected Mel IL, Mel Z, and Mel MTP cells with small
interfering (si)RNA to a scrambled sequence (siCTRL) and GRP78.
According to the data received, treatment of siCTRL-transfected
melanoma cells with 100 µM TMZ had little to no effect compared to
untransfected cells. Knockdown of GRP78 enhanced the cytotoxic
effects of TMZ alone on Mel MTP cells with a high basal level of
autophagy but there was no inhibition effect on Mel IL compared to
untransfected cells and, notably, silencing of GRP78 mitigated TMZ
toxicity on the Mel Z cell line (Fig.
2A and B). Moreover, downregulation of GRP78-dependent
autophagy mitigated the cytotoxic effect of CQ on Mel Z and Mel IL
cell lines, but induced an antiproliferative effect on Mel MTP
(Fig. 2C).
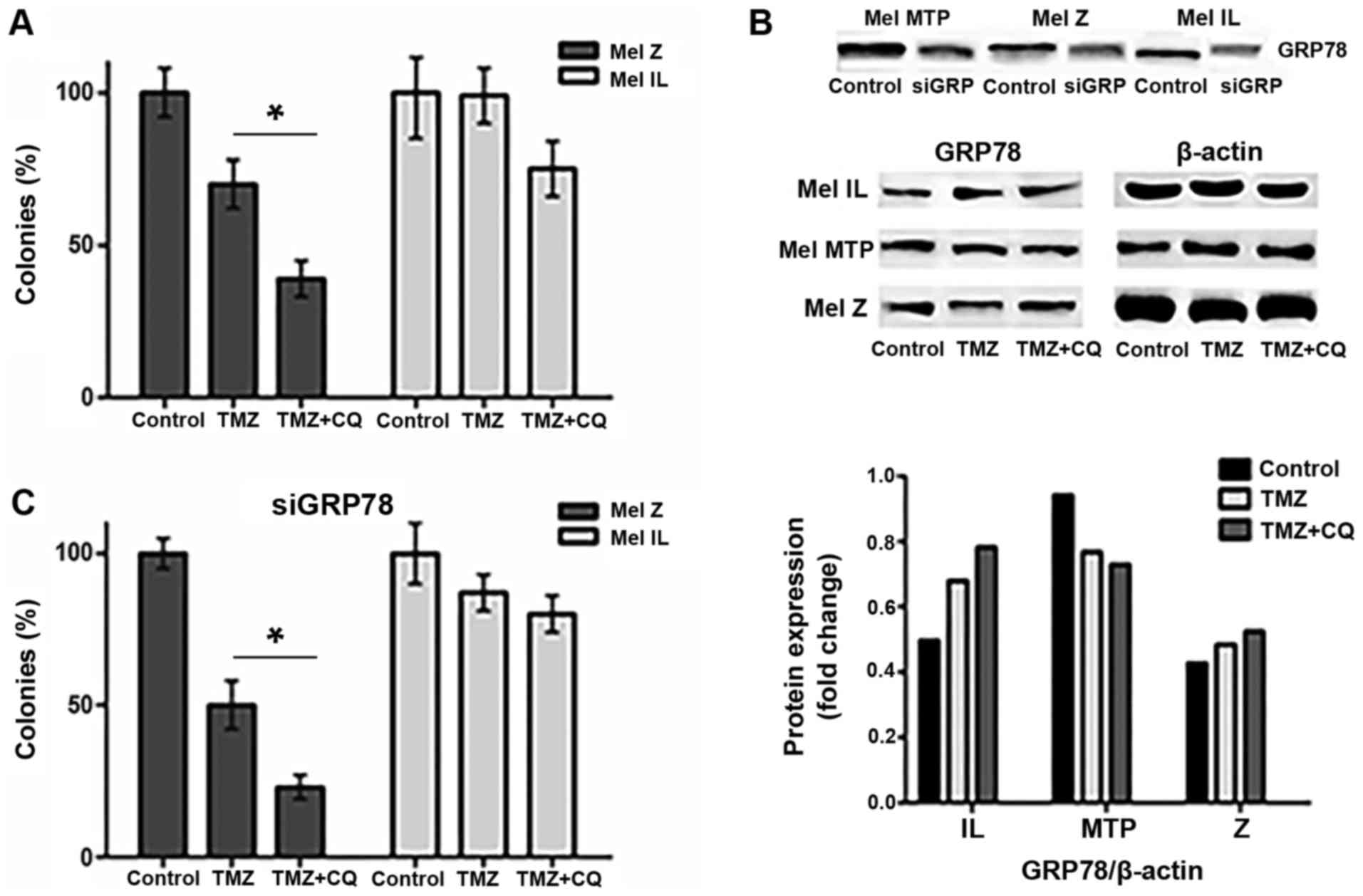
To better understand the mechanism underlying the
synergistic cytotoxic effects of TMZ and CQ in our cells, we
transfected Mel Z, Mel IL, and Mel MTP cells with siCTRL or siRNA
to GRP78. After transfection, cells were cultivated in the presence
or absence of TMZ or TMZ in combination with CQ for 24 h in a
series of colony-forming assays (CFAs). To confirm the knockdown of
the GRP78 protein, we analyzed the protein level of GRP78 by
western blot analysis using β-actin as a loading control (Fig. 3B).
TMZ alone did not affect the viability of Mel IL
cells; there was a slight increase in the number of colonies
relative to the control. Compared to the DMSO-treated control, the
combination of TMZ and CQ reduced the number of colonies by 25%
(P=0.05). The number of colonies in the Mel Z cell line decreased
by 30% with TMZ, the combination of TMZ with CQ reduced the number
of colonies by ~60% compared to the control (P<0.05) (Fig. 3A). Notably, under TMZ treatment
alone or combined with CQ, Mel MTP cells did not form viable
colonies (data not shown).
In Mel IL and Mel Z cells, knockdown of GRP78 with
siGRP78 enhanced the cytotoxic effects of TMZ by further reducing
the percentage of colonies formed by an additional ~20%. Treatment
of siGRP78-transfected melanoma cells with 100 µM TMZ and 20 µM CQ
reduced the percentage of colonies formed by ~70% in Mel Z cells
but did not affect Mel IL. Mel MTP does not form colonies under
siGRP78 transfection (Fig. 3C).
It has been reported that GRP78 maintained ER
integrity and was involved in autophagosome formation independently
of Beclin 1-mediated autophagy (22). Thus, inhibition of GRP78 enhanced
the cytotoxic effects of TMZ, however the combined effect of TMZ
and CQ were detected only in the Mel Z cell line.
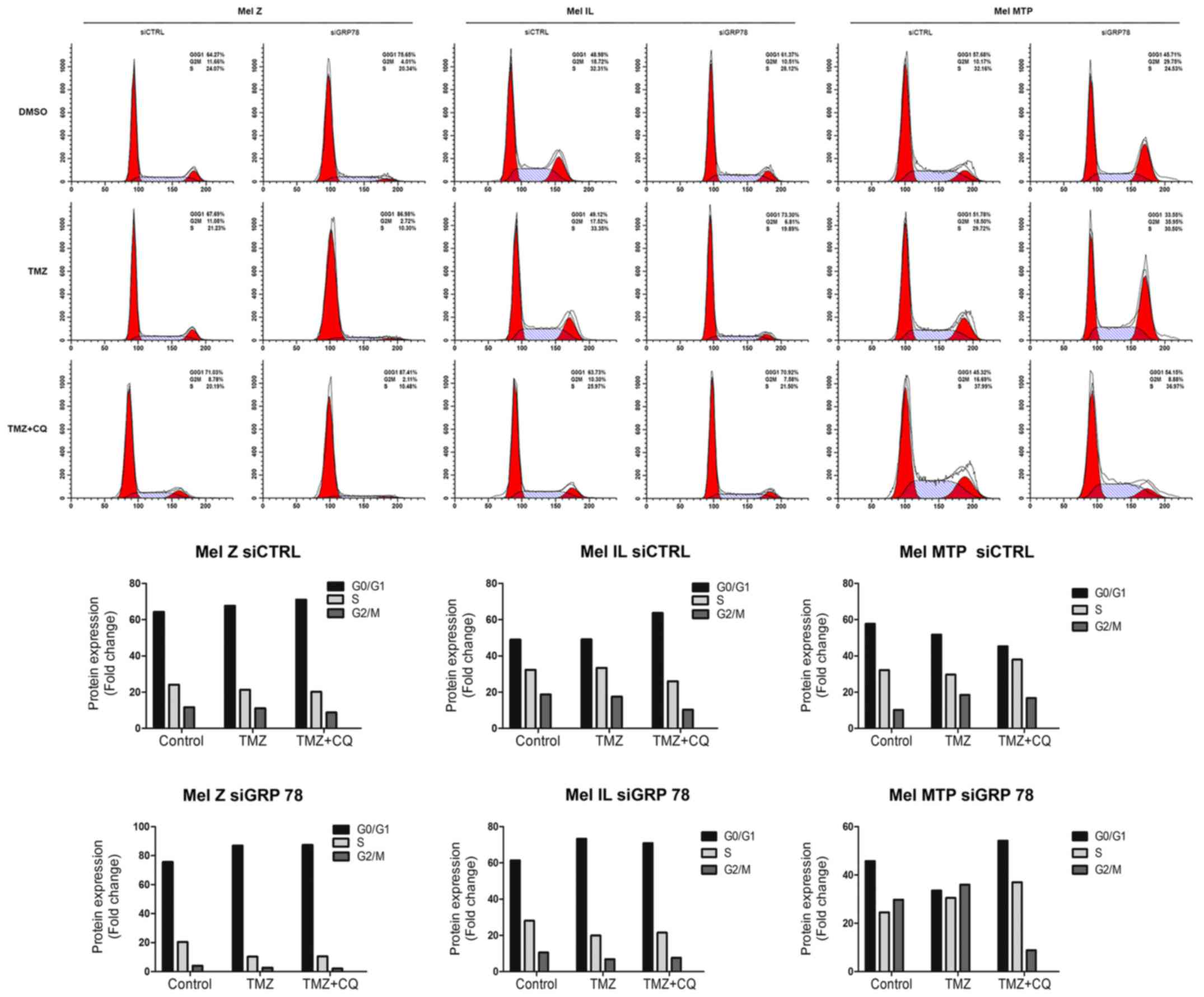
Previously, we demonstrated that TMZ (100 µM)
treatment increased the cell population in the G0/G1 phase in
melanoma cell lines and TMZ and CQ combination further increased
the G0/G1 fraction in BRAF-mutated Mel IL and Mel Z cell lines, but
not in BRAF wild-type Mel MTP (29). Thus, we evaluated the cell cycle
distribution in the siGRP78 transfected cells.
Transfection with siGRP78 resulted in increased
accumulation of cells in the G0/G1 phase compared to the siCTRL
cells under TMZ treatment (49 vs. 73% for Mel IL, and 67 vs. 87%
for Mel Z). However, CQ did not affect the cycle cell distribution
when GRP78 was downregulated as it did in the siCTRL cells
(Fig. 4). Notably, the Mel MTP
line, characterized by a high level of autophagy, was not sensitive
to siGRP78, but the combination with CQ increased in cells in the
G0/G1 phase, which was not observed in the siCTRL cells.
Blockade of GRP78-dependent ER stress
mitigates autophagy and enhances cell death through
caspase-7-mediated apoptosis
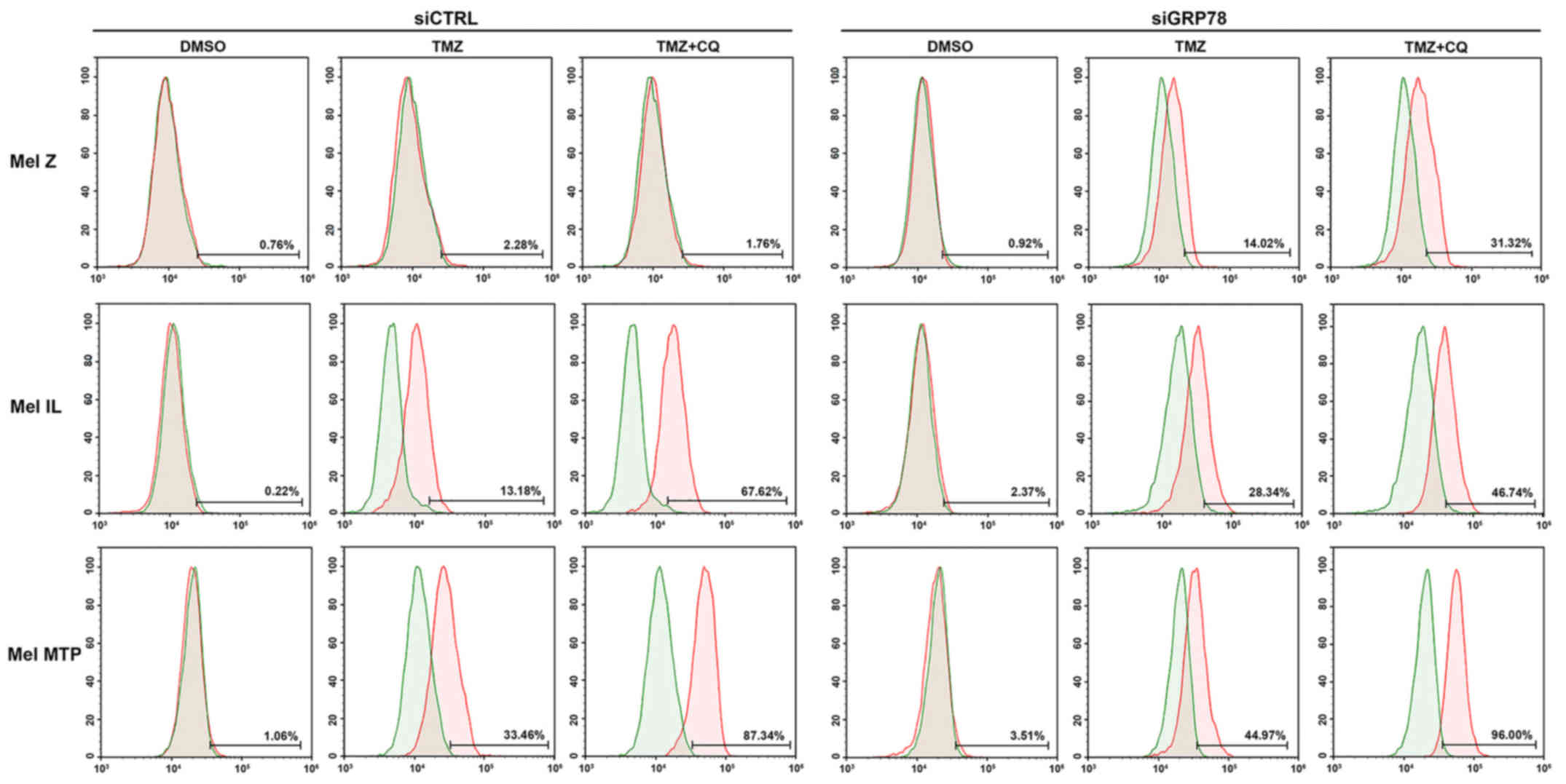
Next, we investigated the activation of caspase-7 by
flow cytometry in melanoma cell lines. We found that, in the Mel Z
cell line, apoptosis was not activated via the caspase pathway.
However, activation of caspase-7 occurred under treatment in cells
transfected with siGRP78. In cell lines with a medium (Mel IL) and
high (Mel MTP) basal level of autophagy, we observed activation of
caspase-7 under TMZ treatment, and its combinations with CQ further
enhanced activation of apoptosis markers. Silencing of GRP78 led to
a more evident activation of caspase-7 in Mel IL and Mel MTP cells
compared to the control. However, downregulation of GRP78 enhanced
apoptosis only in the Mel MTP cell line (up to 96%). Thus, cells
with initially high autophagy were more sensitive to its inhibition
(Fig. 5).
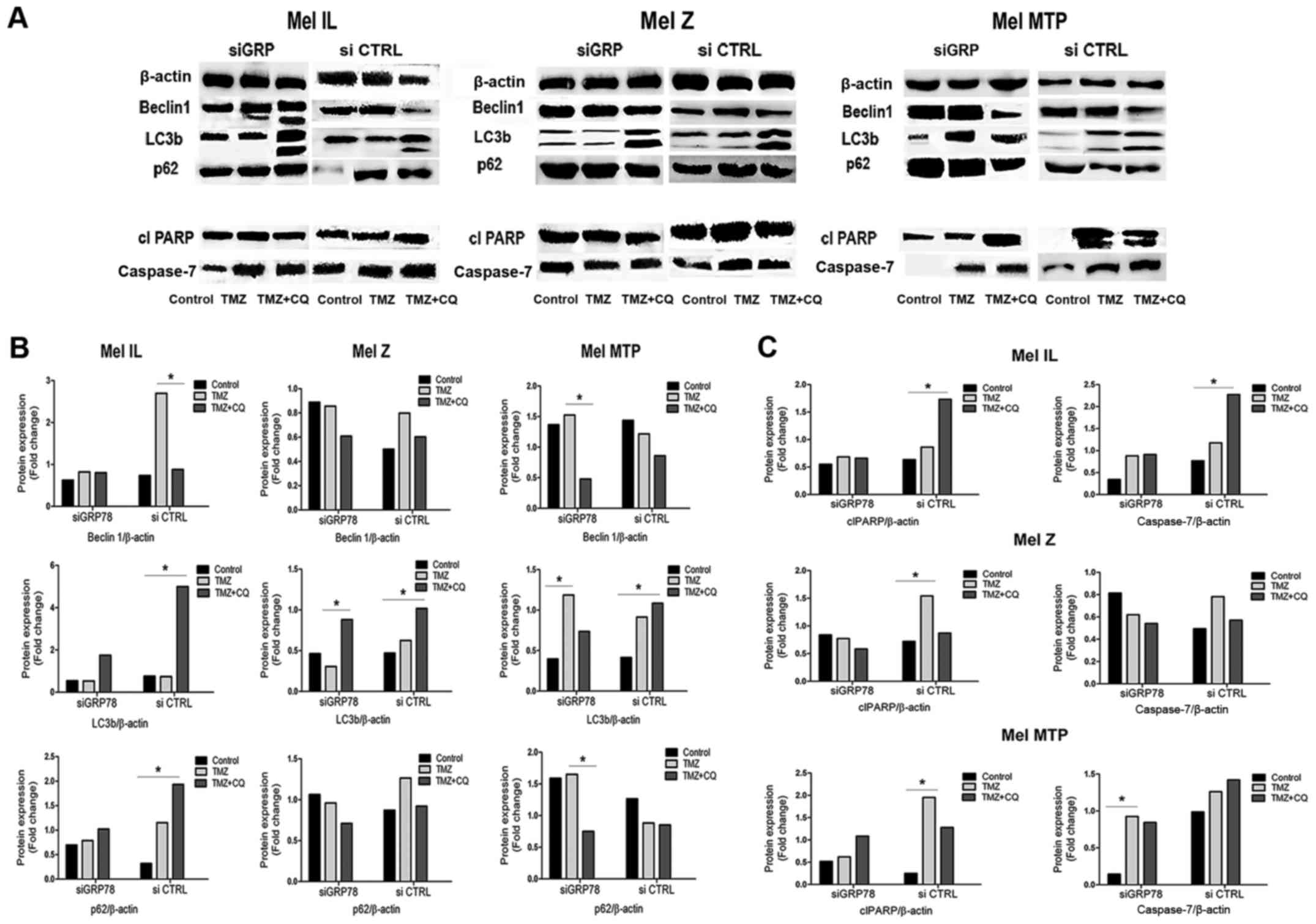
To investigate the mechanistic details of
GRP78-dependent autophagy, we investigated markers of autophagy and
apoptosis. TMZ treatment resulted in a significant increase in the
LC3II/LC3I ratio in the siCTRL cells. In contrast, there was no
TMZ-associated increase in the LC3B-I/LC3B-II ratio in cells
transfected with siGRP78 in either cell line. We found that GRP78
knockdown diminished the ability of TMZ and the TMZ plus CQ
combination to convert LC3B-I to LC3B-II, especially in the Mel MTP
cells. Moreover, in the GRP78-transfected cells, another autophagy
marker, p62, which is involved in trafficking cargo to lysosomes,
was not degraded in autolysosomes compared to the siCTRL cells.
Notably, in the Mel IL cell line, Beclin 1 expression was mitigated
in GRP78-transfected cells but was upregulated in Mel MTP cells.
Thus, GRP78 plays a significant role in TMZ-dependent autophagy
(Fig. 6).
Next, we analyzed the expression of apoptosis
markers caspase-7 and cleaved PARP in melanoma cell lines by
western blotting. In the single treatment, TMZ increased the levels
of apoptosis markers caspase-7 and cleaved PARP in both cell lines.
However, there was a slight increase in caspase-7 and cleaved PARP
activation under combined TMZ and CQ treatment compared to TMZ
alone. Silencing of GRP78 also led to induction of caspase-7 and
caused enhanced cleavage of PARP under the TMZ and CQ combined
treatment in Mel MTP cells compared to control cells, but there
were no differences between Mel IL and Mel Z cells (Fig. 6). Thus, we suggest that the combined
effect of TMZ and CQ was mediated by increased apoptosis.
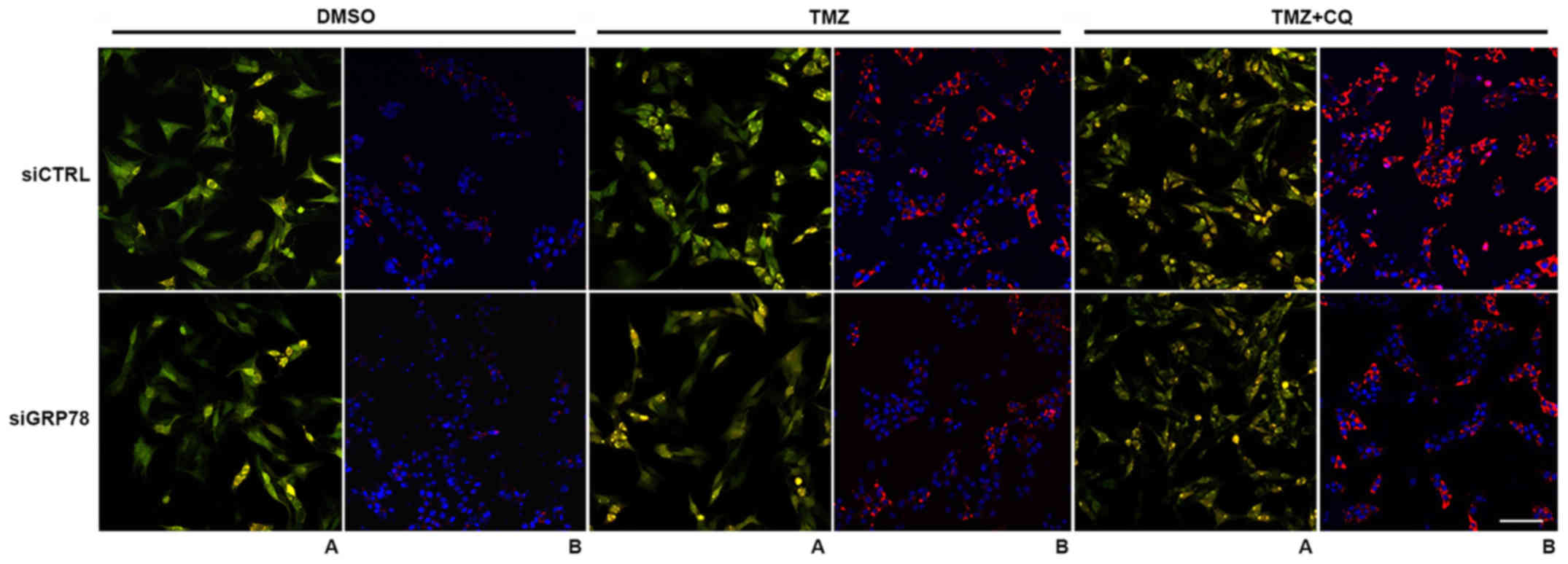
TMZ-induced autophagy is associated
with ER stress
To ensure that TMZ induced autophagy and did not
block clearance of autolysosomes, cells were transfected with
tandem Premo® RFP-GFP-LC3 and Premo RFP-p62 sensors
(Thermo Fisher Scientific, Inc.), as described in the Materials and
methods section. Cells were treated with 100 µM TMZ, and 20 µM CQ
for 24 h and the control cells were treated with equal amounts of
DMSO. In the absence of any treatment, the distribution of green
fluorescence was diffuse. Treatment with TMZ increased the number
of autophagosomes and increased autophagic flux, driven by
functional fusion with the lysosome due to the recruitment of
GFP-LC3 in autophagy. TMZ combined with CQ enhanced the number of
autophagic vacuoles over individual treatment and produced yellow
puncta, reflecting lysosomal impairment, as well as distal
autophagy blockade producing persistence of green and red
fluorescence (31). Under TMZ
treatment, p62 puncta were decreased, and blockade of autophagy led
to an accumulation of p62-positive vesicles. When Mel Z cells were
transfected with GRP78-siRNA for 24 h prior to drug treatment, we
detected impaired autophagic flux, and a smaller amount of
p62-positive cells and combined therapy had no effect (Fig. 7). The same effect was observed in
GRP78-transfected Mel IL and Mel MTP cell lines after 24 h of
treatment with TMZ or TMZ plus CQ (data not shown).
Discussion
Targeting mutant BRAF has been the most promising
breakthrough for melanoma therapy in recent decades (32). However, BRAFV600-targeted therapy
leads to enrichment in resistant cells, resulting in the recurrence
of therapy-resistant disease (33).
Due to this phenomenon, research has focused on understanding the
mechanisms behind the acquired resistance and ways to overcome it.
Moreover, there have been no advantages in the treatment of
patients without BRAF mutation, except alkylating agents and
immunotherapy. There are some drug-resistance mechanisms described
for BRAF mutant and non-mutant melanomas, including activation of
alternative pathways, such as the MAPK (28) and PI3K signaling (34) pathways, as well as receptor tyrosine
kinase activation (35) or
activation of cytoprotective autophagy (36).
Autophagy is known to play a complex role in cancer
and can both suppress and promote tumorigenesis (37). In general, it is thought that
autophagy is used by tumor cells to promote survival, with evidence
supporting the role of dysregulated autophagy in melanoma (38).
Recently, several research groups demonstrated that
targeting autophagy sensitized both BRAF-sensitive and -resistant
melanoma cells to PLX4032 and MEK inhibitors. Thus, autophagy
blockers may represent a novel treatment regime to increase both
cell death and danger-signaling in vemurafenib-resistant metastatic
melanoma (13,39).
Previously, we demonstrated that autophagy
inhibition could enhance melanoma cell death combined with TMZ
therapy on either BRAF-mutated or wild-type cell lines through
induction of apoptosis (30). In
the present study, we evaluated whether inhibition of GRP78, a
Beclin 1-independent activator of autophagy, enhanced cytotoxicity
to TMZ treatment and affected combined TMZ and CQ treatment. GRP78
is an ER molecular chaperone that plays an important role in
protein folding and assembly, targeting misfolded proteins for
degradation, and controlling the activation of transmembrane ER
stress sensors (40).
Several studies have revealed that high levels of
GRP78 expression were correlated with proliferation in glioma and
melanoma cells and that downregulation of GRP78 led to a
significant decrease in cell growth (19,24,41).
Our in vitro research demonstrated that all
tested cell lines with a different basal level of autophagy
expressed GRP78. In the present study, we found that TMZ induced an
increase in GRP78 protein levels. However, TMZ treatment of cells
transfected with siGRP78 led to enhanced cytotoxicity only in the
Mel MTP cell line with high basal autophagy; the Mel IL cell line
was not sensitive to GRP78 silencing. Moreover, compared to the
control, the percent of survived cells was higher in the Mel Z cell
line, which has a low autophagy level.
In a series of colony-forming assays, we
demonstrated that TMZ resulted in a reduction in the percent of
colonies formed under TMZ treatment and that the combination of TMZ
with CQ enhanced the cytotoxic effects of TMZ. We discovered that
knockdown of GRP78 enhanced the cytotoxic effects of TMZ. Treatment
with CQ further enhanced the cytotoxic effects of TMZ on Mel Z and
Mel MTP cells but not Mel IL. We demonstrated that GRP78-dependent
autophagy limited the cytotoxic effects of TMZ.
An investigation of the mechanistic details of
GRP78-dependent autophagy revealed that TMZ treatment resulted in a
significant increase in the LC3II/LC3I ratio in control cells and
that autophagy was mitigated in three melanoma cell lines (with
different basal levels of autophagy) transfected with siGRP78.
GRP78-knockdown diminished the ability of TMZ and the TMZ plus CQ
combination to convert LC3B-I to LC3B-II, particularly in Mel MTP
cells, and accumulation of p62 was significantly compromised. Thus,
GRP78 plays a significant role in TMZ-dependent autophagy.
Furthermore, we also demonstrated that GFP-RFP-LC3
redistribution to autophagosomes and LC3-II accumulation were
decreased after knockdown of GRP78 under TMZ treatment and the
addition of CQ impaired autolysosome degradation. These data
established the role of ER stress as an important driver of
autophagic flux induced by TMZ.
In the single treatment, TMZ increased the levels of
apoptosis markers caspase-7 and cleaved PARP in both of the tested
cell lines. Suppression of ER stress by silencing of GRP78 reduced
TMZ-induced autophagy and cell viability. Activation of caspase-7
demonstrated that TMZ and CQ induced apoptosis in cells with medium
(Mel IL) and high (Mel MTP) basal autophagy levels, and resulted in
cleavage of PARP. Several studies have revealed that ER
stress-mediated autophagy promoted survival in hepatocellular and
colorectal carcinoma cells (42),
pancreatic cancer and melanoma cells (43), and glioma (19). Therefore, those studies and ours
suggest that inhibition of ER stress may be a strategy to enhance
the cytotoxicity of TMZ.
In conclusion, GRP78-dependent autophagy limits the
cytotoxic effects of TMZ. Our data revealed that CQ improved the
cytotoxic effect of TMZ and that autophagy inhibition through
downregulation of ER stress response could overcome resistance to
TMZ treatment in melanoma cells with a high basal level of
autophagy treatment, making this combination applicable as a potent
antitumor treatment of metastatic melanoma.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
Russian Science Foundation (no. 14-35-00107).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
OR and DK conceived and designed the study. OR, DK,
AP and IA performed the experiments. OR and ES wrote the study. OR,
DK and AZ reviewed and edited the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the N.N.
Blokhin National Medical Research Center for oncology ethics
committee (Moscow, Russia).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Saito R de F, Tortelli TC Jr, Jacomassi
MD, Otake AH and Chammas R: Emerging targets for combination
therapy in melanomas. FEBS Lett. 589:3438–3448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luke JJ and Schwartz GK: Chemotherapy in
the management of advanced cutaneous malignant melanoma. Clin
Dermatol. 31:290–297. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boussemart L, Malka-Mahieu H, Girault I,
Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro
N, Thuaud F, et al: eIF4F is a nexus of resistance to anti-BRAF and
anti-MEK cancer therapies. Nature. 513:105–109. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goding CR: The path of least resistance:
Enhancing the effectiveness of BRAF inhibitors. Pigment Cell
Melanoma Res. 26:296–297. 2013. View Article : Google Scholar
|
|
5
|
Iams WT, Sosman JA and Chandra S: Novel
targeted therapies for metastatic melanoma. Cancer J. 23:54–58.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Codogno P and Meijer AJ: Autophagy and
signaling: Their role in cell survival and cell death. Cell Death
Differ. 12 Suppl 2:1509–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Amaravadi RK, Lippincott-Schwartz J, Yin
X-M, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT and White
E: Principles and current strategies for targeting autophagy for
cancer treatment. Clin Cancer Res. 17:654–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lazova R, Klump V and Pawelek J: Autophagy
in cutaneous malignant melanoma. J Cutan Pathol. 37:256–268. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rangwala R, Leone R, Chang YC, Fecher LA,
Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M,
et al: Phase I trial of hydroxychloroquine with dose-intense
temozolomide in patients with advanced solid tumors and melanoma.
Autophagy. 10:1369–1379. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma X-H, Piao S, Wang D, McAfee QW,
Nathanson KL, Lum JJ, Li LZ and Amaravadi RK: Measurements of tumor
cell autophagy predict invasiveness, resistance to chemotherapy,
and survival in melanoma. Clin Cancer Res. 17:3478–3489. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma X-H, Piao S-F, Dey S, McAfee Q,
Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al:
Targeting ER stress-induced autophagy overcomes BRAF inhibitor
resistance in melanoma. J Clin Invest. 124:1406–1417. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levy JMM, Thompson JC, Griesinger AM,
Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH,
Foreman NK, et al: Autophagy inhibition improves chemosensitivity
in BRAF(V600E) brain tumors. Cancer Discov. 4:773–780. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsai YC and Weissman AM: The unfolded
protein response, degradation from endoplasmic reticulum and
cancer. Genes Cancer. 1:764–778. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hersey P and Zhang XD: Adaptation to ER
stress as a driver of malignancy and resistance to therapy in human
melanoma. Pigment Cell Melanoma Res. 21:358–367. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bernales S, McDonald KL and Walter P:
Autophagy counterbalances endoplasmic reticulum expansion during
the unfolded protein response. PLoS Biol. 4:e4232006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pyrko P, Schönthal AH, Hofman FM, Chen TC
and Lee AS: The unfolded protein response regulator GRP78/BiP as a
novel target for increasing chemosensitivity in malignant gliomas.
Cancer Res. 67:9809–9816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee AS: The glucose-regulated proteins:
Stress induction and clinical applications. Trends Biochem Sci.
26:504–510. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Ni M, Lee B, Barron E, Hinton DR and
Lee AS: The unfolded protein response regulator GRP78/BiP is
required for endoplasmic reticulum integrity and stress-induced
autophagy in mammalian cells. Cell Death Differ. 15:1460–1471.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Golden EB, Cho H-Y, Jahanian A, Hofman FM,
Louie SG, Schönthal AH and Chen TC: Chloroquine enhances
temozolomide cytotoxicity in malignant gliomas by blocking
autophagy. Neurosurg Focus. 37:E122014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cook KL, Shajahan AN, Wärri A, Jin L,
Hilakivi-Clarke LA and Clarke R: Glucose-regulated protein 78
controls cross-talk between apoptosis and autophagy to determine
antiestrogen responsiveness. Cancer Res. 72:3337–3349. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cerezo M, Lehraiki A, Millet A, Rouaud F,
Plaisant M, Jaune E, Botton T, Ronco C, Abbe P, Amdouni H, et al:
Compounds triggering ER stress exert anti-melanoma effects and
overcome BRAF inhibitor resistance. Cancer Cell. 29:805–819. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vandewynckel Y-P, Laukens D, Geerts A,
Bogaerts E, Paridaens A, Verhelst X, Janssens S, Heindryckx F and
Van Vlierberghe H: The paradox of the unfolded protein response in
cancer. Anticancer Res. 33:4683–4694. 2013.PubMed/NCBI
|
|
26
|
Mikhaĭlova IN, Lukashina MI, Baryshnikov
AIu, Morozova LF, Burova OS, Palkina TN, Kozlov AM, Golubeva VA,
Cheremushkin EA, Doroshenko MB, et al: Melanoma cell lines as the
basis for antitumor vaccine preparation. Vestn Ross Akad Med Nauk.
60:37–40. 2005.(In Russian).
|
|
27
|
Mikhaylova IN, Kovalevsky DA, Morozova LF,
Golubeva VA, Cheremushkin EA, Lukashina MI, Voronina ES, Burova OS,
Utyashev IA, Kiselev SL, et al: Cancer/testis genes expression in
human melanoma cell lines. Melanoma Res. 18:303–313. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang C, Spevak W, Zhang Y, Burton EA, Ma
Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, et al: RAF
inhibitors that evade paradoxical MAPK pathway activation. Nature.
526:583–586. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ryabaya OO, Inshakov AN, Egorova AV,
Emelyanova MA, Nasedkina TV, Zasedatelev AS, Khochenkov DA and
Stepanova EV: Autophagy inhibitors chloroquine and LY294002 enhance
temozolomide cytotoxicity on cutaneous melanoma cell lines in
vitro. Anticancer Drugs. 28:307–315. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al: BRIM-3 Study Group: Improved survival with vemurafenib in
melanoma with BRAF V600E mutation. N Engl J Med. 364:2507–2516.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Romano E, Pradervand S, Paillusson A,
Weber J, Harshman K, Muehlethaler K, Speiser D, Peters S, Rimoldi D
and Michielin O: Identification of multiple mechanisms of
resistance to vemurafenib in a patient with BRAFV600E-mutated
cutaneous melanoma successfully rechallenged after progression.
Clin Cancer Res. 19:5749–5757. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Caporali S, Alvino E, Lacal PM, Levati L,
Giurato G, Memoli D, Caprini E, Cappellini Antonini GC and D'Atri
S: Targeting the PI3K/AKT/mTOR pathway overcomes the stimulating
effect of dabrafenib on the invasive behavior of melanoma cells
with acquired resistance to the BRAF inhibitor. Int J Oncol.
49:1164–1174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanomas
acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Armstrong JL, Corazzari M, Martin S,
Pagliarini V, Falasca L, Hill DS, Ellis N, Al Sabah S, Redfern CP,
Fimia GM, et al: Oncogenic B-RAF signaling in melanoma impairs the
therapeutic advantage of autophagy inhibition. Clin Cancer Res.
17:2216–2226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Corazzari M, Fimia GM, Lovat P and
Piacentini M: Why is autophagy important for melanoma? Molecular
mechanisms and therapeutic implications. Semin Cancer Biol.
23:337–343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Martin S, Dudek-Perić AM, Maes H, Garg AD,
Gabrysiak M, Demirsoy S, Swinnen JV and Agostinis P: Concurrent MEK
and autophagy inhibition is required to restore cell death
associated danger-signalling in Vemurafenib-resistant melanoma
cells. Biochem Pharmacol. 93:290–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin C-J, Lee C-C, Shih Y-L, Lin C-H, Wang
S-H, Chen T-H and Shih C-M: Inhibition of mitochondria- and
endoplasmic reticulum stress-mediated autophagy augments
temozolomide-induced apoptosis in glioma cells. PLoS One.
7:e387062012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang P-M, Liu Y-L, Lin Y-C, Shun C-T, Wu
M-S and Chen C-C: Inhibition of autophagy enhances anticancer
effects of atorvastatin in digestive malignancies. Cancer Res.
70:7699–7709. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xi H, Kurtoglu M, Liu H, Wangpaichitr M,
You M, Liu X, Savaraj N and Lampidis TJ: 2-Deoxy-D-glucose
activates autophagy via endoplasmic reticulum stress rather than
ATP depletion. Cancer Chemother Pharmacol. 67:899–910. 2011.
View Article : Google Scholar : PubMed/NCBI
|