Introduction
Heat shock proteins (HSPs) can be found in all cells
and are key elements in the process of protein synthesis. When
cells are exposed to stressors, protein misfolding, aggregation or
denaturation may occur resulting in cell death. Under such
difficult conditions, the expression of HSPs are increased and
facilitate cell survival (1,2).
Hsp90 is a member of the HSPs and is important in
protein post-translational maturation and disposition. Its
N-terminal domain contains a characteristic Bergerat fold (unique
structural ATP-binding domain). Binding and hydrolysis of ATP
induces conformational changes to Hsp90, and this allows for the
binding of Hsp90 to its client proteins to help them fold their
active conformation. Hsp90 inhibitor targets the ATP domain and
disrupts the exchange of ADP for ATP of Hsp90 protein, thereby
causing client proteins to undergo misfolding, ubiquitination and
subsequent degradation by the proteasome pathway.
Hsp90 is evolutionarily conserved and ubiquitously
expressed in normal cells, accounting for as much as 1–2% of total
cellular protein (3). Cancer cells,
particularly hematological malignant cells, express 2- to 10-fold
higher levels of Hsp90 than normal cells. Many of its client
proteins are signal transducers that are essential for tumor cell
proliferation, survival and generation (3). In fact, Hsp90 is important in the
acquisition and maintenance of the malignant phenotype for its
unique function in transformation maintanence and growth
facilitation. Moreover, cancer cells are exposed to numerous harsh
conditions and rely on the function of Hsp90 to survive. As a
consequence, it is rational to apply Hsp90 inhibitors in the
treatment of malignancies (4,5). Hsp90
inhibitors can target estrogen and progesterone receptors in breast
cancer (6,7), ERBB2 in ERBB2-driven xenograft models
(8,9), androgen receptor in hormone-sensitive
metastatic prostate cancer (10,11),
BRAF in melanoma and colon cancer (12–15),
EGFR in non-small cell lung cancer (16), Bcr-Abl in chronic myeloid leukemia
(CML) (17), ZAP-70 in chronic
lymphocytic leukemia (CLL) (18)
and c-KIT in gastrointestinal stromal tumors (19).
Over the past few years, numerous efforts have been
made to discover Hsp90 inhibitors from derivatives of natural
products to fully synthetic small molecules. Geldanamycin (GA),
first isolated from Streptomyces hygroscopicus as an
antibiotic in 1970, can bind specifically to the ATP pocket of
Hsp90 by positioning the benzoquinone ring to the entrance of the
binding pocket and the ansa ring towards the bottom of the pocket
(20). Once appropriately
positioned, GA forms hydrogen bonds with the pocket and restrains
Hsp90 in its ADP-bound conformation. This leads to immaturation and
degradation of the client proteins by the proteasomal pathway
(21,22). GA has exhibited effective anticancer
potency in pre-clinical studies. Yet, it has never been evaluated
at the clinical level as it displays severe hepatotoxicity
(stemming from the benzoquinone moiety), metabolical and chemical
unstability and very low solubility in aqueous solution (23).
17-Allylamino-17-desmethoxy-geldanamycin (17-AAG)
inhibited Her2 in breast cancer cells with IC50=31 nM
and is the first GA derivative that has proceeded to clinical
trials (24). Disappointingly, the
toxic profile of 17-AAG restricts its application (25). Moreover, this agent has limited
solubility in water and shows reduced activity in some
multidrug-resistant cells.
Radicicol (RD) is a macrocyclic lactone antibiotic
originally isolated from the fungus Monosporium bonorden in
1953 (26). The co-crystal
structure of RD with yeast Hsp90 shows that RD can exhibit a
C-shaped conformation similar to ADP and then competitively bind to
the N-terminal ATP pocket of Hsp90 (21). Thus, it restrains Hsp90 in its
ADP-bound conformation and leads to degradation of client proteins,
similar to GA. It has antitumor effects in vitro, but no
in vivo efficacy has been noted due to its instability in
serum (27). A series of RD analogs
have been synthesized and evaluated at preclinical and clinical
levels.
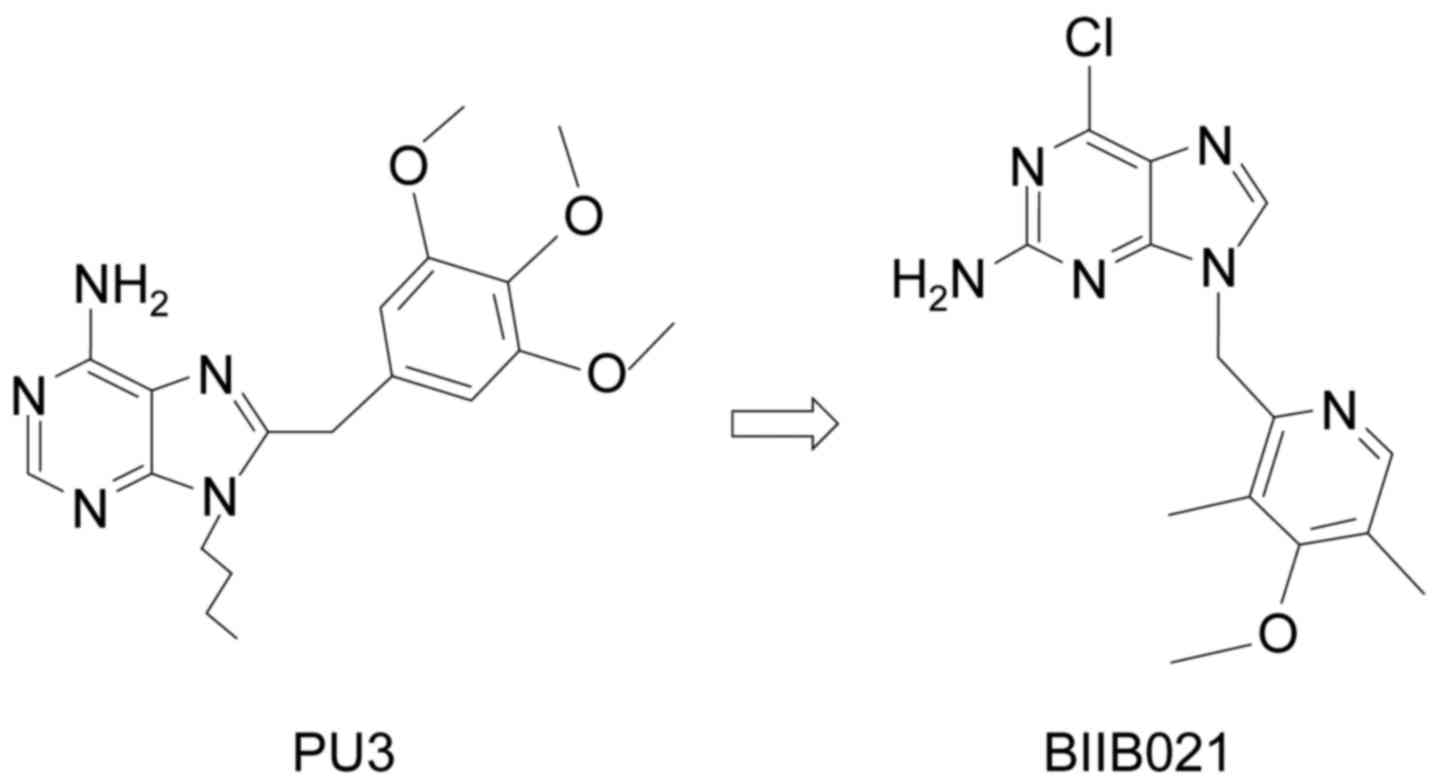
PU3
[9-butyl-8(3,4,5-trimethoxy-benzyl)-9H-purin-6-ylamine], the first
reported synthetic purine-scaffold inhibitor, was designed by
Chiosis and colleagues (28)
(Fig. 1). In a manner similar to
GA, PU3 can induce protein degradation. Yet, PU3 is not as potent
as 17-AAG.
Based on the structure of PU3, the aryl substituent
was shifted from 8- to the 9-position along with the NH2 from the
6- to the 2-position. Since the 9-benzyl series had poor aqueous
solubility, the phenyl ring was replaced with a 2′-pyridyl group
(29). BIIB021, the
3′,5′-dimethyl-4′-methoxy-2′-pyridyl derivative, is soluble in
biological fluids (Fig. 1). The
binding affinity of BIIB021 was found to be 1.7±0.4, better than
that of 17-AAG (4.6±0.5 nmol/l) (30). The drug is selective for Hsp90 over
kinases and another ATPase. Even at a low nanomolar concentration,
BIIB021 can inhibit the growth of cancer cells, such as N87, MCF-7
and BT474, with IC50 values ranging from 60 to 310
nmol/l. It also exhibited antitumor effects in a xenograft model.
As long as 48 h after treatment, the compound can be detected in
tumor tissues despite its short half-life in serum. The effects of
BIIB021 can last for more than 24 h. This provides the possibility
of flexible dose regimen to lessen dose-limiting hepatotoxicity.
Furthermore, the cytotoxic activity of BIIB021 is not influenced by
efflux pump, loss of NQO1 or Bcl-2 overexpression, avoiding the
limitations of 17-AAG (31).
In conclusion, BIIB021 is a promising Hsp90
inhibitor. This review focuses on the utility of BIIB021 in a wide
variety of cancers, especially in hematologic malignancies, and its
underlying mechanisms. In addition, this review is followed by a
discussion of current research on the application of Hsp90
inhibitors in blood malignancies.
Effects of BIIB021 in hematologic
malignancies
Chronic myeloid leukemia
The majority of chronic myeloid leukemia (CML) cases
are characterized by a fusion oncoprotein (Bcr-Abl) derived from
the reciprocal translocation between chromosomes 9 and 22.
Protein-tyrosine kinase expressed by this fusion protein activates
several intracellular signaling pathways that prevent cells from
apoptosis and promote cell proliferation. The introduction of
tyrosine kinase inhibitors (TKIs) has dramatically improved the
prognosis of CML patients. Yet, following the wide use of TKIs,
almost 30% of patients have developed resistance to the standard
treatment of imatinib. This phenomenon is attributed to mutations
in the kinase domain, genomic instability and Bcr-Abl
amplification, especially the T315I mutation even insensitive to
the first and second generation of TKIs (32). The third generation TKIs, ponatinib,
has antileukemia effect against CML cells with mutated Bcr-Abl
including T315I. However, the risk of serious thromboembolic events
restricts its application (33).
Novel approaches therefore should be developed to override TKI
resistance. Indeed, Bcr-Abl has been confirmed as a client protein
of Hsp90 (34). Inhibitors of Hsp90
can destabilize the binding of Bcr-Abl protein thus resulting in
formation of heteroprotein that is degraded via the ubiquitin
proteasomal pathway (34).
A series of CML cell lines including T315I mutant
were quite sensitive to BIIB021 (35). This drug inhibited cell
proliferation and induced caspase-dependent apoptosis. Moreover,
the oncoprotein Bcr-Abl was degraded and several signaling pathways
were downregulated, including JAK/STAT and Akt. BIIB021 decreased
the nuclear and cytoplasmic levels of β-catenin, a factor essential
for survival and self-renewal of leukemic stem cells and blockage
for the achievement of molecular remission in CML patients
(36). Notably, BIIB021 triggered
autophagy in CML cells independent of Beclin-1. Following exposure
to BIIB021, the expression of Beclin-1 was decreased. This was
contributed to activated caspase which cleaved Beclin-1 and the
N-terminal fragment of Beclin-1 in turn suppressing autophagy
(37,38). In fact, BIIB021 targeted mTOR
complex 1 to influence Ulk1 phosphorylation and eventually
initiated autophagy. Pretreatment of autophagy inhibitor
(3-methyladenine or bafilomycin A1) effectively increased the rate
of BIIB021-mediated cell death and apoptosis. This suggested a
possibility: BIIB021 combined with autophagy inhibitors in a
regimen would achieve optimized therapeutic effect against
imatinib-sensitive and -resistant CML, including cells harboring
T315I-mutant Bcr-Abl.
Lymphomas
Lymphoma is one of the most frequently diagnosed
human cancer. It may be divided into non-Hodgkin (90%) and Hodgkin
(10%) subtypes. A total of 90% of lymphomas are of B-cell origin
while some can be derived from T-cell or natural killer cells. Even
though treatment of Hodgkin's lymphoma (HL) has already achieved
great success, ~2% of patients with HL are refractory to
traditional therapy and 13% of patients suffer relapse (39). Hence, novel therapies are urgently
needed. Current research indicates that Hsp90 is highly expressed
in HL cells and promotes tumor survival by supporting the
activation of NF-κB signaling (40–42).
One previous study illustrated that BIIB021 induces a decrease in
NF-κB activity by 14–70% in HL cells with mutated IκB or functional
IκB. This means that BIIB021 inhibits the constitutionally active
NF-κB in HL cells despite IκB mutations. Moreover, Böll et
al (43) observed that BIIB021
decreased HL cell viability and had additive effects with
traditional chemotherapy (doxorubicin and gemcitabine).
Furthermore, this type of Hsp90 inhibitor increased the
susceptibility of HL cells against NK cell attack. Indeed, in
response to BIIB021, researchers observed enhanced expression of
NKG2D-specific ligands (MICA/B and ULBP2) on tumor cells. This in
turn increased NK cell-related cytotoxicity of HL cells and may
have a profound impact on the immune system and antitumor effects
(44,45). In accordance with the in
vitro effects, BIIB021 also inhibited tumor growth in
vivo.
In addition to HL, BIIB021 has shown efficacy
against Kaposi sarcoma-associated herpes virus (KSHV)-associated
primary effusion lymphoma (PEL) cells. PEL is a type of aggressive
non-HL with poor prognosis. It is characterized by KSHV infection
and frequently occurs in immunocompromised patients (46). Although a cytotoxic chemotherapeutic
regimen is available to cure PEL, the prognosis of this disease is
extremely poor and new therapeutic strategies must be found.
Treatment with BIIB021 induced cell cycle arrest and apoptosis in
PEL cells, by decreasing the level of several proteins critical for
cell cycle regulation (CDK, c-MYC and cyclin) and pathogenesis of
the disease (AKT, GSK3β and survivin) (47). As NF-κB activity is essential for
the survival and proliferation of PEL cells, researchers
demonstrated that BIIB021 can block the constitutive NF-κB pathway.
On the one hand, BIIB021 reduced the level and activity of
IKKα/IKKβ to influence the classical NF-κB pathway. On the other
hand, the Hsp90 inhibitor disrupted the K13-IKK complex resulting
in downregulation of vFLIP and K13 and blockage of K13-related
NF-κB activation. Moreover, the inability of BIIB021 to induce
expression of lytic genes and inhibition of their expression
alleviated the safety concern of lytic reactivation for KSHV lytic
gene replication and transcription which are crucial to KSHV
tumorigenesis. The antiproliferative effects were also evaluated in
a mouse xenograft model. Compared with control vehicle, BIIB021
significantly reduced tumor volume and prevented development of
splenomegaly.
More recently, it has emerged that BIIB021 is a
candidate suppressor of latent membrane protein 1 (LMP1) expression
which is a major oncogene encoded by the Epstein-Barr virus (EBV)
(48). EBV can infect B cells, T
cells and NK cells and is closely connected with immune cell
malignancies. BIIB021 decreased the viability, induced apoptosis
and caused cell cycle arrest of T and NK cell lines, including
EBV-positive T cell lines (SNT13 and SNT16) and NK cell lines (KAI3
and SNK6). The drug also had a similar effect on Jurkat and KHYG1
which are EBV-negative counterparts. The protein level of LMP1
downstream targets was decreased, such as NF-κB, JNK and Akt. A
murine xenograft model was used to demonstrate that BIIB021 can
inhibit the growth of EBV-positive NK cell lymphomas in
vivo.
T-cell acute lymphoblastic
leukemia
T-cell acute lymphoblastic leukemia (T-ALL) is a
clinically aggressive hematologic malignancy that accounts for 25%
of adult ALL and 15% of pediatric cases (49). Limited targeting therapies are
available for this type of disease at present. Li et al
(50) reported that BIIB021 can
inhibit the growth and induce the apoptosis of Molt-4 cells (a
human T-ALL cell line) at low nanomolar concentrations. Notably,
the drug disrupted the interaction between p53-MDM2 by suppressing
the expression of MDM2 while increasing the level of p53. This
resulted in p53-mediated apoptosis. Co-treatment with BIIB021 and
triptolide (TPL) exhibited a synergetic inhibitory effect on the
proliferation of Molt-4 cells. This phenomenon may be because TPL
can activate p53 without influence on MDM2 levels. Therefore, the
co-treatment markedly enhanced p53 activation and upregulated the
expression of several Bcl-2 family members (Bak and Bim).
Myelodysplastic syndrome
Researchers have previously reported that, compared
with healthy subjects and low-risk myelodysplastic syndrome (MDS)
patients, the level of Hsp90 is overexpressed in high-risk
counterparts and is associated with a poor outcome. A preclinical
trial examined the antitumor activity of BIIB021 on an MDS cell
line (SKM-1) (51). Following
BIIB021 treatment, SKM-1 cells were arrested in the G1 phase of the
cell cycle and underwent apoptosis. Furthermore, the study also
indicated that the mechanisms of apoptosis were attributed to a
decrease in phosphatidylinositide 3-kinase/Akt and nuclear
factor-κB pathway. All of the above findings imply that BIIB021 may
be a new available strategy for high-risk MDS.
Effects of BIIB021 in solid tumors
Gastrointestinal stromal tumors
Gastrointestinal stromal tumors (GISTs) are one of
the most common mesenchymal cancers of the digestive system
(52). Activating mutations in KIT
and PDGFRα, two receptor tyrosine kinases, are the key element in
the development and progression of GISTs. Specific mutations are
associated with the therapeutic response to imatinib and sunitinib;
however, most tumors ultimately become resistant to these drugs due
to secondary kinase mutations or alternative activated pathways
(53,54). KIT and PDGFRα are client proteins of
Hsp90 for their mutated forms rely on Hsp90 to stabilize (55). Inhibition of Hsp90 can result in the
degradation of any form of these kinases. A phase II study
evaluated the antitumor activity of BIIB021 in GIST patients
(56). Twenty-three patients were
stratified into two groups: 12 subjects received 600 mg twice
weekly (b.i.w.) while 11 subjects were administered 400 mg three
times weekly (t.i.w.). All patients had received prior treatment
and had acquired resistance to imatinib and sunitinib. By
evaluating the change from baseline (before day 1) to the end of
one cycle (day 29), 5 patients had a decline of >25% in SUVmax
and achieved a partial response (PR). Among them, 3 of 12 patients
(25%) were from 600 mg b.i.w. cohort and 2 of 11 patients (18%)
received 400 mg t.i.w. Another 9 patients suffered a lower decrease
in SUVmax although they did not meet the PR criterion. Moreover,
the study also suggested that a more frequent dose of BIIB021 may
induce better antitumor effect. The agent was generally well
tolerated as most of the drug-related adverse events were less than
grade 2. Compared with ansamycin derivatives such as IPI-504,
treatment of BIIB021 did not induce severe hepatotoxicity.
Advanced solid tumors
Patients were treated with BIIB021 in a phase I
dose-escalation trial with advanced solid tumors refractory to
standard treatment (57). A total
of 60 patients were enrolled in the study. The study determined the
maximum tolerated dose (MTD) and safety of this Hsp90 inhibitor on
two schedules: Twice a week for 3 weeks followed by 1 week off and
twice a week for 4 weeks in continuous 28 day cycles. In schedule
1, 50 subjects were given a dose ranging from 25 to 800 mg and
BIIB021 was found to be well tolerated at doses up to 700 mg twice
a week. Dosed with 800 mg, two cases of dose-limiting toxicity
(DLT) were observed: Syncope and dizziness. Based on the clinical
information collected in schedule 1, 6 subjects received a dose of
600 and 700 mg in schedule 2. An MTD was not established in this
schedule formally, whereas the 6 patients tolerated these doses.
The most common adverse events, defined as occurring in >20% of
patients, were nausea, hot flashes, vomiting and dizziness. These
events were mild or moderate.
Kaposi sarcoma
Latency-associated nuclear antigen (LANA) is
essential for Kaposi's sarcoma-associated herpesvirus (KSHV) genome
persistence and Kaposi sarcoma (KS) tumorigenesis (58,59).
Recently, Chen et al (60)
determined that KSHV LANA is a client protein of Hsp90 and
ATP-competitive Hsp90 inhibitors disrupted the association between
Hsp90 and LANA for Hsp90 bound the N-terminal domain of LANA. This
led to degradation of LANA through the ubiquitin-based proteasome
pathway. Depletion of Hsp90 by shRNA induced the apoptosis of PEL
cells. In vitro studies demonstrated that BIIB021 suppressed
the proliferation of KS cells (SLK-KSHV, L1T2, SLK and KS-IMM) and
decreased the expression of ephrin-B2 and EphA2 at low nanomolar
concentrations. Remarkably, compared with KSHV-negative SLK cells,
KSHV-positive counterparts were more sensitive to the Hsp90
inhibitor. Colony formation assays and cell cycle analysis further
verified the antitumor potency of BIIB021. In addition,
KSHV-infected L1T2 cells were injected into SCID mice to establish
a xenograft KSHV tumor model. Compared to the negative control, the
growth of tumors was notably retarded following treatment of Hsp90
inhibitor (AUY922) at a dose of 50 mg/kg. Immunohistochemistry
showed declined levels of LANA and ephrin-B2.
Squamous cell carcinoma
Every year, there are ~600,000 newly diagnosed cases
of head and neck squamous cell carcinoma (HNSCC) reported. At
present, the standard treatment includes surgery and/or
radiotherapy. Radiotherapy is mainly applied to patients with
advanced disease and chemotherapy is concomitantly used to increase
the efficacy. Recently, BIIB021 has been found as a new adjuvant
agent that enhances the sensitivity of HNSCC cell lines to
radiation (61).
BIIB021 showed better anti-proliferative effects in
4 cell lines with a mean IC50 value of 250 nM, superior
to 17-AAG. Compared with each treatment alone, co-treatment with
BIIB021 effectively increased the radiation-related cell death and
apoptosis even in radiation-resistant cell lines, by downregulating
several oncogenic proteins including EGFR, c-Raf-1 and Akt.
Moreover, BIIB021 enhanced the G2/M cell population in response to
radiotherapy independent of the p53 status.
In xenograft models, the tumors grew much smaller in
the combination arm. Tumor sizes remained stabilized following 3
weeks of treatment with BIIB021 or radiation alone while the volume
of tumors showed regression after 4 weeks with the combination
treatment.
Furthermore, BIIB021 potentiated a significant
therapeutic window of radiation to esophageal squamous cell
carcinoma cell lines by influencing apoptosis and the cell cycle
(62). BIIB021 dramatically
decreased the levels of radio-resistance-related proteins such as
EGFR, Akt and Raf-1.
Thyroid carcinoma
Although well-differentiated thyroid carcinoma
(WDTC) is a type of tumor associated with a good prognosis, 2/3 of
cases of metastatic WDTC are refractory. Meanwhile, patients with
anaplastic thyroid carcinoma exhibit poor outcome as the tumors are
highly aggressive. Recently, research illustrated that BIIB021
induced the cell death of thyroid carcinoma cell lines (8505C and
TPC-1) by degrading Hsp90 client proteins. Furthermore, cotreatment
with BIIB021 and histone acetyltransferase inhibitor triptolide
demonstrated a combined effect in regards to cytotoxicity
induction. This synergism was contributed to inhibition of the
PI3K/Akt/mTOR and NF-κB signaling pathways, a decrease of survivin,
xIAP and cIAP and promotion of DNA damage (63).
Targeting Hsp90 in hematologic
malignancies
The promising antitumor potency of BIIB021 in
leukemia and lymphoma motivates the focus of this inhibitor for the
treatment of blood malignancies by targeting Hsp90.
A high level of Hsp90 has been observed in leukemia
and myeloma (64–67). The Hsp90 level in plasma can be used
as a biomarker of leukemia engraftment and progression (68). Overexpression of the protein is
correlated with poor prognosis and chemotherapy resistance in acute
myeloid leukemia (AML) (69).
Furthermore, Hsp90 is required to maintain the stability and
function of oncogenic proteins such as c-KIT and FLT3-ITD in AML
and Bcr-Abl in CML (70).
Inhibition of Hsp90 extensively influences a variety of signaling
proteins involved in cell apoptosis, survival and differentiation.
Hence, it is reasonable to use Hsp90 inhibitors for the treatment
of hematologic malignancies.
Preclinical studies
Control of apoptosis and cell
cycle
Treatment with Hsp90 inhibitors could induce
apoptosis by activating the mitochondrial caspase pathway and
regulating Bcl-2 family proteins (50,71).
In diffuse large B-cell lymphoma (DLBCL), endogenous
Hsp90 interacts with Bcl6 and stabilizes Bcl6 at both the
transcription and protein level. Apoptosis was observed after
treatment with the Hsp90 inhibitor, PU-H71. The drug preferentially
accumulated in lymphomas and suppressed Bcl6-dependent DLBCL
xenografts (72). Recently, a novel
oncogenic pathway was found which promotes the aberrant survival of
T-ALL cells, i.e. tyrosine kinase 2 (TYK2)/phospho-STAT1/Bcl2
pathway (73). TYK2, as a member of
the JAK tyrosine family, is a client protein of Hsp90 (74,75).
An Hsp90 inhibitor NVP-AUY922 effectively degraded TYK2 and
resultantly decreased phospho-STAT1 and Bcl2, a pro-survival
protein. Meanwhile, the drug increased pro-apoptotic proteins Bim
and Bad (76).
Furthermore, Hsp90 inhibitors are able to regulate
the cell cycle. Mantle cell lymphoma (MCL) is characterized by the
overexpression of cyclin D1 and abnormal regulation of the cell
cycle. Treatment with Hsp90 inhibitors induced G0/1 arrest in MCL
cells and decreased cell cycle regulatory proteins, including
cyclin D1, cdk4, p21 and CHK1 (77,78).
Degradation of oncoprotein and
disruption of signaling transduction
FLT3-ITD and point mutation occur in 25–30% of AML
patients and are associated with poor prognosis. Meanwhile,
FLT3-ITD is a client protein of Hsp90. 17-AAG induced
polyubiquitination and proteasomal degradation of FLT3-ITD and
mutants by disrupting its association with Hsp90 (79,80).
Downstream signaling of FLT3-ITD such as JAK-STAT and PI3K/AKT was
also decreased (81). In addition,
5% of AML patients have KIT mutations and activated KIT kinase
plays an essential role in the pathophysiology of the disease
(82). 17-AAG and GA were found to
suppress the growth of cells expressing D816V-KIT (83).
The oncoprotein Bcr-Abl is involved in the
pathogenesis of Bcr-Abl+ human leukemia including CML
and Ph+ ALL. Bcr-Abl is a client protein of Hsp90, and
Hsp90 inhibitors have been developed as novel approaches for the
treatment of Bcr-Abl+ leukemia especially in relapsed or
IM-resistant cases. Hsp90 inhibitors were found to induce Bcr-Abl
degradation and this was accompanied by inhibition of downstream
signaling (JAK/STAT, Akt and β-catenin) including cells expressing
T315I mutation (35,84,85). A
novel Hsp90 inhibitor IPI-504 was found to effectively inhibit the
survival and proliferation of leukemic stem cells which is a
potential reason for relapse (86).
The drug prolonged the survival period of mice bearing Bcr-Abl
T315I-induced leukemia. In addition, Hsp90 inhibitors exerted a
combined effect with TKIs (87,88).
Bcr-Abl-negative myeloproliferative neoplasms (MPNs)
are a group of stem cell diseases that include polycythemia vera,
essential thrombocytosis and primary myelofibrosis. In the majority
of patients with MPNs, a mutation in the JAK2 kinase
(JAK2V617V) is always detected with constitutive activation
of the JAK2-STAT pathway which is independent of growth factors.
This allows hematopoietic cell proliferation in the absence of
cytokines. However, JAK2 inhibitors show limited efficacy in the
clinic. Hsp90 inhibitors have been evaluated for the treatment of
MPNs in preclinical studies, considering JAK2 is a client protein
of Hsp90. PU-H71, a new Hsp90 inhibitor, induced degradation of
JAK2, inhibited JAK-STAT signaling and triggered cell apoptosis in
JAK2 mutant cell lines and primary patient samples. PU-H71 also had
the potency to improve the survival period in mouse bone marrow
transplant models by disrupting JAK2 stability, without toxic
effects on normal hematopoiesis (89).
As the most common leukemia in the Western world,
B-cell CLL is a malignancy of mature B cells expressing T-cell
antigen CD5. The disease is characterized by elevated expression of
several Hsp90 client proteins making Hsp90 a potential therapeutic
target. At the molecular level, Hsp90 inhibitors led to depletion
of Akt, IKK and NF-κB, accompanied by a decline in NF-κB target
gene (MCL1, CFLAR, BIRC5 and BCL2) transcription and apoptosis in a
caspase-dependent manner (90,91).
Compared with normal B lymphocytes, Lyn is overexpressed,
abnormally exists in the cytosol of B-CLL and shows high activity
which mediates signaling cascade triggered by BCR. As Hsp90 binds
to the catalytic domain of Lyn, treatment with GA triggers the
cytosolic Lyn complex destabilizes in the early phases of apoptosis
and resultantly inactivates cytosolic Lyn (92). Recently, research indicated that
SOCS3 acts as a regulator of important cell survival pathways in
CLL. By activation of p38 signaling, 17-DMAG increased the SOCS3
level and in turn prohibited phosphorylation of AKT and STAT3, thus
inducing blockage of cell migration and survival in CLL (93).
Adult T-cell leukemia-lymphoma (ATL) is a
chemoresistant malignancy with an origin from
CD4+CD25+ T lymphocytes linked to HLTV-1.
Tax, encoded by the HTLV-1 genome, can control HTLV-1 replication
and advance oncogenic transformation of T lymphocytes. Recently,
research indicated that Hsp90 is a binding partner of Tax.
Downregulation of Hsp90 by 17-DMAG or shRNAs provoked Tax
degradation and this was accompanied by attenuation of NF-κB and
HTLV-1 LTR activation. Thus, 17-DMAG suppressed HTLV-1 replication
and led to apoptosis in cell lines and primary ATL cell samples
(94,95). The drug has no apparent effects on
normal PBMCs. In addition, PIM kinases and the β-catenin/TCF7L2
pathway underwent a decrease and resultantly these contributed to
Hsp90 inhibitor-associated cell apoptosis in ALT cells (95,96).
In an ATL mouse model, 17-DMAG administration reduced infiltration
of tumor cells into organs, inhibited de novo viral
production and prolonged the survival period (97).
Abrogation of micro-environment
protection
Hematological malignancies always develop in the
bone marrow and secondary lymphoid organs. These microenvironments
are characterized by various stromal and T cells that are essential
to cancer cell survival and drug resistance.
Microenvironment-targeted treatment has emerged and gained
attention in hemato-oncology.
In multiple myeloma (MM), 17-AAG suppressed the
expression of IGF1R and IL-6R on the cell surface and their
downstream signaling including IKK/NF-κB, PI-3K/Akt and Raf/MAPK.
Such effects abrogated the protection of bone marrow stromal cells
(BMSCs) on MM tumor cells and made them sensitive to other
anticancer agents (66).
Furthermore, treatment with SNX-2112 was able to overcome the
protective effects derived from cytokines and BMSCs. This is
because SNX-2112 inhibited Akt and MEK/ERK pathway even in the
presence of exogenous IL-6, IGF-1, or BMSCs and block the formation
of capillary-like tubes by suppression of eNOS/Akt. In addition, as
osteolytic bone destruction is a common complication of MM,
SNX-2112 has the potency to markedly inhibit osteoclastogenesis by
downregulation of ERK/c-fos and PU.1 (98). Exposure of Hsp90 inhibitor (KW-2478)
to MM cells induced a decrease in IgH translocation products
(FGFR3, c-Maf and cyclin D1). In a MM orthotopic model, KW-2478 not
only decreased the M protein level in serum but also reduced tumor
burden in bone marrow (99).
Within bone marrow and lymph nodes, CLL cells are
mixed with numerous T lymphocytes expressing CD40 ligand and IL-4.
Together with BMSCs and follicular dendritic cells, these cells can
protect CLL cells from chemotherapy-related apoptosis in
vitro. Co-cultured CLL cells with NTL or CD40L cells abrogated
fludarabine's ability to kill cells and dasatinib resistance also
occurred in NTL or CD154L/IL-4 co-culture system. By comparison,
Hsp90 inhibitor NVP-AUY922-AG retained its toxicity under the same
condition by decreasing IKKα, IKKβ, regulators of NF-κB, and
retarding the transcription of NF-κB target genes MCL1, CFLAR and
BIRC5. Considering fludarabine activation of MCL1 and BIRC5, Walsby
et al (91) treated
NVP-AUY922 with fludarabine and found that this Hsp90 inhibitor
potentiated CLL cell sensitivity to fludarabine. This combination
maintained the net transcriptional repression of MCL1, CFLAR and
BIRC5, providing a potential explanation for the synergy. The same
synergistic effect was also observed following cotreatment of
17-DMAG with dasatinib (100).
Overcoming drug resistance
Initially, there is only a minority of CLL patients
at diagnosis who present with TP53 mutation or deletion. However,
such defects are frequently obtained during the disease course and
induced p53 defects are strongly associated with resistance to
alkylating agents and purine analogues, the mainstay of current
treatment. Treatment with GA depressed the overexpressed mutant p53
protein while increased the level of wt counterparts. These
phenomena were ATM-independent and linked to a decrease in Akt and
activation of MDM2. p21, a potent inducer of cell cycle arrest, was
upregulation without dependence of p53/ATM. Cytotoxicity studies
demonstrated that Hsp90 inhibitors prohibited the proliferation of
cell lines and patient samples (101,102). It was suggested that Hsp90
inhibitor abrogated chemo-resistance in CLL with TP53 defects for
it killed cells independent of ATM or TP53 mutations. Furthermore,
SNX-7081, a synthetic Hsp90 inhibitor, synergized with fludarabine
against CLL cells as evidenced by a significant reduction in the
IC50 value of fludarabine to within a clinically
achievable range and a decrease in cell viability (103).
As described above, inhibitors of JAK2 have been
developed for the treatment of MPNs, CRLF2-rearranged B-ALL
and other tumors with activated JAK2 signaling. Research has
indicated that cells expressing G935R, Y931C and E864K mutations
near the ATP-binding region of the JAK2 kinase are resistance to a
panel of JAK inhibitors, whereas, these mutations had little
influence on tumor cell sensitivity to Hsp90 inhibitors. In fact,
AUY922 degraded both wild-type and mutant type of JAK2. AUY922 also
exhibited 100- to 1,000-fold more potency against B-ALL cells
harboring CRLF2 rearrangements than an enzymatic JAK2
inhibitor for the Hsp90 inhibitor has multi-targets (104).
Eradication of stem cells
Cancer stem cells (CSCs) are a subpopulation of
cancer cells with properties of quiescence, self-renewal and
persistent proliferation. CSCs are the key contributors to drug
resistance as well as relapse and metastasis (105).
At low concentrations, 17-AAG has the ability to
eliminate lymphoma stem cells in vitro and in vivo,
as the drug disrupts Hsp90-mediated mRNA expression and
transcriptional activity of HSF1α (106). Peng et al (86) isolated bone marrow cells from mice
with CML expressing T315I and showed that treatment with IPI-504
had a marked inhibitory effect on stem cells while IM had little
influence. Efficacious anti-CSC potency of IPI-504 was also
observed in mouse models without inhibition of normal CSCs. Hsp90
inhibitor decreased β-catenin, a factor essential for survival and
self-renewal of leukemic stem cells (36). In addition, co-treatment with Hsp90
inhibitor and SIRT1 inhibitor exhibited a combined effect on
chemo-resistant stem-like cells of CML (107). Depletion of SIRT1 prohibited the
17-AAG-mediated induction of Hsp70/Hsp27 and BCRP-mediated activity
of 17-AAG efflux and reduced the levels of CD44, Oct-4, β-catenin,
c-Myc and mut-p53.
Combined effects
Hsp90 inhibitors exert a synergistic effect together
with traditional or targeted chemotherapeutic agents including
doxorubicin, bortezomib, PI3K inhibitor, Akt inhibitor, IBP
inhibitor, rapamycin and HDACi (108–113). Recently, it was found that PU-H71
can target BCR signaling, and subsequently attenuate kinase
phosphorylation, calcium signaling and NF-κB activity. Combined
exposure to PU-H71 and ibrutinib, a BCR pathway inhibitor, induced
synergistic killing of DLBCL cell lines (114).
Bortezomib has been introduced to the treatment of
relapsed/refractory MCL for it can induce cell death through
upregulation of the BH3-only proapoptotic protein Noxa and
generation of reactive oxygen species rather than dependence on the
NF-κB pathway, whereas, not all patients respond to the drug and
resistance often appears. Based on an analysis of 18 MCL samples,
Roue et al (115) found
that these phenomena may result from an increase in pro-survival
chaperone Bip/Grp78 of which stabilization is dependent on Hsp90.
Simultaneous exposure to IPI-504 and bortezomib abrogated the
association between Hsp90 and Bip, then activated the ER stress
pathway and ultimately restored MCL cell sensitivity to bortezomib
both in vitro and in vivo. This combination deserves
further research at the clinical level.
In primary AML blasts, the 50% lethal dose of Hsp90
inhibitor (NVP-AUY922-AG) was observed at concentrations 2 logs
lower than cytarabine (Ara-C) which is a traditional chemotherapy
agent. There was a synergistic decrease in the proliferation of AML
cells and 20/25 primary samples following co-treatment with the two
agents (116,117). This was due to the fact that
Ara-C, even at low concentrations, induced activation of Chk1 which
facilitated cell survival. Conversely, following treatment of
17-AAG for 24 h, the expression of Chk1 was depleted. This was
accompanied by diminished Ara-C-related S phase accumulation and
decreased Cdc25A degradation (118).
Clinical trials
As the preclinical data suggest that Hsp90
inhibitors have anticancer activity via multiple mechanisms and are
an attractive therapeutic strategy, clinical trials have been
conducted to evaluate their MTD, safety and pharmacokinetic
properties (Table I).
 | Table I.Reported clinical studies of Hsp90
inhibitors in blood malignancies. |
Table I.
Reported clinical studies of Hsp90
inhibitors in blood malignancies.
| Blood
malignancies | Hsp90
inhibitor | Phase | Patient no. | DLTs | Findings |
|---|
| Acute leukemia | 17-DMAG | I | 24 | Cardiac
ischemia | MTD was 24
mg/m2 twice weekly; 3/17 achieved CR with incomplete
blood count recovery |
| B cell
malignancies | KW-2478 | I | 27 (22 MM and 5
nNL) | No DLTs | MTD was not
reached; 24/25 (96%) achieved SD with 5 being free of disease
progression for ≥6 months |
| Relapsed
lymphoma | 17-AAG | II | 22 (13 cHL and 9
MCL) | – | 7/18 (39 %)
demonstrated tumor reduction, 2/18 achieved PR |
| CLL | 17-DMAG | I | 15 | No DLTs | MTD was not
reached, 3/15 achieved SD |
| nHL | AUY922 | II | 20 (14 DLBCL and 6
PTCL) | – | 1/14 achieved CR in
DLBCL and 1/6 achieved PR in PTCL; 13/20 received only 1 cycle of
AUY922 or less due to apparent disease progression |
| MM | NVP-AUY922 | I | 24 | Blurred vision | No MTD was reached,
16/24 had SD |
| MM | 17-AAG+
bortezomib | I/II | 72 | Hepatotoxicity,
renal failure, metabolic acidosis | MTD was 17-AAG 340
mg/m2 and bortezomib 1.3 mg/m2; 2/67 achieved
CR, 8/67 achieved PR, 8/67 had a minimal response and 22/67 had
SD |
| MM |
NVP-AUY922+bortezomib | IB | 5 | Musculoskeletal
pain, diarrhea, atypical noncardiac chest and musculoskeletal
pain | 1/5 had PR, 4/5 had
SD |
| Acute leukemia | 17-AAG+ Ara-C | I | 26 | DIC, ARDS,
myocardial infarction | MTD was Ara-C 400
mg/m2/day for 5 days along with 17-AAG 300
mg/m2 on days 3 and 6; 2/21 had CR, 4/21 had PR |
In a phase I study, 24 patients with advanced AML
were enrolled and received escalating doses of 17-DMAG. This type
of Hsp90 inhibitor was well tolerated with toxicities of
neutropenic, fever, fatigue, nausea and diarrhea. The MTD
recommended was 24 mg/m2 on a twice-weekly dosing
schedule. Two cases of cardiac DLT were observed at 32
mg/m2. Compared with baseline, the apoptosis increased
within leukemic marrow CD34+ cells at day 15. Evidence
of 17-DMAG antitumor activity included three patients which
achieved a complete response (CR) with incomplete blood count
recovery (CRi) and one had a >50% bone marrow blast reduction.
Notably, among the 3 patients who had CRi, 2 showed del(7q)
karyotype (119). Whereas, another
phase I study indicated that 17-AAG had limited synergistic
activity with Ara-C at clinically tolerable doses. Among 21
patients, 2 achieved CR and 4 had PR. The drug combination induced
serious adverse effects: Disseminated intravascular coagulation
(DIC), acute respiratory distress syndrome (ARDS) and myocardial
infarction. A modest downregulation of Chk1 and other Hsp90 client
proteins was observed. This may have resulted from Hsp70
upregulation caused by the two drugs and, at clinical tolerated
dose, time was limited to sustain effective concentrations of
17-AAG that were able to downregulate substantial client proteins
in vivo (120). Other Hsp90
inhibitors with reduced toxicity, better solubility and improved
pharmacokinetics and pharmacodynamics should be developed and used
in the treatment of AML.
The oncogenic process of multiple myeloma (MM) is
not driven by classical client proteins of Hsp90. However, Hsp90
plays a critical role in the regulation of MM cell proliferation,
survival, drug resistance and microenvironmental interactions. In a
phase I/IB study, 24 patients with relapsed or refractory MM
received NVP-AUY922 at doses ranging from 8 to 70 mg/m2.
At 70 mg/m2, grade 3 blurred vision was observed while
no MTD was reached. None of the patients achieved CR or PR. A total
of 16/24 of the subjects had stable disease (SD). Another
non-ansamycin, non-purine Hsp90 inhibitor, KW-2478, was
administered intravenously in a phase I study in a dose range of 14
to 176 mg/m2 and was well tolerated without DLTs. A
total of 20 (95.2%) patients achieved SD among 24 evaluable
patients (121). As in
vitro and animal experiments suggest that 17-AAG synergizes
with standard therapy bortezomib, a phase I/II clinical trial
enrolled 72 patients with relapsed or refractory MM to assess the
combined effect. On days 1, 4, 8 and 11 in each 21-day cycle,
patients received 17-AAG (100–340 mg/m2) and bortezomib
(0.7–1.3 mg/m2) in a phase I study while 17-AAG (340
mg/m2) plus bortezomib (1.3 mg/m2) was
administered during the phase II expansion. Among the evaluable 67
patients, 2 patients (3%) had CR, 8 patients (12%) had PR, 8
patients (12%) had a minimal response and 22 patients (33%)
experienced SD (122). Prior
exposure and response to bortezomib dramatically influenced the
response rates; the highest rates were in bortezomib-naive patients
(10/21; 48%).
Hsp90 is commonly expressed in many types of B- and
T-cell lymphoma and its client proteins are critical in cell
proliferation and survival (40,67).
Hence, many lymphoma types are suitable targets for Hsp90
inhibitors and clinical trials were also conducted to examine the
effects of Hsp90 inhibitors on lymphoma. In a phase II study, 22
patients with relapsed lymphoma were enrolled and received 17-AAG
at 220 mg/m2. Seven of 18 patients suffered tumor
reduction of whom 2 had PR (123).
The biopsy specimens of MCL showed, after treatment of 17-AAG, the
expression of p-AKT, cyclin D1 and Ki-67 declined while activation
of caspase-3 increased. In addition, another open-label, single arm
phase II study evaluated the efficacy of AUY922 against DLBCL and
peripheral T-cell lymphoma (PTCL). After 2 cycles, 1/14 DLBCL
patients achieved a CR and 1/6 PTCL patients achieved PR (124).
Conclusion
In an era of precision medicine, clinicians are able
to design optimal therapies for patients with malignant tumors
according to molecular analyses. Hsp90 inhibitors are a type of
agents that target oncogenic proteins and signaling networks in
several tumors by influencing protein post-translational maturation
and disposition. Unfortunately, the clinical potency of first
generation Hsp90 inhibitors is limited as natural product
derivatives are toxic and have unfavorable pharmacokinetic
properties.
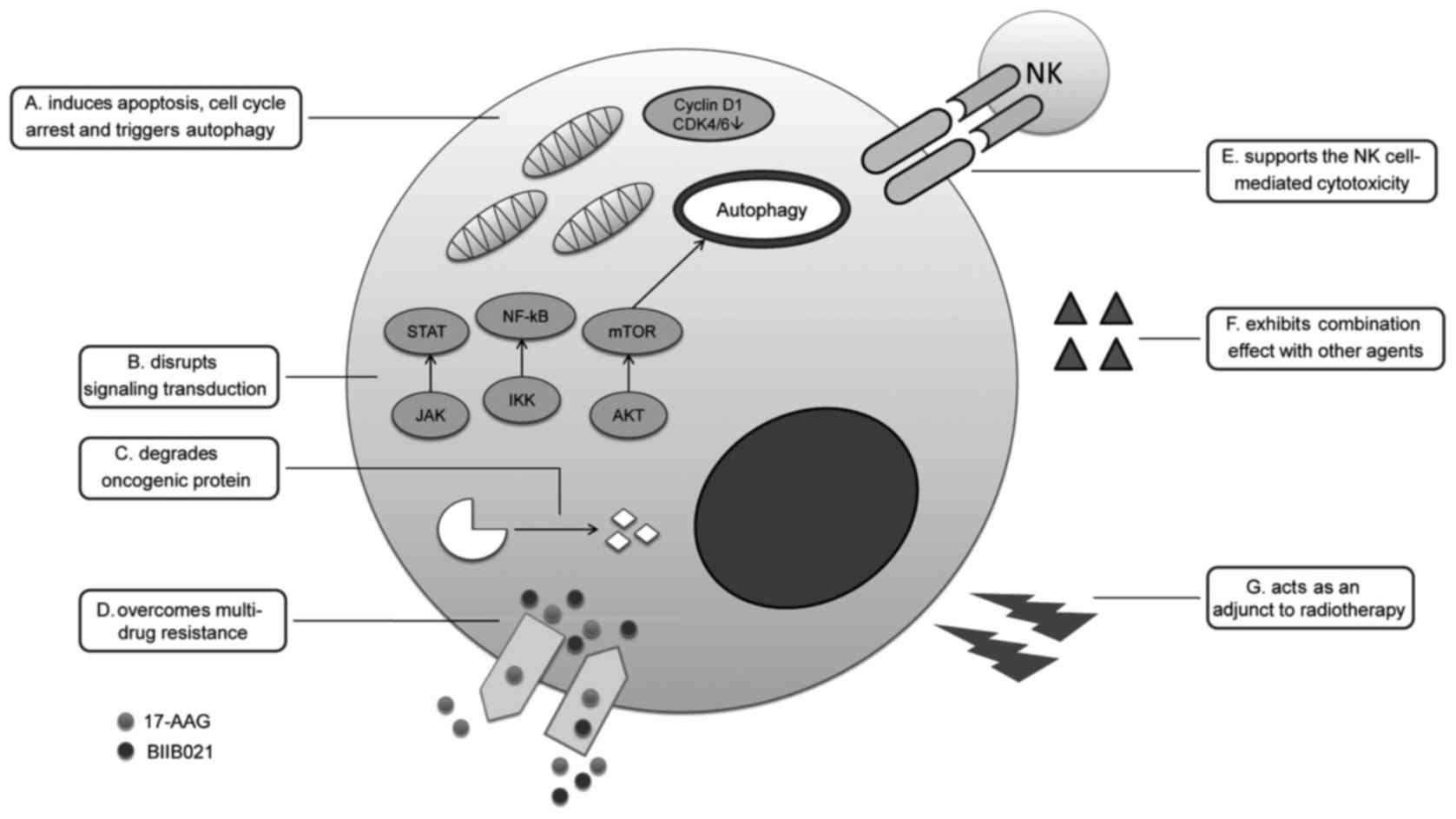
BIIB021, as a fully synthetic small-molecule
inhibitor, has shown effective antitumor potency. In vitro
experiments have demonstrated that it induces apoptosis, cell cycle
arrest and triggers autophagy of tumor cells. Treatment with
BIIB021 led to degradation of oncogenic proteins, disrupted
signaling transduction, supported NK cell-mediated cytotoxicity,
acted as an adjunct to radiotherapy, overcame multi-drug resistance
and exhibited combined effects with other agents (Fig. 2). Furthermore, this agent has
advantages of good solubility and low toxicity. phase I/II trials
indicate that the drug is active in serum, PBMCs and tumor tissue,
is well tolerated and could lead to objective responses in
refractory GIST patients (56,57).
BIIB021 is a promising Hsp90 inhibitor that deserves further
studies in regards to the treatment of patients with refractory or
relapsed tumors especially in blood malignancies.
The efficacy of BIIB021 against leukemia and
lymphoma allows us to focus on using this Hsp90 inhibitor in the
treatment of blood malignancies. Indeed, the expression level of
Hsp90 is high and is correlated with poor prognosis in leukemia and
myeloma (64–67). Inhibition of Hsp90 can result in
degradation of its client protein including Bcr-Abl, c-KIT and
FLT3-ITD which are oncoproteins or constitutively activated in
leukemia.
Pre-clinical data demonstrated that Hsp90 inhibitors
lead to apoptosis of cancer cells by varied mechanisms. Clinical
trials have demonstrated that Hsp90 inhibitors can enable a
population of patients with relapsed or refractory blood
malignancies to achieve CR, PR or SD (119,123,125) (Table
I). However, single treatment with an Hsp90 inhibitor showed
limited activity against various diseases (124). One potential solution may be
combination therapy. Several in vitro studies have provided
rationale for combining Hsp90 inhibitor with a number of other
agents, including Ara-C, bortezomib, imatinib, histone deacetylase
inhibitor and FLT3 inhibitor. In a phase I/II trial, 17-AAG +
bortezomib were well tolerated and 27% patients with relapsed or
refractory myeloma achieved objective response (122). In another phase I study, after
exposure to 17-AAG + Ara-C, the overall response rate was 23% among
26 patients with relapsed and refractory acute leukemia (120). At tolerable doses, the time of
17-AAG for effective concentration was insufficient to decrease
client proteins. This may contribute to the limited combination
activity. It is expected that more clinical studies can be
conducted to search for synergistic effects by using novel Hsp90
inhibitors with favorable pharmacologic properties.
In summary, BIIB021 is a novel agent in the fight
against cancer and targeting Hsp90 is a promising therapeutic
strategy for the management of blood malignancies.
Acknowledgements
Not applicable.
Funding
The present study was supported by Funds of the
Medical and Health Science Project of Zhejiang Province (no.
2017ZD029).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WH conceived and designed the study. WH and HXH
researched the literature, performed analysis of data and drafted
the manuscript. Both authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HSPs
|
heat shock proteins
|
|
CML
|
chronic myeloid leukemia
|
|
CLL
|
chronic lymphocytic leukemia
|
|
GA
|
geldanamycin
|
|
17-AAG
|
17-allylamino-17-desmethoxy-geldanamycin
|
|
RD
|
radicicol
|
|
TKIs
|
tyrosine kinase inhibitors
|
|
HL
|
Hodgkin's lymphoma
|
|
KSHV
|
Kaposi sarcoma-associated herpes
virus
|
|
PEL
|
primary effusion lymphoma
|
|
LMP1
|
latent membrane protein 1
|
|
EBV
|
Epstein-Barr virus
|
|
T-ALL
|
T-cell acute lymphoblastic
leukemia
|
|
TPL
|
triptolide
|
|
MDS
|
myelodysplastic syndrome
|
|
GISTs
|
gastrointestinal stromal tumors
|
|
PR
|
partial response
|
|
MTD
|
maximum tolerated dose
|
|
DLT
|
dose-limiting toxicity
|
|
LANA
|
latency-associated nuclear
antigen
|
|
KS
|
Kaposi sarcoma
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
WDTC
|
well-differentiated thyroid
carcinoma
|
|
AML
|
acute myeloid leukemia
|
|
DLBCL
|
diffuse large B-cell lymphoma
|
|
TYK2
|
tyrosine kinase 2
|
|
MCL
|
mantle cell lymphoma
|
|
MPNs
|
myeloproliferative neoplasms
|
|
ATL
|
adult T-cell leukemia-lymphoma
|
|
MM
|
multiple myeloma
|
|
BMSCs
|
bone marrow stromal cells
|
|
CSCs
|
cancer stem cells
|
|
Ara-C
|
cytarabine
|
|
CR
|
complete response
|
|
CRi
|
incomplete blood count recovery
|
|
DIC
|
disseminated intravascular
coagulation
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
SD
|
stable disease
|
|
PTCL
|
peripheral T-cell lymphoma
|
References
|
1
|
Amolins MW and Blagg BS: Natural product
inhibitors of Hsp90: Potential leads for drug discovery. Mini Rev
Med Chem. 9:140–152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang X, Chen M, Zhou J and Zhang X: HSP27,
70 and 90, anti-apoptotic proteins, in clinical cancer therapy
(Review). Int J Oncol. 45:18–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scaltriti M, Dawood S and Cortes J:
Molecular pathways: Targeting hsp90-who benefits and who does not.
Clin Cancer Res. 18:4508–4513. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mori M, Hitora T, Nakamura O, Yamagami Y,
Horie R, Nishimura H and Yamamoto T: Hsp90 inhibitor induces
autophagy and apoptosis in osteosarcoma cells. Int J Oncol.
46:47–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hernandez MP, Chadli A and Toft DO: HSP40
binding is the first step in the HSP90 chaperoning pathway for the
progesterone receptor. J Biol Chem. 277:11873–11881. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pratt WB, Galigniana MD, Morishima Y and
Murphy PJ: Role of molecular chaperones in steroid receptor action.
Essays Biochem. 40:41–58. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chandarlapaty S, Scaltriti M, Angelini P,
Ye Q, Guzman M, Hudis CA, Norton L, Solit DB, Arribas J, Baselga J
and Rosen N: Inhibitors of HSP90 block p95-HER2 signaling in
Trastuzumab-resistant tumors and suppress their growth. Oncogene.
29:325–334. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Scaltriti M, Serra V, Normant E, Guzman M,
Rodriguez O, Lim AR, Slocum KL, West KA, Rodriguez V, Prudkin L, et
al: Antitumor activity of the Hsp90 inhibitor IPI-504 in
HER2-positive trastuzumab-resistant breast cancer. Mol Cancer Ther.
10:817–824. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Y, Sawyers CL and Scher HI: Targeting
the androgen receptor pathway in prostate cancer. Curr Opin
Pharmacol. 8:440–448. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vanaja DK, Mitchell SH, Toft DO and Young
CY: Effect of geldanamycin on androgen receptor function and
stability. Cell Stress Chaperones. 7:55–64. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al: Improved survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
da Rocha Dias S, Friedlos F, Light Y,
Springer C, Workman P and Marais R: Activated B-RAF is an Hsp90
client protein that is targeted by the anticancer drug
17-allylamino-17-demethoxygeldanamycin. Cancer Res. 65:10686–10691.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haigis KM, Kendall KR, Wang Y, Cheung A,
Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A,
Sebolt-Leopold J, Shannon KM, et al: Differential effects of
oncogenic K-Ras and N-Ras on proliferation, differentiation and
tumor progression in the colon. Nat Genet. 40:600–608. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shimamura T, Lowell AM, Engelman JA and
Shapiro GI: Epidermal growth factor receptors harboring kinase
domain mutations associate with the heat shock protein 90 chaperone
and are destabilized following exposure to geldanamycins. Cancer
Res. 65:6401–6408. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shiotsu Y, Soga S and Akinaga S: Heat
shock protein 90-antagonist destabilizes Bcr-Abl/HSP90 chaperone
complex. Leuk Lymphoma. 43:961–968. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Castro JE, Prada CE, Loria O, Kamal A,
Chen L, Burrows FJ and Kipps TJ: ZAP-70 is a novel conditional heat
shock protein 90 (Hsp90) client: Inhibition of Hsp90 leads to
ZAP-70 degradation, apoptosis, and impaired signaling in chronic
lymphocytic leukemia. Blood. 106:2506–2512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bauer S, Yu LK, Demetri GD and Fletcher
JA: Heat shock protein 90 inhibition in imatinib-resistant
gastrointestinal stromal tumor. Cancer Res. 66:9153–9161. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stebbins CE, Russo AA, Schneider C, Rosen
N, Hartl FU and Pavletich NP: Crystal structure of an
Hsp90-geldanamycin complex: Targeting of a protein chaperone by an
antitumor agent. Cell. 89:239–250. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roe SM, Prodromou C, O'Brien R, Ladbury
JE, Piper PW and Pearl LH: Structural basis for inhibition of the
Hsp90 molecular chaperone by the antitumor antibiotics radicicol
and geldanamycin. J Med Chem. 42:260–266. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Prodromou C, Roe SM, O'Brien R, Ladbury
JE, Piper PW and Pearl LH: Identification and structural
characterization of the ATP/ADP-binding site in the Hsp90 molecular
chaperone. Cell. 90:65–75. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neckers L, Schulte TW and Mimnaugh E:
Geldanamycin as a potential anti-cancer agent: Its molecular target
and biochemical activity. Invest New Drugs. 17:361–373. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Solit DB, Zheng FF, Drobnjak M, Münster
PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB,
et al: 17-Allylamino-17-demethoxygeldanamycin induces the
degradation of androgen receptor and HER-2/neu and inhibits the
growth of prostate cancer xenografts. Clin Cancer Res. 8:986–993.
2002.PubMed/NCBI
|
|
25
|
Solit DB, Ivy SP, Kopil C, Sikorski R,
Morris MJ, Slovin SF, Kelly WK, DeLaCruz A, Curley T, Heller G, et
al: Phase I trial of 17-allylamino-17-demethoxygeldanamycin in
patients with advanced cancer. Clin Cancer Res. 13:1775–1782. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Delmotte P and Delmotte-Plaque J: A new
antifungal substance of fungal origin. Nature. 171:3441953.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soga S, Neckers LM, Schulte TW, Shiotsu Y,
Akasaka K, Narumi H, Agatsuma T, Ikuina Y, Murakata C, Tamaoki T
and Akinaga S: KF25706, a novel oxime derivative of radicicol,
exhibits in vivo antitumor activity via selective depletion of
Hsp90 binding signaling molecules. Cancer Res. 59:2931–2938.
1999.PubMed/NCBI
|
|
28
|
Chiosis G, Timaul MN, Lucas B, Munster PN,
Zheng FF, Sepp-Lorenzino L and Rosen N: A small molecule designed
to bind to the adenine nucleotide pocket of Hsp90 causes Her2
degradation and the growth arrest and differentiation of breast
cancer cells. Chem Biol. 8:289–299. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Taldone T and Chiosis G: Purine-scaffold
Hsp90 inhibitors. Curr Top Med Chem. 9:1436–1446. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lundgren K, Zhang H, Brekken J, Huser N,
Powell RE, Timple N, Busch DJ, Neely L, Sensintaffar JL, Yang YC,
et al: BIIB021, an orally available, fully synthetic small-molecule
inhibitor of the heat shock protein Hsp90. Mol Cancer Ther.
8:921–929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang H, Neely L, Lundgren K, Yang YC,
Lough R, Timple N and Burrows F: BIIB021, a synthetic Hsp90
inhibitor, has broad application against tumors with acquired
multidrug resistance. Int J Cancer. 126:1226–1234. 2010.PubMed/NCBI
|
|
32
|
Karvela M, Helgason GV and Holyoake TL:
Mechanisms and novel approaches in overriding tyrosine kinase
inhibitor resistance in chronic myeloid leukemia. Expert Rev
Anticancer Ther. 12:381–392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jain P, Kantarjian H, Jabbour E, Gonzalez
GN, Borthakur G, Pemmaraju N, Daver N, Gachimova E, Ferrajoli A,
Kornblau S, et al: Ponatinib as first-line treatment for patients
with chronic myeloid leukaemia in chronic phase: A phase 2 study.
Lancet Haematol. 2:e376–e383. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khajapeer KV and Baskaran R: Hsp90
inhibitors for the treatment of chronic myeloid leukemia. Leuk Res
Treatment. 2015:7576942015.PubMed/NCBI
|
|
35
|
He W, Ye X, Huang X, Lel W, You L, Wang L,
Chen X and Qian W: Hsp90 inhibitor, BIIB021, induces apoptosis and
autophagy by regulating mTOR-Ulk1 pathway in imatinib-sensitive and
-resistant chronic myeloid leukemia cells. Int J Oncol.
48:1710–1720. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Heidel FH, Bullinger L, Feng Z, Wang Z,
Neff TA, Stein L, Kalaitzidis D, Lane SW and Armstrong SA: Genetic
and pharmacologic inhibition of β-catenin targets
imatinib-resistant leukemia stem cells in CML. Cell Stem Cell.
10:412–424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li H, Wang P, Sun Q, Ding WX, Yin XM,
Sobol RW, Stolz DB, Yu J and Zhang L: Following cytochrome c
release, autophagy is inhibited during chemotherapy-induced
apoptosis by caspase 8-mediated cleavage of Beclin 1. Cancer Res.
71:3625–3634. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wirawan E, Vande Walle L, Kersse K,
Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R,
Verspurten J, Declercq W, et al: Caspase-mediated cleavage of
Beclin-1 inactivates Beclin-1-induced autophagy and enhances
apoptosis by promoting the release of proapoptotic factors from
mitochondria. Cell Death Dis. 1:e182010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Glimelius I and Diepstra A: Novel
treatment concepts in Hodgkin lymphoma. J Intern Med. 281:247–260.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Georgakis GV, Li Y, Rassidakis GZ,
Martinez-Valdez H, Medeiros LJ and Younes A: Inhibition of heat
shock protein 90 function by
17-allylamino-17-demethoxy-geldanamycin in Hodgkin's lymphoma cells
down-regulates Akt kinase, dephosphorylates extracellular
signal-regulated kinase, and induces cell cycle arrest and cell
death. Clin Cancer Res. 12:584–590. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Broemer M, Krappmann D and Scheidereit C:
Requirement of Hsp90 activity for IkappaB kinase (IKK) biosynthesis
and for constitutive and inducible IKK and NF-kappaB activation.
Oncogene. 23:5378–5386. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Janz M, Stühmer T, Vassilev LT and Bargou
RC: Pharmacologic activation of p53-dependent and p53-independent
apoptotic pathways in Hodgkin/Reed-Sternberg cells. Leukemia.
21:772–779. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Boll B, Eltaib F, Reiners KS, von Tresckow
B, Tawadros S, Simhadri VR, Burrows FJ, Lundgren K, Hansen HP,
Engert A, et al: Heat shock protein 90 inhibitor BIIB021 (CNF2024)
depletes NF-kappaB and sensitizes Hodgkin's lymphoma cells for
natural killer cell-mediated cytotoxicity. Clin Cancer Res.
15:5108–5116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Strid J, Roberts SJ, Filler RB, Lewis JM,
Kwong BY, Schpero W, Kaplan DH, Hayday AC and Girardi M: Acute
upregulation of an NKG2D ligand promotes rapid reorganization of a
local immune compartment with pleiotropic effects on
carcinogenesis. Nat Immunol. 9:146–154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Friese MA, Platten M, Lutz SZ, Naumann U,
Aulwurm S, Bischof F, Bühring HJ, Dichgans J, Rammensee HG, Steinle
A and Weller M: MICA/NKG2D-mediated immunogene therapy of
experimental gliomas. Cancer Res. 63:8996–9006. 2003.PubMed/NCBI
|
|
46
|
Nador RG, Cesarman E, Chadburn A, Dawson
DB, Ansari MQ, Sald J and Knowles DM: Primary effusion lymphoma: A
distinct clinicopathologic entity associated with the Kaposi's
sarcoma-associated herpes virus. Blood. 88:645–656. 1996.PubMed/NCBI
|
|
47
|
Gopalakrishnan R, Matta H and Chaudhary
PM: A purine scaffold HSP90 inhibitor BIIB021 has selective
activity against KSHV-associated primary effusion lymphoma and
blocks vFLIP K13-induced NF-kB. Clin Cancer Res. 19:5016–5026.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Suzuki M, Takeda T, Nakagawa H, Iwata S,
Watanabe T, Siddiquey MN, Goshima F, Murata T, Kawada J, Ito Y, et
al: The heat shock protein 90 inhibitor BIIB021 suppresses the
growth of T and natural killer cell lymphomas. Front Microbiol.
6:2802015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ferrando AA, Neuberg DS, Staunton J, Loh
ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland
DG, et al: Gene expression signatures define novel oncogenic
pathways in T cell acute lymphoblastic leukemia. Cancer Cell.
1:75–87. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li M, Zhang X, Zhou WJ, Chen YH, Liu H,
Liu L, Yang CM and Qan WB: Hsp90 inhibitor BIIB021 enhances
triptolide-induced apoptosis of human T-cell acute lymphoblastic
leukemia cells in vitro mainly by disrupting p53-MDM2 balance. Acta
Pharmacol Sin. 34:1545–1553. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lin S, Li J, Zhou W, Qian W, Wang B and
Chen Z: BIIB021, an Hsp90 inhibitor, effectively kills a
myelodysplastic syndrome cell line via the activation of caspases
and inhibition of PI3K/Akt and NF-kB pathway proteins. Exp Ther
Med. 7:1539–1544. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rubin BP, Heinrich MC and Corless CL:
Gastrointestinal stromal tumour. Lancet. 369:1731–1741. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen LL, Trent JC, Wu EF, Fuller GN,
Ramdas L, Zhang W, Raymond AK, Prieto VG, Oyedeji CO, Hunt KK, et
al: A missense mutation in KIT kinase domain 1 correlates with
imatinib resistance in gastrointestinal stromal tumors. Cancer Res.
64:5913–5919. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gramza AW, Corless CL and Heinrich MC:
Resistance to tyrosine kinase inhibitors in gastrointestinal
stromal tumors. Clin Cancer Res. 15:7510–7518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Solit DB and Rosen N: Hsp90: A novel
target for cancer therapy. Curr Top Med Chem. 6:1205–1214. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dickson MA, Okuno SH, Keohan ML, Maki RG,
D'Adamo DR, Akhurst TJ, Antonescu CR and Schwartz GK: phase II
study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal
tumors. Ann Oncol. 24:252–257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Saif MW, Takimoto C, Mita M, Banerji U,
Lamanna N, Castro J, O'Brien S, Stogard C and Von Hoff D: A phase
1, dose-escalation, pharmacokinetic and pharmacodynamic study of
BIIB021 administered orally in patients with advanced solid tumors.
Clin Cancer Res. 20:445–455. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ballestas ME, Chatis PA and Kaye KM:
Efficient persistence of extrachromosomal KSHV DNA mediated by
latency-associated nuclear antigen. Science. 284:641–644. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ballestas ME and Kaye KM: Kaposi's
sarcoma-associated herpesvirus latency-associated nuclear antigen 1
mediates episome persistence through cis-acting terminal repeat
(TR) sequence and specifically binds TR DNA. J Virol. 75:3250–3258.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chen W, Sin SH, Wen KW, Damania B and
Dittmer DP: Hsp90 inhibitors are efficacious against Kaposi Sarcoma
by enhancing the degradation of the essential viral gene LANA, of
the viral co-receptor EphA2 as well as other client proteins. PLoS
Pathog. 8:e10030482012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yin X, Zhang H, Lundgren K, Wilson L,
Burrows F and Shores CG: BIIB021, a novel Hsp90 inhibitor,
sensitizes head and neck squamous cell carcinoma to radiotherapy.
Int J Cancer. 126:1216–1225. 2010.PubMed/NCBI
|
|
62
|
Wang XT, Bao CH, Jia YB, Wang N, Ma W, Liu
F, Wang C, Wang JB, Song QX and Cheng YF: BIIB021, a novel Hsp90
inhibitor, sensitizes esophageal squamous cell carcinoma to
radiation. Biochem Biophys Res Commun. 452:945–950. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kim SH, Kang JG, Kim CS, Ihm SH, Choi MG,
Yoo HJ and Lee SJ: Synergistic cytotoxicity of BIIB021 with
triptolide through suppression of PI3K/Akt/mTOR and NF-kB signal
pathways in thyroid carcinoma cells. Biomed Pharmacother. 83:22–32.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yufu Y, Nishimura J and Nawata H: High
constitutive expression of heat shock protein 90 alpha in human
acute leukemia cells. Leuk Res. 16:597–605. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chant ID, Rose PE and Morris AG: Analysis
of heat-shock protein expression in myeloid leukaemia cells by flow
cytometry. Br J Haematol. 90:163–168. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Mitsiades CS, Mitsiades NS, McMullan CJ,
Poulaki V, Kung AL, Davies FE, Morgan G, Akiyama M, Shringarpure R,
Munshi NC, et al: Antimyeloma activity of heat shock protein-90
inhibition. Blood. 107:1092–1100. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Valbuena JR, Rassidakis GZ, Lin P, Atwell
C, Georgakis GV, Younes A, Jones D and Medeiros LJ: Expression of
heat-shock protein-90 in non-Hodgkin's lymphomas. Mod Pathol.
18:1343–1349. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Milani M, Laranjeira AB, de Vasconcellos
JF, Brandalise SR, Nowill AE and Yunes JA: Plasma Hsp90 level as a
marker of early acute lymphoblastic leukemia engraftment and
progression in mice. PLoS One. 10:e01292982015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Flandrin P, Guyotat D, Duval A, Cornillon
J, Tavernier E, Nadal N and Campos L: Significance of heat-shock
protein (HSP) 90 expression in acute myeloid leukemia cells. Cell
Stress Chaperones. 13:357–364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Reikvam H, Hatfield KJ, Ersvaer E, Hovland
R, Skavland J, Gjertsen BT, Petersen K and Bruserud O: Expression
profile of heat shock proteins in acute myeloid leukaemia patients
reveals a distinct signature strongly associated with FLT3 mutation
status-consequences and potentials for pharmacological
intervention. Br J Haematol. 156:468–480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Tsai HJ, Shih NY, Kuo SH, Cheng AL, Lin
HY, Chen TY, Chang KC, Lin SF, Chang JS and Chen LT: AUY922
effectively targets against activated B cell subtype of diffuse
large B-cell lymphoma and low-grade lymphoma cells harboring
genetic alteration-associated nuclear factor-kB activation. Leuk
Lymphoma. 56:2674–2682. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Cerchietti LC, Lopes EC, Yang SN, Hatzi K,
Bunting KL, Tsikitas LA, Mallik A, Robles AI, Walling J,
Varticovski L, et al: A purine scaffold Hsp90 inhibitor
destabilizes BCL-6 and has specific antitumor activity in
BCL-6-dependent B cell lymphomas. Nat Med. 15:1369–1376. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sanda T, Tyner JW, Gutierrez A, Ngo VN,
Glover J, Chang BH, Yost A, Ma W, Fleischman AG, Zhou W, et al:
TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic
leukemia. Cancer Discov. 3:564–577. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Taipale M, Krykbaeva I, Koeva M, Kayatekin
C, Westover KD, Karras GI and Lindquist S: Quantitative analysis of
HSP90-client interactions reveals principles of substrate
recognition. Cell. 150:987–1001. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Caldas-Lopes E, Cerchietti L, Ahn JH,
Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A,
Guo Y, et al: Hsp90 inhibitor PU-H71, a multimodal inhibitor of
malignancy, induces complete responses in triple-negative breast
cancer models. Proc Natl Acad Sci USA. 106:8368–8373. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Akahane K, Sanda T, Mansour MR, Radimerski
T, DeAngelo DJ, Weinstock DM and Look AT: HSP90 inhibition leads to
degradation of the TYK2 kinase and apoptotic cell death in T-cell
acute lymphoblastic leukemia. Leukemia. 30:219–228. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Georgakis GV, Li Y and Younes A: The heat
shock protein 90 inhibitor 17-AAG induces cell cycle arrest and
apoptosis in mantle cell lymphoma cell lines by depleting cyclin
D1, Akt, Bid and activating caspase 9. Br J Haematol. 135:68–71.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sugimoto K, Sasaki M, Isobe Y, Tsutsui M,
Suto H, Ando J, Tamayose K, Ando M and Oshimi K: Hsp90-inhibitor
geldanamycin abrogates G2 arrest in p53-negative leukemia cell
lines through the depletion of Chk1. Oncogene. 27:3091–3101. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
George P, Bali P, Annavarapu S, Scuto A,
Fiskus W, Guo F, Sigua C, Sondarva G, Moscinski L, Atadja P and
Bhalla K: Combination of the histone deacetylase inhibitor LBH589
and the hsp90 inhibitor 17-AAG is highly active against human
CML-BC cells and AML cells with activating mutation of FLT-3.
Blood. 105:1768–1776. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yu C, Kancha RK and Duyster J: Targeting
oncoprotein stability overcomes drug resistance caused by FLT3
kinase domain mutations. PLoS One. 9:e971162014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Al Shaer L, Walsby E, Gilkes A, Tonks A,
Walsh V, Mills K, Burnett A and Rowntree C: Heat shock protein 90
inhibition is cytotoxic to primary AML cells expressing mutant FLT3
and results in altered downstream signalling. Br J Haematol.
141:483–493. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Beghini A, Peterlongo P, Ripamonti CB,
Larizza L, Cairoli R, Morra E and Mecucci C: C-kit mutations in
core binding factor leukemias. Blood. 95:726–727. 2000.PubMed/NCBI
|
|
83
|
Tsujimura A, Kiyoi H, Shiotsu Y, Ishikawa
Y, Mori Y, Ishida H, Toki T, Ito E and Naoe T: Selective KIT
inhibitor KI-328 and HSP90 inhibitor show different potency against
the type of KIT mutations recurrently identified in acute myeloid
leukemia. Int J Hematol. 92:624–633. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Barnes DJ, De S, van Hensbergen P,
Moravcsik E and Melo JV: Different target range and cytotoxic
specificity of adaphostin and
17-allylamino-17-demethoxygeldanamycin in imatinib-resistant and
sensitive cell lines. Leukemia. 21:421–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Jin L, Xiao CL, Lu CH, Xia M, Xing GW,
Xiong S, Liu QY, Liu H, Li YC, Ge F, et al: Transcriptomic and
proteomic approach to studying SNX-2112-induced K562 cells
apoptosis and anti-leukemia activity in K562-NOD/SCID mice. FEBS
Lett. 583:1859–1866. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Peng C, Brain J, Hu Y, Goodrich A, Kong L,
Grayzel D, Pak R, Read M and Li S: Inhibition of heat shock protein
90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia
and suppresses leukemic stem cells. Blood. 110:678–685. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Radujkovic A, Schad M, Topaly J, Veldwijk
MR, Laufs S, Schultheis BS, Jauch A, Melo JV, Fruehauf S and Zeller
WJ: Synergistic activity of imatinib and 17-AAG in
imatinib-resistant CML cells overexpressing BCR-ABL-Inhibition of
P-glycoprotein function by 17-AAG. Leukemia. 19:1198–1206. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Tauchi T, Okabe S, Ashihara E, Kimura S,
Maekawa T and Ohyashiki K: Combined effects of novel heat shock
protein 90 inhibitor NVP-AUY922 and nilotinib in a random
mutagenesis screen. Oncogene. 30:2789–2797. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Marubayashi S, Koppikar P, Taldone T,
Abdel-Wahab O, West N, Bhagwat N, Caldas-Lopes E, Ross KN, Gonen M,
Gozman A, et al: HSP90 is a therapeutic target in JAK2-dependent
myeloproliferative neoplasms in mice and humans. J Clin Invest.
120:3578–3593. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hertlein E, Wagner AJ, Jones J, Lin TS,
Maddocks KJ, Towns WH III, Goettl VM, Zhang X, Jarjoura D, Raymond
CA, et al: 17-DMAG targets the nuclear factor-kappaB family of
proteins to induce apoptosis in chronic lymphocytic leukemia:
Clinical implications of HSP90 inhibition. Blood. 116:45–53. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Walsby E, Pearce L, Burnett AK, Fegan C
and Pepper C: The Hsp90 inhibitor NVP-AUY922-AG inhibits NF-kB
signaling, overcomes microenvironmental cytoprotection and is
highly synergistic with fludarabine in primary CLL cells.
Oncotarget. 3:525–534. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Trentin L, Frasson M, Donella-Deana A,
Frezzato F, Pagano MA, Tibaldi E, Gattazzo C, Zambello R, Semenzato
G and Brunati AM: Geldanamycin-induced Lyn dissociation from
aberrant Hsp90-stabilized cytosolic complex is an early event in
apoptotic mechanisms in B-chronic lymphocytic leukemia. Blood.
112:4665–4674. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen TL, Gupta N, Lehman A, Ruppert AS, Yu
L, Oakes CC, Claus R, Plass C, Maddocks KJ, Andritsos L, et al:
Hsp90 inhibition increases SOCS3 transcript and regulates migration
and cell death in chronic lymphocytic leukemia. Oncotarget.
7:28684–28696. 2016.PubMed/NCBI
|
|
94
|
Gao L and Harhaj EW: HSP90 protects the
human T-cell leukemia virus type 1 (HTLV-1) tax oncoprotein from
proteasomal degradation to support NF-kB activation and HTLV-1
replication. J Virol. 87:13640–13654. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Taniguchi H, Hasegawa H, Sasaki D, Ando K,
Sawayama Y, Imanishi D, Taguchi J, Imaizumi Y, Hata T, Tsukasaki K,
et al: Heat shock protein 90 inhibitor NVP-AUY922 exerts potent
activity against adult T-cell leukemia-lymphoma cells. Cancer Sci.
105:1601–1608. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kurashina R, Ohyashiki JH, Kobayashi C,
Hamamura R, Zhang Y, Hirano T and Ohyashiki K: Anti-proliferative
activity of heat shock protein (Hsp) 90 inhibitors via
beta-catenin/TCF7L2 pathway in adult T cell leukemia cells. Cancer
Lett. 284:62–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ikebe E, Kawaguchi A, Tezuka K, Taguchi S,
Hirose S, Matsumoto T, Mitsui T, Senba K, Nishizono A, Hori M, et
al: Oral administration of an HSP90 inhibitor, 17-DMAG, intervenes
tumor-cell infiltration into multiple organs and improves survival
period for ATL model mice. Blood Cancer J. 3:e1322013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Okawa Y, Hideshima T, Steed P, Vallet S,
Hall S, Huang K, Rice J, Barabasz A, Foley B, Ikeda H, et al:
SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell
growth, angiogenesis, and osteoclastogenesis in multiple myeloma
and other hematologic tumors by abrogating signaling via Akt and
ERK. Blood. 113:846–855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Nakashima T, Ishii T, Tagaya H, Seike T,
Nakagawa H, Kanda Y, Akinaga S, Soga S and Shiotsu Y: New molecular
and biological mechanism of antitumor activities of KW-2478, a
novel nonansamycin heat shock protein 90 inhibitor, in multiple
myeloma cells. Clin Cancer Res. 16:2792–2802. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
McCaig AM, Cosimo E, Leach MT and Michie
AM: Dasatinib inhibits B cell receptor signalling in chronic
lymphocytic leukaemia but novel combination approaches are required
to overcome additional pro-survival microenvironmental signals. Br
J Haematol. 153:199–211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lin K, Rockliffe N, Johnson GG,
Sherrington PD and Pettitt AR: Hsp90 inhibition has opposing
effects on wild-type and mutant p53 and induces p21 expression and
cytotoxicity irrespective of p53/ATM status in chronic lymphocytic
leukaemia cells. Oncogene. 27:2445–2455. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Best OG, Singh N, Forsyth C and Mulligan
SP: The novel Hsp-90 inhibitor SNX7081 is significantly more potent
than 17-AAG against primary CLL cells and a range of haematological
cell lines, irrespective of lesions in the TP53 pathway. Br J
Haematol. 151:185–188. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Best OG, Che Y, Singh N, Forsyth C,
Christopherson RI and Mulligan SP: The Hsp90 inhibitor SNX-7081
synergizes with and restores sensitivity to fludarabine in chronic
lymphocytic leukemia cells with lesions in the TP53 pathway: A
potential treatment strategy for fludarabine refractory disease.
Leuk Lymphoma. 53:1367–1375. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Weigert O, Lane AA, Bird L, Kopp N, Chapuy
B, van Bodegom D, Toms AV, Marubayashi S, Christie AL, McKeown M,
et al: Genetic resistance to JAK2 enzymatic inhibitors is overcome
by HSP90 inhibition. J Exp Med. 209:259–273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ghia P, Chiorazzi N and Stamatopoulos K:
Microenvironmental influences in chronic lymphocytic leukaemia: The
role of antigen stimulation. J Intern Med. 264:549–562. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Newman B, Liu Y, Lee HF, Sun D and Wang Y:
HSP90 inhibitor 17-AAG selectively eradicates lymphoma stem cells.
Cancer Res. 72:4551–4561. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kim HB, Lee SH, Um JH, Kim MJ, Hyun SK,
Gong EJ, Oh WK, Kang CD and Kim SH: Sensitization of
chemo-resistant human chronic myeloid leukemia stem-like cells to
Hsp90 inhibitor by SIRT1 inhibition. Int J Biol Sci. 11:923–934.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Born EJ, Hartman SV and Holstein SA:
Targeting HSP90 and monoclonal protein trafficking modulates the
unfolded protein response, chaperone regulation and apoptosis in
myeloma cells. Blood Cancer J. 3:e1672013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Huston A, Leleu X, Jia X, Moreau AS, Ngo
HT, Runnels J, Anderson J, Alsayed Y, Roccaro A, Vallet S, et al:
Targeting Akt and heat shock protein 90 produces synergistic
multiple myeloma cell cytotoxicity in the bone marrow
microenvironment. Clin Cancer Res. 14:865–874. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ishii T, Seike T, Nakashima T, Juliger S,
Maharaj L, Soga S, Akinaga S, Cavenagh J, Joel S and Shiotsu Y:
Anti-tumor activity against multiple myeloma by combination of
KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J.
2:e682012. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Chatterjee M, Andrulis M, Stühmer T,
Müller E, Hofmann C, Steinbrunn T, Heimberger T, Schraud H,
Kressmann S, Einsele H and Bargou RC: The PI3K/Akt signaling
pathway regulates the expression of Hsp70, which critically
contributes to Hsp90-chaperone function and tumor cell survival in
multiple myeloma. Haematologica. 98:1132–1141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Kaiser M, Lamottke B, Mieth M, Jensen MR,
Quadt C, Garcia-Echeverria C, Atadja P, Heider U, von Metzler I,
Türkmen S and Sezer O: Synergistic action of the novel HSP90
inhibitor NVP-AUY922 with histone deacetylase inhibitors,
melphalan, or doxorubicin in multiple myeloma. Eur J Haematol.
84:337–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Francis LK, Alsayed Y, Leleu X, Jia X,
Singha UK, Anderson J, Timm M, Ngo H, Lu G, Huston A, et al:
Combination mammalian target of rapamycin inhibitor rapamycin and
HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin has
synergistic activity in multiple myeloma. Clin Cancer Res.
12:6826–6835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Goldstein RL, Yang SN, Taldone T, Chang B,
Gerecitano J, Elenitoba-Johnson K, Shaknovich R, Tam W, Leonard JP,
Chiosis G, et al: Pharmacoproteomics identifies combinatorial
therapy targets for diffuse large B cell lymphoma. J Clin Invest.
125:4559–4571. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Roué G, Pérez-Galan P, Mozos A,
López-Guerra M, Xargay-Torrent S, Rosich L, Saborit-Villarroya I,
Normant E, Campo E and Colomer D: The Hsp90 inhibitor IPI-504
overcomes bortezomib resistance in mantle cell lymphoma in vitro
and in vivo by down-regulation of the prosurvival ER chaperone
BiP/Grp78. Blood. 117:1270–1279. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Walsby EJ, Lazenby M, Pepper CJ, Knapper S
and Burnett AK: The HSP90 inhibitor NVP-AUY922-AG inhibits the PI3K
and IKK signalling pathways and synergizes with cytarabine in acute
myeloid leukaemia cells. Br J Haematol. 161:57–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lazenby M, Hills R, Burnett AK and
Zabkiewicz J: The HSP90 inhibitor ganetespib: A potential effective
agent for Acute Myeloid Leukemia in combination with cytarabine.
Leuk Res. 39:617–624. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Mesa RA, Loegering D, Powell HL, Flatten
K, Arlander SJ, Dai NT, Heldebrant MP, Vroman BT, Smith BD, Karp
JE, et al: Heat shock protein 90 inhibition sensitizes acute
myelogenous leukemia cells to cytarabine. Blood. 106:318–327. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lancet JE, Gojo I, Burton M, Quinn M,
Tighe SM, Kersey K, Zhong Z, Albitar MX, Bhalla K, Hannah AL, et
al: Phase I study of the heat shock protein 90 inhibitor
alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice
weekly to patients with acute myeloid leukemia. Leukemia.
24:699–705. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Kaufmann SH, Karp JE, Litzow MR, Mesa RA,
Hogan W, Steensma DP, Flatten KS, Loegering DA, Schneider PA,
Peterson KL, et al: Phase I and pharmacological study of cytarabine
and tanespimycin in relapsed and refractory acute leukemia.
Haematologica. 96:1619–1626. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Yong K, Cavet J, Johnson P, Morgan G,
Williams C, Nakashima D, Akinaga S, Oakervee H and Cavenagh J:
phase I study of KW-2478, a novel Hsp90 inhibitor, in patients with
B-cell malignancies. Br J Cancer. 114:7–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Richardson PG, Chanan-Khan AA, Lonial S,
Krishnan AY, Carroll MP, Alsina M, Albitar M, Berman D, Messina M
and Anderson KC: Tanespimycin and bortezomib combination treatment
in patients with relapsed or relapsed and refractory multiple
myeloma: Results of a phase 1/2 study. Br J Haematol. 153:729–740.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Oki Y, Copeland A, Romaguera J, Fayad L,
Fanale M, Faria Sde C, Medeiros LJ, Ivy P and Younes A: Clinical
experience with the heat shock protein-90 inhibitor, tanespimycin,
in patients with relapsed lymphoma. Leuk Lymphoma. 53:990–992.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Oki Y, Younes A, Knickerbocker J,
Samaniego F, Nastoupil L, Hagemeister F, Romaguera J, Fowler N,
Kwak L and Westin J: Experience with HSP90 inhibitor AUY922 in
patients with relapsed or refractory non-Hodgkin lymphoma.
Haematologica. 100:e272–e274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Maddocks K, Hertlein E, Chen TL, Wagner
AJ, Ling Y, Flynn J, Phelps M, Johnson AJ, Byrd JC and Jones JA: A
phase I trial of the intravenous Hsp90 inhibitor alvespimycin
(17-DMAG) in patients with relapsed chronic lymphocytic
leukemia/small lymphocytic lymphoma. Leuk Lymphoma. 57:2212–2215.
2016. View Article : Google Scholar : PubMed/NCBI
|