Introduction
Cervical cancer remains the second leading cause of
cancer-related deaths among women worldwide, accounting for 275,000
deaths annually (1). It is
estimated that 12,990 new cases of cervical cancer were diagnosed
in the United States in 2016, and 4120 people succumbed to the
disease (2). Although cervical
cancer death rates are decreasing among women in some developed
countries, the incidence remains high among Southeast Asia, Central
and Eastern Europe, the Caribbean, sub-Saharan Africa, and Latin
America populations due to persistent human papillomavirus
infections that lead to the occurrence of cancerous lesions
(3–5).
The approaches currently used to treat cancer, such
as radiation therapy, chemotherapy, gene therapy, or immune-based
treatments, usually kill cancer cells by inducing apoptosis.
Apoptosis is an intrinsic form of programmed cell death that occurs
in response to various physiological and pathological processes.
Apoptosis is identified by typical changes in cell morphology and
biology, including cell shrinkage, nuclear chromatin condensation
and DNA fragmentation (6,7).
Apoptosis can be triggered via both the intrinsic
(mitochondrial) and extrinsic (Fas receptor) pathways, with all
signaling converging on key effectors that include caspase-3 or −7
(8). In the intrinsic apoptotic
pathway, caspase activation is tightly linked to permeabilization
of the outer mitochondrial membrane by pro-apoptotic members of the
Bcl-2 family (6,9). Cells typically die via apoptosis in a
healthy organism. However, in tumor tissues, there is an
insufficient number of apoptotic cells resulting in uncontrolled
cell proliferation. For this reason, drugs that selectively trigger
apoptosis of cancer cells by targeting the extrinsic pathway offer
promise for cancer treatment.
Targeting the extrinsic pathway to trigger apoptosis
in cancer cells is an attractive approach for cancer therapy since
death receptors are a key factor for the cell death process
(7,10). Activation of the intrinsic pathway
in cancer cells is another way to trigger apoptosis. For example,
some chemotherapeutic agents, such as doxorubicin, cisplatin or
paclitaxel, can enhance mitochondrial permeabilization by
increasing the concentration of pro-apoptotic second messengers or
by triggering perturbations of intermediary metabolism in an
indirect fashion (6). However,
cancer cells often develop resistance to these drugs during these
types of therapies (11,12). Thus, searching for new alternatives
remains urgent in cancer therapy.
Taspine, a natural alkaloid that exists in many
types of plants, was identified in our laboratory from Radix et
Rhizoma Leonticis by using cell membrane chromatography. This
compound exhibited inhibitory effects on some cancer cells
(14–16). As one of the novel biphenyl urea
taspine derivatives, TPD7
(N-(4′-acetyl-3′,5,6-trimethoxybiphenyl-3-yl)-N'-[4-(3-morpholin-4-ylpropoxy)phenyl]urea)
(13) (Fig. 1A) significantly inhibited the
proliferation of different cancer cell lines. In the present study,
we demonstrated that TPD7 effectively decreased the growth of HeLa
cervical cancer cells. Mechanistically, we determined that TPD7
caused cell cycle blockade and triggered apoptosis in these
cells.
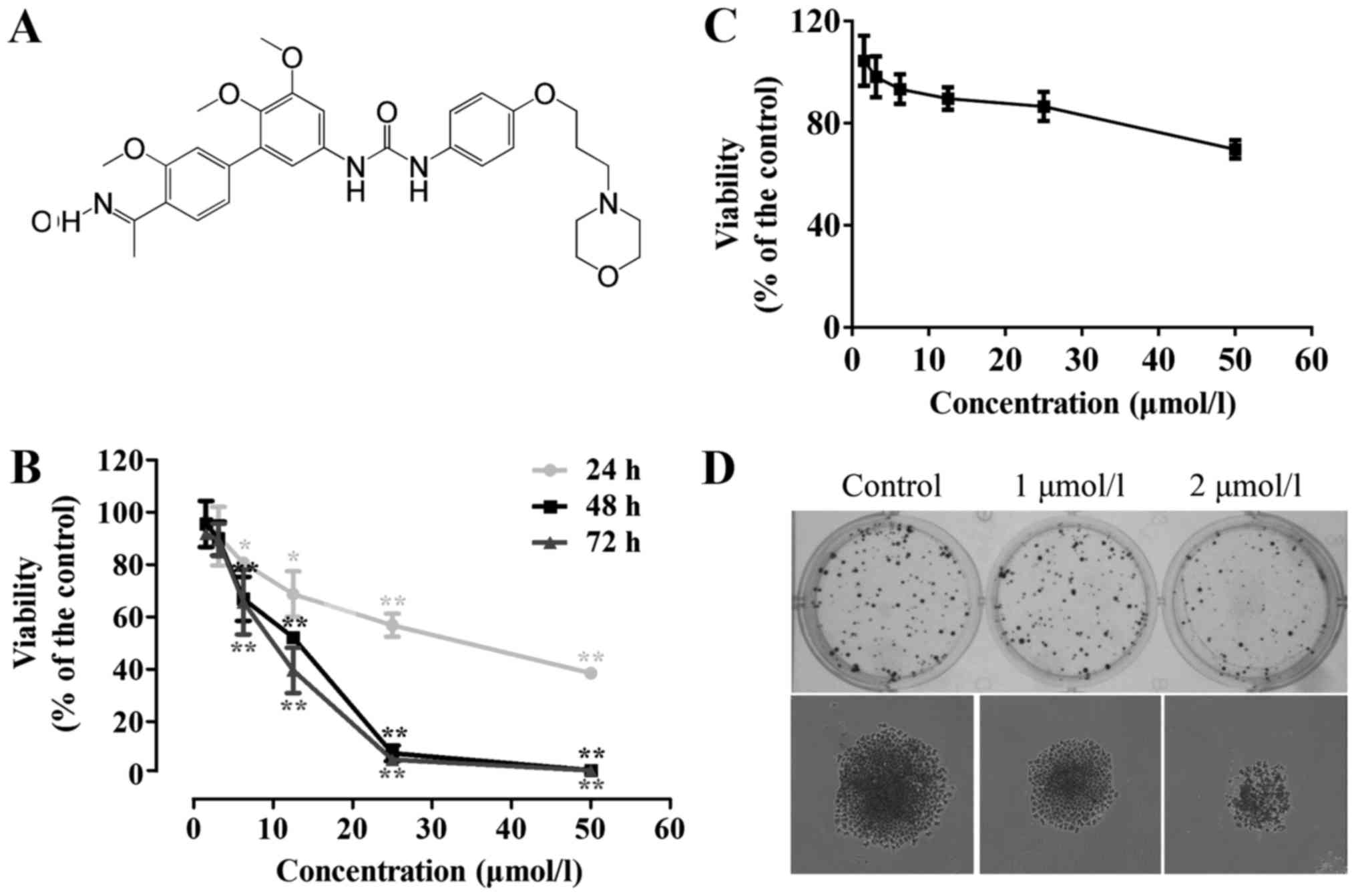 | Figure 1.TPD7 suppresses the proliferation and
colony formation of HeLa cells. (A) Chemical structure of TPD7. (B)
HeLa cells were treated with 1.56, 3.13, 6.25, 12.5, 25 and 50
µmol/l TPD7 for 24, 48 and 72 h. *P<0.05; **P<0.01 compared
with the control. (C) Human primary endocervical epithelial cells
were treated with 1.56, 3.13, 6.25, 12.5, 25 and 50 µmol/l TPD7 for
24, 48 and 72 h. (D) TPD7 suppressed the colony formation of HeLa
cells. The upper image is of a single well of a 6-well plate
photographed after staining with 0.2% crystal violet solution. The
lower image is of a representative single colony. Quantitative data
are presented as the means ± SEM for triplicate experiments. |
Materials and methods
Animals and cell culture
The use of Balb/c mice for the purpose of this study
was approved by the Animal Ethics Committee of Xi'an Jiaotong
University (Permission no. XJTULAC2013-031). All experiments were
conducted in accordance with the approved guidelines of the
regional authorities according to Xi'an Jiaotong University animal
care regulations. Female immunodeficient Balb/c mice (4–6 weeks of
age) were obtained from the Shanghai Laboratory Animal Center of
the Chinese Academy of Sciences and housed under aseptic and
ventilated conditions.
The HeLa cervical cancer cell line, obtained from
the Shanghai Institute of Cell Biology at the Chinese Academy of
Sciences, was cultured in RPMI-1640 medium (Sigma-Aldrich; Merck
KGaA, St. Louis, MO, USA) supplemented with 10% (v/v) fetal bovine
serum (FBS; Hyclone Laboratories; GE Healthcare Life Sciences,
Logan, UT, USA). Human primary endocervical epithelial cells were
obtained from CHI Scientific, Ltd. (Wuxi, China), and maintained in
DMEM/F12 medium (Sigma-Aldrich; Merck KGaA) supplemented with 10%
(v/v) FBS (Hyclone Laboratories; GE Healthcare Life Sciences), EGF,
insulin and hydrocortisone. All cell lines were incubated at 37°C
in a 5% CO2 incubator with saturated humidity. The
cumulative culture length of HeLa cells was fewer than six months
after resuscitation.
Cell viability assay
For the HeLa cell line, cells were seeded in a
96-well plate at a density of 4000 cells/well and treated with
1.56, 3.13, 6.25, 12.5, 25 and 50 µmol/l TPD7 for 24, 48 and 72 h.
Then, 0.5 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma-Aldrich; Merck KGaA) solution was added to each well. After
4 h of incubation, the absorbance of each well was measured at 490
nm with a microplate reader (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). For human primary endocervical epithelial cells, cells
were seeded in a 96-well plate at a density of 4000 cells/well and
treated with 1.56, 3.13, 6.25, 12.5, 25 and 50 µmol/l TPD7 for 48
h. Then, an MTT assay was performed to evaluate the effect of TPD7
on cell viability.
Colony formation assay
HeLa cells were trypsinized and seeded in a 6-well
plate at a density of 400 cells/well. After overnight adherence,
cells were treated with 1 or 2 µmol/l TPD7 for 10–15 days. Cell
numbers >50 were counted as colonies after staining with 0.2%
crystal violet solution (Beijing Chemical Works, Beijing,
China).
Cell cycle analysis
For the cell cycle analysis, HeLa cells were
trypsinized and seeded in a 6-well plate at a density of
6×105 cells/well. After overnight adherence, the cells
were incubated with 2, 4 and 8 µmol/l TPD7 for 48 h. The cells were
then harvested with trypsin/EDTA, resuspended and fixed overnight
with ice-cold 70% ethanol at 4°C. RNase and propidium iodide (PI)
were then added and the mixture was incubated for 30 min away from
light. Cell samples were then analyzed for the cell cycle
distribution using a flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA).
Hoechst staining assay
Hoechst staining was used to observe apoptotic
changes in nuclear morphology. HeLa cells were cultured in a 6-well
plate at a density of 2×105 cells/well and then treated
with 2, 4 and 8 µmol/l TPD7 for 48 h. After incubation, the cells
were washed three times with phosphate-buffered saline (PBS) and
fixed with paraformaldehyde at 4°C for 30 min. Cells were then
washed with PBS and allowed to dry on the bottom of the well.
Hoechst 33342 was added to each well and incubated at 37°C for 10
min. The dye was then removed and cells were washed with PBS prior
to observation of the cellular morphology under a microscope (Nikon
Eclipse Ti-E; Nikon, Tokyo, Japan). The number of apoptotic cells
was determined by assessing the percentage of cells displaying
chromatin condensation compared to the total number of cells.
Analysis of apoptosis by flow
cytometry
HeLa cells seeded in a 6-well plate at a density of
6×105 cells/well were incubated with 2, 4 and 8 µmol/l
TPD7 for 48 h, then trypsinized with trypsin-EDTA, centrifuged and
suspended in PBS. Following double staining with Annexin V-FITC (5
µl) and 20 µg/ml PI (10 µl) for 15 min away from light, the samples
were analyzed for apoptosis by flow cytometry.
Cell migration assays
To determine the effect of TPD7 on HeLa cell
migration, wound healing and Millicell (EMD Millipore, Billerica,
MA, USA) chamber assays were used. HeLa cells were seeded at
2×105 cells/well in a 12-well plate and allowed to grow
overnight up to 70% confluency in complete medium. Cells were then
incubated in serum-free medium for 24 h. Wounds were made by
scratching the cell monolayers with a 10–100 µl pipette tip.
Wounded monolayers were washed several times with serum-free medium
to remove floating cells and photographed under a microscope. Cells
were then treated with 2, 4 and 8 µmol/l TPD7 for 48 and 72 h and
the average distance the cells migrated into the scratched area was
determined under an inverted microscope as an index of cell
migration ability.
To conduct the chamber assay, HeLa cells suspended
in culture medium were seeded at a density of 4×104
cells/well in an upper chamber (8-µm pore size) placed in a 12-well
plate. Vehicle- or TPD7-containing medium supplemented with 10% FBS
was added to the upper chamber the following day. Medium
supplemented with 30% FBS was added to the lower chamber as a
chemoattractant. After 48 h of treatment, the cells on the upper
surface of the chamber were carefully removed and migrating cells
on the lower surface were fixed with 100% methanol and stained with
0.2% crystal violet (Beijing Chemical Works). Stained cells on the
lower surface were visualized and images were captured in five
microscopic fields that were selected randomly (magnification,
×100).
Invasion assay
HeLa cells were seeded at a density of
4×104 cells/well in an upper Millicell chamber (8-µm
pore size; EMD Millipore) coated with 100 µl Matrigel
(Becton-Dickinson; BD Biosciences, San Jose, CA, USA) at a
concentration of 1 mg/ml, which was placed in a 12-well plate.
Vehicle- or TPD7-containing medium supplemented with 10% FBS was
added to the upper chamber the following day. Medium supplemented
with 30% FBS was added to the lower chamber as a chemoattractant.
Following 48 h of treatment, the cells on the upper surface of the
chamber were removed and invading cells were fixed with 100%
methanol and stained with 0.2% crystal violet (Beijing Chemical
Works), and images were captured in five randomly selected
microscopic fields (magnification, ×100).
Immunoblotting analysis
HeLa cells were seeded at a density of
6×105 cells/well in a 6-well plate and incubated with 2,
4, and 8 µmol/l TPD7 for 48 h. Cells were then washed with PBS to
remove the medium and lysed with RIPA lysis buffer (Applygen
Technologies, Inc., Beijing, China) supplemented with protease
inhibitor cocktail tablets and phosphatase inhibitor cocktail
tablets (Roche Diagnostics, Basel, Switzerland). The cell lysates
were spinned at 12,000 × g at 4°C for 10 min and the supernatants
were collected. Protein concentration in supernatants was assessed
using a BCA protein quantification kit (Bio-Rad Laboratories,
Inc.). An equal amount of proteins was separated by 10% SDS-PAGE,
transferred to PVDF membranes (EMD Millipore). The membranes were
then incubated with primary and secondary antibodies, respectively.
The blots were detected using ECL reagents (Thermo Fisher
Scientific, Inc.). The GAPDH antibody (1:1,000 dilution; cat. no.
10494-1-AP; ProteinTech Group, Inc., Chicago, IL, USA) was used as
internal control. The primary antibodies including cyclin D1 (cat.
no. 2978), CDK2 (cat. no. 2546), Apaf-1 (cat. no. 8969), Bak (cat.
no. 6947), Bax (cat. no. 5023), Bcl-2 (cat. no. 4223), Bad (cat.
no. 9239), Mcl-1 (cat. no. 94296), Fas (cat. no. 4233), cleaved
caspase-3 (Asp175) (cat. no. 9664), cleaved PARP (cat. no. 5625)
(all from Cell Signaling Technology, Danvers, MA, USA, 1:1,000
dilution before use), cyclin A2 (cat. no. 18202-1-AP), CDK1 (cat.
no. 19532-1-AP), FADD (cat. no. 14906-1-AP) (all from ProteinTech
Group, 1:1,000 dilution before use), p53 (cat. no. 1026-1), NF-κB
p65 (cat. no. 1546-1), CXCR4 (cat. no. 3108-1), MMP-2 (cat. no.
1948-1), MMP-9 (cat. no. 1939-1) (all from Epitomics; Abcam
Cambridge, MA, USA, 1:1,000 dilution before use) were detected in
this assay.
Animal experiments
For acute toxicity assessment, Balb/c female mice
were divided randomly into five groups of eight. Mice received a
single dose of TPD7 at 100, 500, 1,000, 2,000 and 5,000 mg/kg,
while the control animals received equivalent volumes of solvent.
General behavioral and body weight changes, hazardous symptoms and
mortality were observed the next day and for 21 days.
For xenograft studies, female nude Balb/c mice at
4–6 weeks of age were implanted subcutaneously in the right flank
with HeLa cells resuspended in 5% saline. Mice were observed daily
for the presence of palpable tumors. Tumor volumes (in
cm3) were determined on alternate days by using a
vernier caliper and the formula: (length × width2)/2.
Body weights were assessed daily to monitor overall health. Mice
were divided randomly into four groups of eight when tumor volumes
reached ~0.1 cm3. In all studies, animals received
either vehicle (equivalent volumes of solvent) or TPD7 daily at 50,
100, or 200 mg/kg for 21 days. For assessment of the effect of TPD7
treatment in HeLa xenograft-bearing mice, tumor tissues were
excised from the injection site and weighed after mice were
sacrificed. The percentage inhibition of tumor growth was
calculated with the following formula: [(A - B)/A] × 100, where A
and B were the mean tumor weights of the control and treatment
groups, respectively.
Statistical analyses
Quantitative data are presented as the means ±
standard error of the mean (SEM). All data analyses were performed
with the Prism software (GraphPad, Inc., La Jolla, CA, USA), and
statistical comparisons were performed with SPSS 19.0 (IBM Corp.,
Armonk, NY, USA). ANOVA was used to determine statistical
significance of the data compared with the control group. P<0.05
was considered to indicate a statistically significant
difference.
Results
TPD7 suppresses viability and colony
formation of HeLa cells
An MTT assay was used to evaluate the effect of TPD7
on the proliferation of human cervical cancer HeLa cells. TPD7
exhibited significant inhibitory effects on HeLa cell proliferation
in a dose- and time-dependent fashion (Fig. 1B). The 50% inhibitory concentration
(IC50) of TPD7 at 48 h was 9.95 µmol/l, whereas the
IC50 value of TPD7 to inhibit human primary endocervical
epithelial cells at 48 h was higher than 50 µmol/l (Fig. 1B and C), suggesting that TPD7 was
considerably less active in human primary endocervical epithelial
cells. Concentrations lower than the IC50 value were
considered less toxic, and were used in further studies. In
addition, the IC50 value in HeLa cells was not
significantly different between TPD7 at 48 and 72 h of treatment.
Thus, 2, 4 and 8 µmol/l of TPD7 at 48 h of treatment was adopted to
investigate the anticancer potential of TPD7 on HeLa cells in
subsequent mechanism studies.
The colony formation assay revealed that TPD7
treatment generated a significantly lower number of colonies
compared with the vehicle (Fig.
1D). These results demonstrated that TPD7 exhibited potential
anticancer properties in human cervical cancers in
vitro.
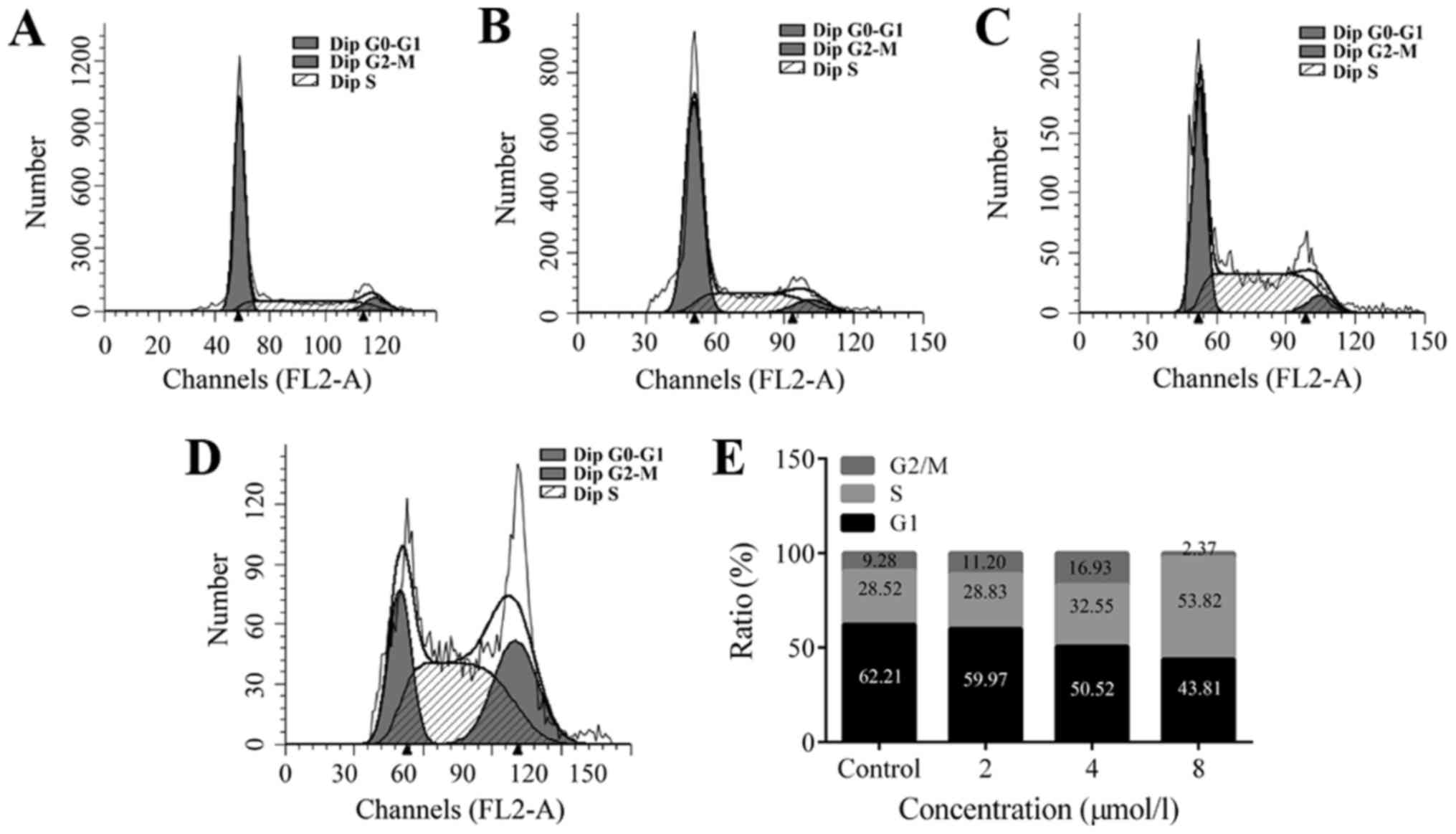
TPD7 induces S phase arrest in HeLa
cells
To assess the mechanisms underlying the suppressive
effects of TPD7 on HeLa cell growth, we evaluated cell cycle
progression after TPD7 treatment. Cells treated with TPD7 for 48 h
displayed S phase arrest (Fig. 2).
The percentages of cells in the S phase were 28.83, 32.55 and
58.82% following treatment with 2, 4 and 8 µmol/l TPD7,
respectively. Accordingly, the number of cells in the G2/M phase
decreased from 62.21 to 59.97, 50.52, and 43.81% with 2, 4 and 8
µmol/l TPD7, respectively. These data indicated that TPD7 regulated
the distribution of cells by blocking the cell cycle in the S
phase.
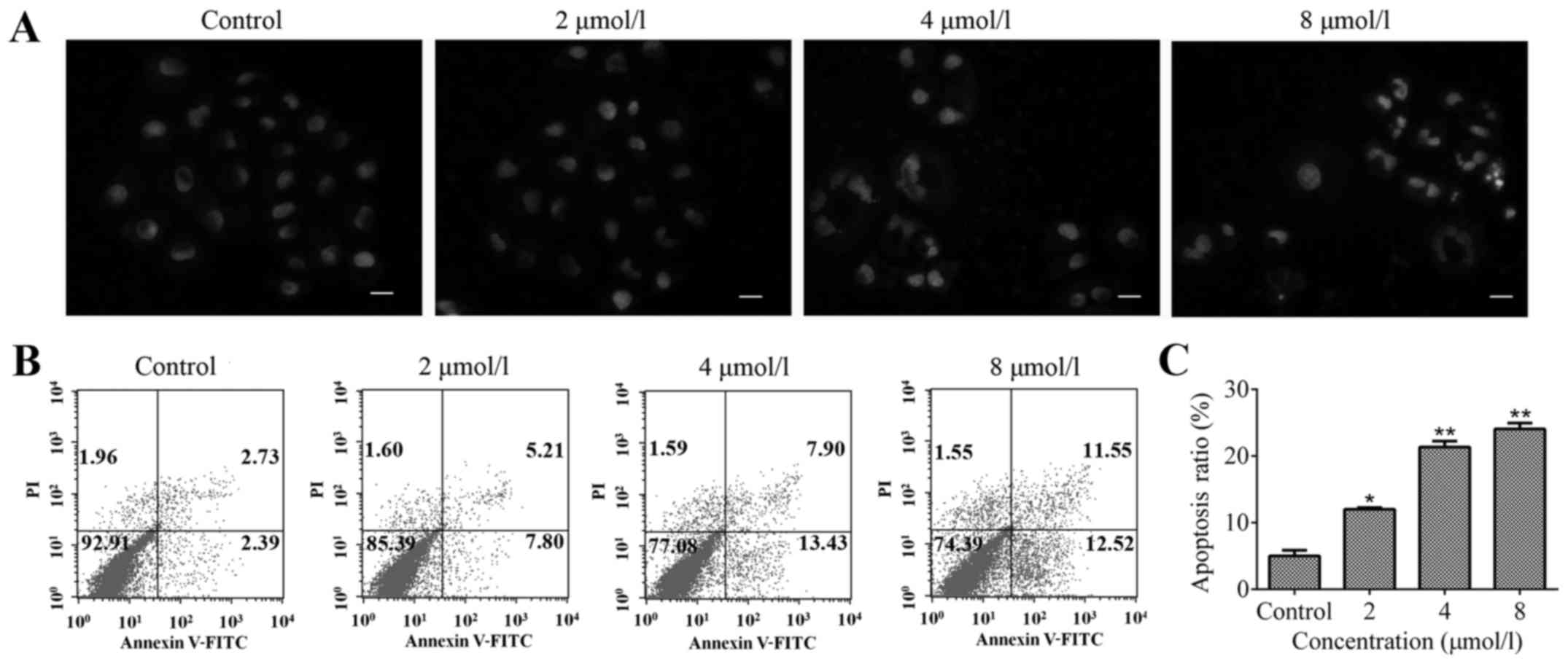
TPD7 induces apoptosis of HeLa cells. Hoechst
staining was used to determine nuclear morphological changes of
cells exposed to various concentrations of TPD7. Treatment with 2,
4 and 8 µmol/l TPD7 for 48 h induced evident changes in nuclear
morphology such as nuclear condensation or fragmentation (Fig. 3A).
Flow cytometric analysis was used to investigate the
percentage of apoptotic cells induced by TPD7. The results revealed
that, compared with the control, the percentage of apoptotic cells
was elevated in the TPD7 treatment groups (Fig. 3B and C). Collectively, the results
in Fig. 3 indicated that TPD7 could
induce HeLa cell apoptosis.
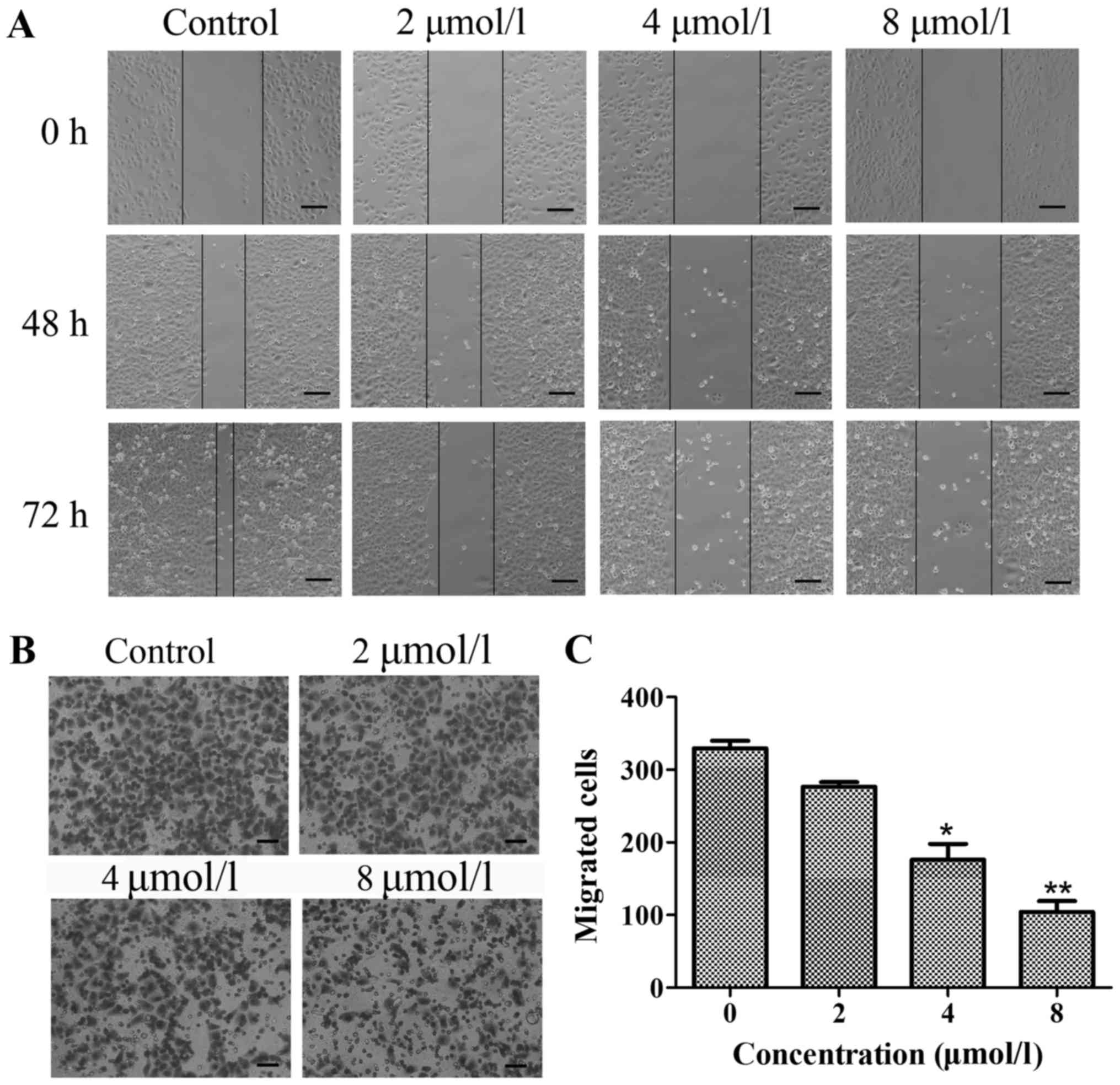
TPD7 inhibits the migration and
invasion of HeLa cells
Wound healing and chamber assays were used to
determine the effects of TPD7 on HeLa cell mobility. For wound
healing, confluent cell monolayers were scratched to form a wound
and then cultured in medium with various concentrations of TPD7 (2,
4 and 8 µmol/l) for 48 or 72 h. The movement of cells treated with
TPD7 was much slower than the control cells (Fig. 4A).
The Millicell chamber assay was a second approach
used to asceertain the inhibitory effect of TPD7 on HeLa cell
mobility. This assay revealed that the cell number on the lower
surface of the insert was decreased following 48 h of TPD7
treatment (Fig. 4B and C).
Collectively, these results revealed that TPD7 impaired HeLa cell
migration.
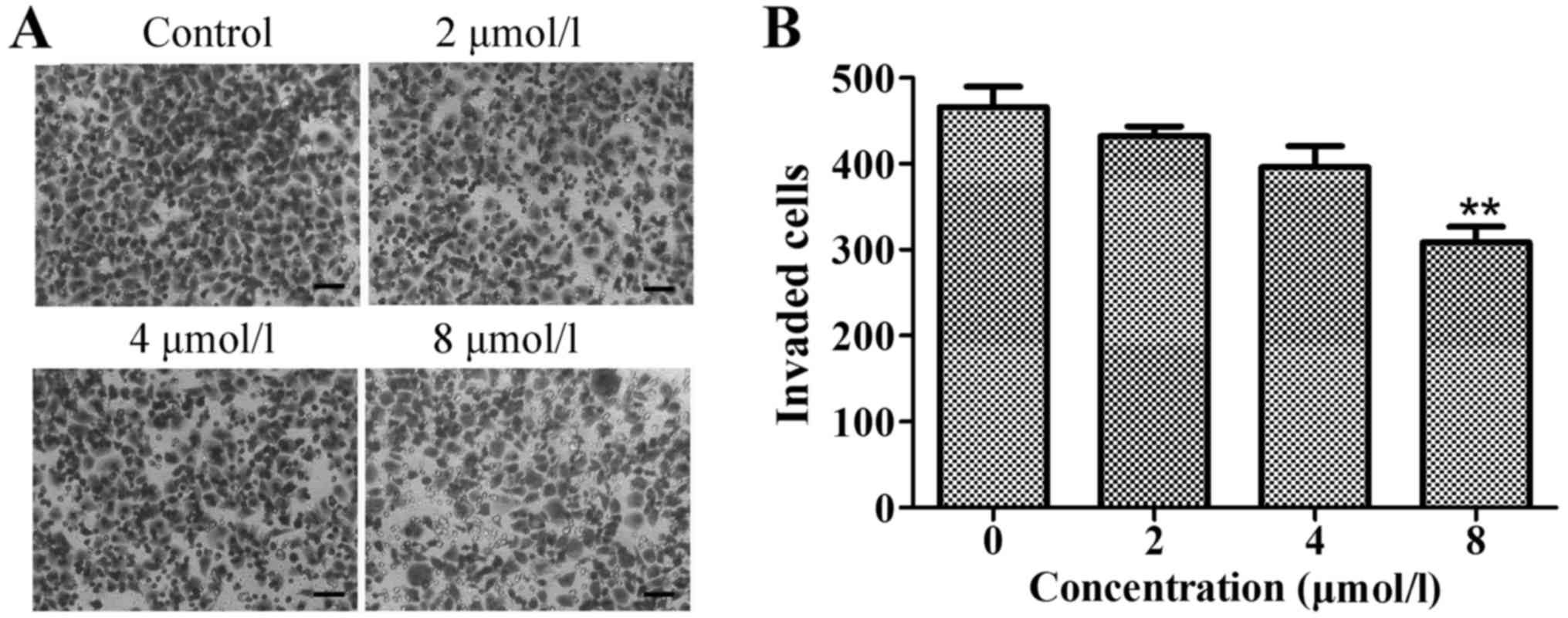
We also evaluated cancer cell invasion through
Matrigel-coated chambers. As shown in Fig. 5, a similar effect of TPD7 on
invasiveness was also observed in this system. These results
indicated that TPD7 has anticancer properties that can
significantly suppress cell invasion in a concentration-dependent
manner.
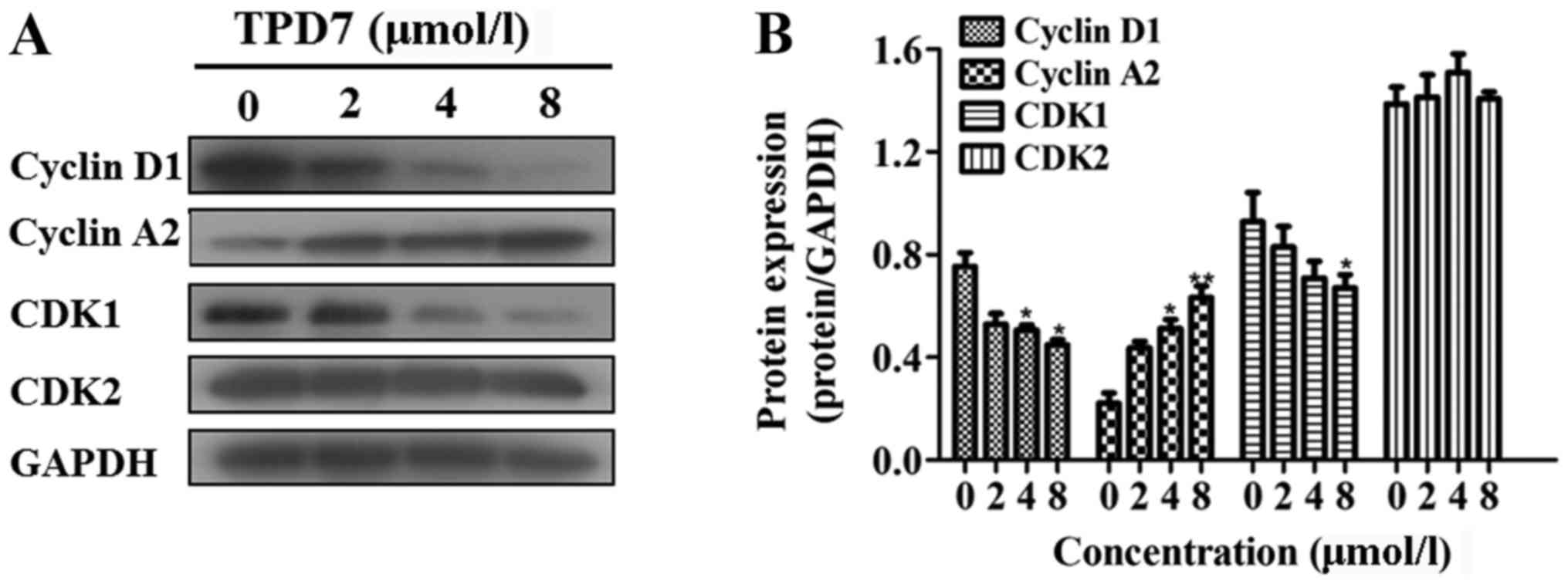
TPD7 alters the expression of proteins
related to the cell cycle and apoptosis
Multiple signal transduction pathways take part in
regulating the cell cycle distribution, apoptosis, and cell
invasion. Since TPD7 treatment induced S-phase arrest in HeLa
cells, we evaluated the expression of proteins regulating the cell
cycle. Treatment with TPD7 for 48 h led to a significant increase
in cyclin A2, and a subsequent decrease in cyclin D1 and CDK1
protein expression. There was no change in CDK2 expression
(Fig. 6).
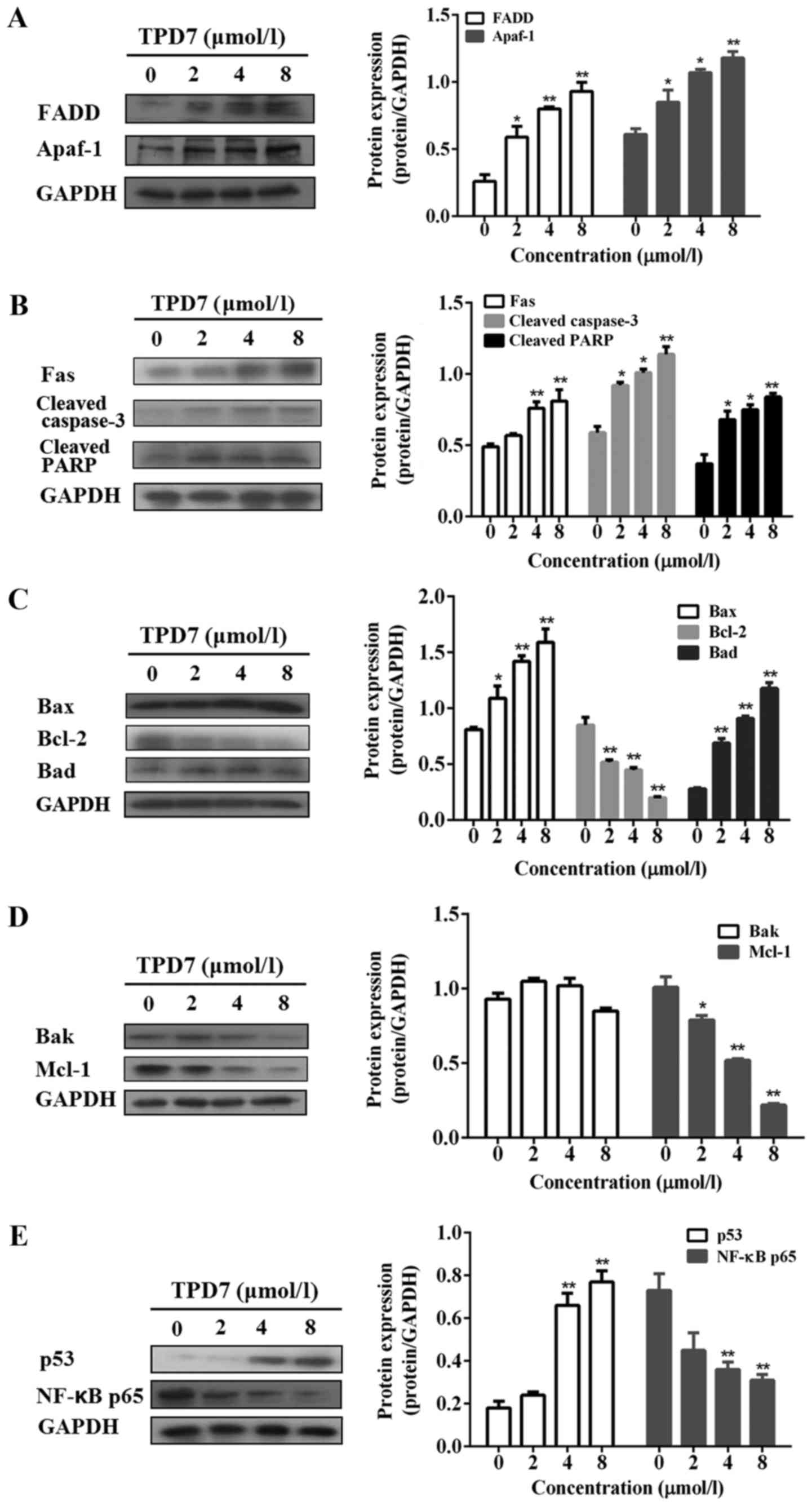
Next, we examined the expression of proteins related
to apoptosis including Fas receptor signaling and Bcl-2 family
proteins in HeLa cells. TPD7 treatment upregulated Apaf-1, Bax and
Bad and significantly downregulated Bcl-2 Mcl-1 and NF-κB p65
protein levels. In addition, TPD7 upregulated the expression of
Fas, FADD, cleaved caspase-3, cleaved PARP and p53 (Fig. 7A-E).
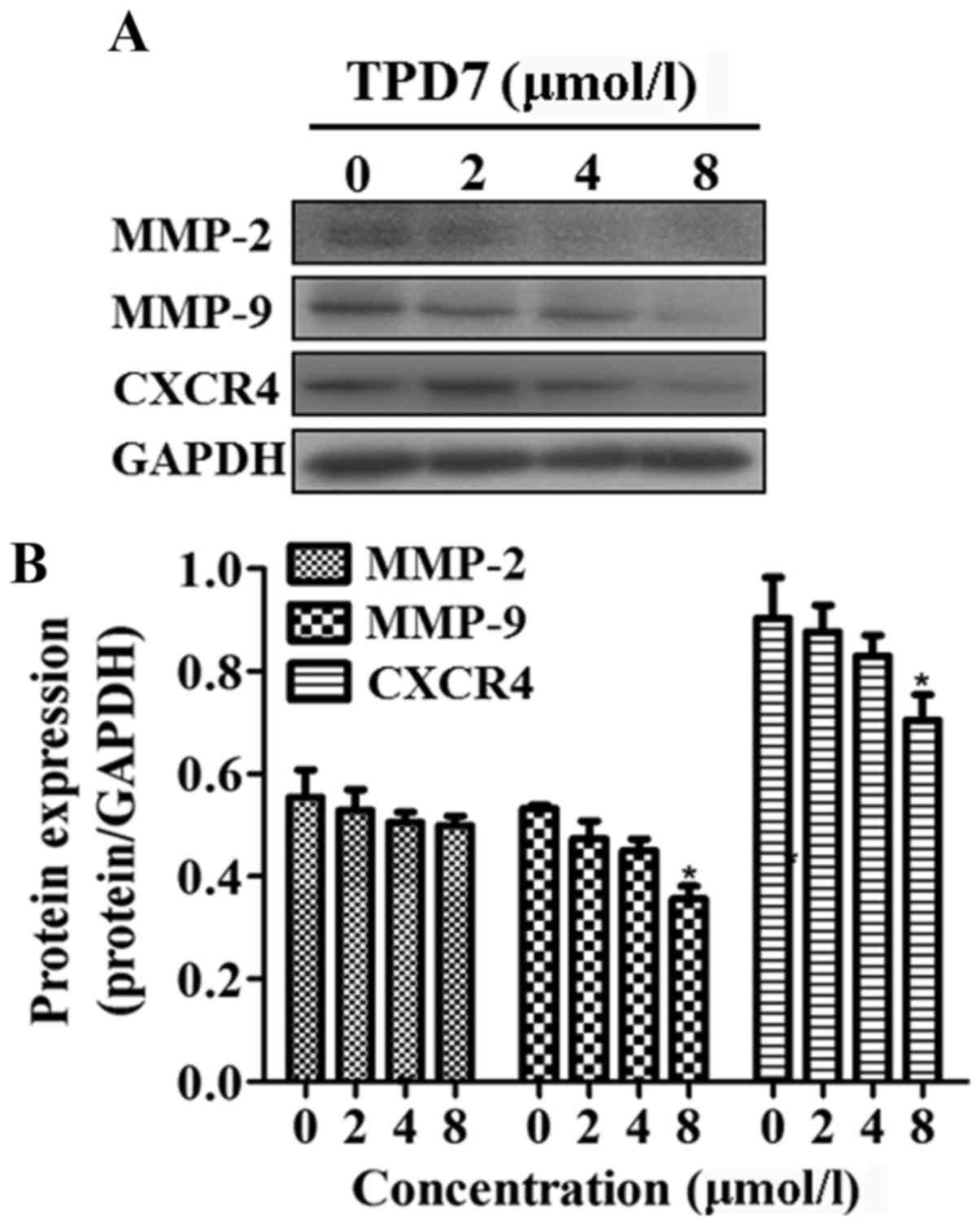
MMP-2, MMP-9 and CXCR4 are key molecules in the
processes of cancer cell invasion and metastasis. The potential
effect of TPD7 treatment on the expression of these proteins in
HeLa cells was investigated. HeLa cells treated with TPD7 exhibited
significant inhibitory effects on MMP-9 and CXCR4 expression, but
no effect on MMP-2 (Fig. 8).
TPD7 is efficacious against HeLa cell
xenograft tumors in vivo
A mouse model was used to evaluate the ability of
TPD7 to suppress tumor growth in vivo. Prior to xenograft
studies, we determined toxicity of TPD7 in mice by acute toxicity
test. Receiving TPD7 at a dosage of 1,000 mg/kg did not lead to
toxic effects in mice, and the TPD7 dose toxic to 10% of the
animals (LD10) was 2,000 mg/kg, therefore, one-tenth of the LD10,
200 mg/kg, was considered to be a safe dose, and was used as the
maximum tested dose in the xenograft studies. The medium tested
dose and the minimum tested dose were 100 and 50 mg/kg,
respectively. Table I revealed the
tumor growth in animals treated with vehicle or TPD7. TPD7
significantly decreased tumor weight in HeLa cell xenografted
athymic mice compared with vehicle-treated animals. Furthermore,
mice receiving TPD7 had no evident loss of body weight during the
experiment, suggesting that TPD7 was nontoxic in athymic mice at
the dose used.
 | Table I.Effect of treatment with TPD7 in nude
mice bearing HeLa human cervical cancer xenografts. |
Table I.
Effect of treatment with TPD7 in nude
mice bearing HeLa human cervical cancer xenografts.
| Treatment | Initial body weight
(g) | Initial tumor
volume (cm3) | Final body weight
(g) | Final tumor volume
(cm3) | Final tumor weight
(g) | Tumor growth
inhibition (%) |
|---|
| Control | 18.85±1.31 | 0.108±0.02 | 17.68±0.97 | 1.95±0.12 | 0.74±0.14 | – |
| TPD7 |
|
|
|
|
|
|
| (50
mg/kg) | 19.95±1.17 | 0.107±0.02 | 18.55±1.35 | 1.75±0.33 | 0.68±0.16 |
8.37 |
| (100
mg/kg) | 19.55±2.09 | 0.104±0.01 | 19.00±2.21 |
1.32±0.26a |
0.53±0.28a | 28.73 |
| (200
mg/kg) | 19.53±1.73 | 0.110±0.03 | 17.33±2.22 |
0.88±0.31b |
0.36±0.20b | 51.58 |
Discussion
Despite the increasing number of novel anticancer
drugs being applied clinically to treat cancer, the development of
acquired resistance ultimately limits the use of these agents.
Thus, novel therapeutic strategies targeting cancer cells are
urgently needed. The induction of apoptosis in cancer cells has
revealed promising antitumor effects in many cancer models.
Apoptosis is a normal physiological process that functions to
orderly maintain the balance of cell numbers in multicellular
organisms. However, when the balance is disrupted, human cancers
may occur (7,17).
Our research focused on identifying novel taspine
derivatives that exhibit anticancer activity in HeLa cells. We
previously revealed tumor inhibitory effects in breast cancer
(18). In the present study, TPD7
decreased the viability and arrested the cell cycle of HeLa cells.
We hypothesized that TPD7 treatment induced apoptosis and thereby
inhibited cell proliferation. Apoptosis was assessed in
TPD7-treated HeLa cells and we determined that TPD7 induced this
form of cell death.
Cell cycle dysregulation may result in the
uncontrolled proliferation that underlies the malignant phenotype
(19). Therefore, blockade of cell
cycle progression is regarded as a useful approach to shrink or
kill tumors. Progression through the cell cycle is also tightly
controlled by CDKs 1–6, whose activity requires association with
specific cyclin subunits (20). G1
progression and the transition between the G1 and S phases requires
depletion in cyclin D, while CDK2 controls the S phase when
associated with cyclin A (21–23).
In this study, we demonstrated that TPD7 induced S-phase blockade
in HeLa cells, resulting in a corresponding reduction of cells in
both the G1 and G2/M phases. Downregulation of cyclin D1 and CDK1,
and upregulation of cyclin A2 after TPD7 treatment was subsequent
to the G1/S phase transition and followed by a reduction of cells
in the G2/M phase.
To ascertain TPD7-induced cell apoptosis in HeLa
cells, we determined the presence of apoptotic cells following TPD7
treatment. As predicted, more apoptotic cells could be observed in
TPD7 treatment groups by Hoechst staining. Additional verification
for TPD7-triggered HeLa cell apoptosis involved FITC-labeled
Annexin V/PI staining and flow cytometry. HeLa cells exposed to
TPD7 exhibited increases in both early and late apoptotic
cells.
Apoptosis can be triggered through two key molecular
signaling pathways. One is the death receptor (Fas/FasL) pathway,
known as the extrinsic apoptosis pathway. This pathway is initiated
when death receptors on the cell surface interact with specialized
ligands and is closely related to tumor progression (24). Fas-associated protein death domain
(FADD) is a protein that plays a crucial role in the apoptotic
pathway of Fas. It recruits procaspase-8 to promote formation of
the death-inducing signaling complex (DISC) and mediates
receptor-induced apoptosis (25).
The intrinsic apoptosis pathway is another signaling system
involved in this form of cell death. This pathway can be triggered
by members of the Bcl-2 family of proteins and downstream
mitochondrial signals (6,26,27).
Both pathways converge to activate caspases, which are the final
step carrying out numerous proteolytic events that mediate the
apoptotic program (28). Among the
caspases, caspase-3 is a crucial protease activated by death
signals to hydrolyze many key specific substrates (29). In this study, Fas, FADD, caspase-3,
and the cleavage of PARP were upregulated in HeLa cells treated
with TPD7.
Pro- and anti-death proteins of the Bcl-2 family are
the major molecules involved in the intrinsic apoptosis pathway
(30). Mcl-1, Bcl-2, Bcl-XL and
Bcl-W function as anti-apoptotic molecules, and their expression
can be elevated in many tumors. Bax, Bak and Bad, identified as
cell-death mediators of the Bcl-2 family, accelerate apoptosis
induced by external stimuli (31).
We detected the key Bcl-2 family molecules involved in apoptosis
and found that the pro-apoptotic proteins, such as Bad and Bax,
were upregulated in TPD7-treated cells. In contrast, the
anti-apoptotic proteins, Mcl-1 and Bcl-2, were downregulated in
TPD7-treated cells. Apaf-1, a cell-death effector, functions
downstream of Bcl-2 but upstream of caspase-3. It binds to
cytochrome c to trigger the activation of caspase-3, leading to
apoptosis (32). The amount of
Apaf-1 protein was markedly increased by TPD7 treatment. Restoring
physiological levels of Apaf-1 by TPD7 may contribute to enhance
sensitivity to apoptosis signals.
p53 serves as a tumor suppressor that affects the
progression of apoptosis primarily through modulating key
functional proteins in the intrinsic pathway. The pro-apoptotic
activity of p53 occurs by directly activating the transcription of
genes that initiate the apoptotic program (33). TPD7 upregulated the expression of
p53 in HeLa cells and, accordingly, promoted apoptosis thereby
decreasing HeLa cell viability and proliferation.
To characterize the in vivo inhibitory
activity of TPD7 on cancer, we conducted studies using xenograft
tumors in mice. We consistently observed an inhibition of tumor
growth in TPD7-treated mice, suggesting that TPD7 may be a good
candidate for cervical cancer intervention.
In conclusion, our findings revealed that
TPD7-mediated cell cycle arrest was associated with downregulation
of cyclin D1 and CDK1, along with an increase of cyclin A2.
Furthermore, TPD7-induced apoptosis in cervical cancer cells was
mediated through both the intrinsic and extrinsic pathways. These
findings demonstrated that TPD7, a novel taspine derivative, is a
potential agent against cervical carcinoma in vitro and
in vivo.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81302800 and
81773772), the Natural Science Basic Research Plan of Shaanxi
Province in China (grant no.2015JQ8296) and the Basic Research
Project in Shaanxi Administration of Traditional Chinese Medicine
(grant no. JCMS028).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YiZ, YaZ and LH conceived and designed the study.
YiZ, HZ, BD and JZ contributed to the acquisition of the data. YiZ,
YaZ, JZ and LH wrote, reviewed and revised the manuscript. LH
supervised the study. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The use of Balb/c mice for the purpose of this study
was approved by the Animal Ethics Committee of Xi'an Jiaotong
University (Permission no. XJTULAC2013-031). All experiments were
conducted in accordance with the approved guidelines of the
regional authorities according to Xi'an Jiaotong University animal
care regulations.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wright AA, Howitt BE, Myers AP, Dahlberg
SE, Palescandolo E, Van Hummelen P, MacConaill LE, Shoni M, Wagle
N, Jones RT, et al: Oncogenic mutations in cervical cancer: Genomic
differences between adenocarcinomas and squamous cell carcinomas of
the cervix. Cancer. 119:3776–3783. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koh WJ, Greer BE, Abu-Rustum NR, Apte SM,
Campos SM, Chan J, Cho KR, Cohn D, Crispens MA, DuPont N, et al:
National Comprehensive Cancer Network: Cervical cancer. J Natl
Compr Canc Netw. 11:320–343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Torre LA, Siegel RL, Ward EM and Jemal A:
Global cancer incidence and mortality rates and trends - An update.
Cancer Epidemiol Biomarkers Prev. 25:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siegel RL, Fedewa SA, Miller KD,
Goding-Sauer A, Pinheiro PS, Martinez-Tyson D and Jemal A: Cancer
statistics for Hispanics/Latinos, 2015. CA Cancer J Clin.
65:457–480. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fulda S and Debatin KM: Sensitization for
anticancer drug-induced apoptosis by the chemopreventive agent
resveratrol. Oncogene. 23:6702–6711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Green DR and Kroemer G: The
pathophysiology of mitochondrial cell death. Science. 305:626–629.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ashkenazi A: Targeting death and decoy
receptors of the tumour-necrosis factor superfamily. Nat Rev
Cancer. 2:420–430. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen J, Solomides C, Parekh H, Simpkins F
and Simpkins H: Cisplatin resistance in human cervical, ovarian and
lung cancer cells. Cancer Chemother Pharmacol. 75:1217–1227. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen Y, Wang P, Li Y, Ye F, Wang F, Wan X,
Cheng X, Lu W and Xie X: miR-375 is upregulated in acquired
paclitaxel resistance in cervical cancer. Br J Cancer. 109:92–99.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang C, Dong J, Zhang Y, Wang F, Gao H, Li
P, Wang S and Zhang J: Design, synthesis and biological evaluation
of biphenyl urea derivatives as novel VEGFR-2 inhibitors. Bioorg
Med Chem. 4:1434–1438. 2013.
|
|
14
|
Li Y and He L: Establishment of the model
of vascular endothelial cell membrane chromatography and its
preliminary application. Chin Sci Bull. 52:922–928. 2007.
View Article : Google Scholar
|
|
15
|
Zhan Y, Zhang Y, Chen Y, Wang N, Zheng L
and He L: Activity of taspine isolated from Radix et Rhizoma
Leonticis against estrogen-receptor-positive breast cancer.
Fitoterapia. 82:896–902. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, He L, Meng L and Luo W: Taspine
isolated from Radix et Rhizoma Leonticis inhibits proliferation and
migration of endothelial cells as well as chicken chorioallantoic
membrane neovascularisation. Vascul Pharmacol. 48:129–137. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhan Y, Zhang H, Li J, Zhang Y, Zhang J
and He L: A novel biphenyl urea derivate inhibits the invasion of
breast cancer through the modulation of CXCR4. J Cell Mol Med.
19:1614–1623. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Williams GH and Stoeber K: The cell cycle
and cancer. J Pathol. 226:352–364. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Malumbres M, Harlow E, Hunt T, Hunter T,
Lahti JM, Manning G, Morgan DO, Tsai LH and Wolgemuth DJ:
Cyclin-dependent kinases: A family portrait. Nat Cell Biol.
11:1275–1276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Diaz-Moralli S, Tarrado-Castellarnau M,
Miranda A and Cascante M: Targeting cell cycle regulation in cancer
therapy. Pharmacol Ther. 138:255–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Casagrande F and Darbon JM: Effects of
structurally related flavonoids on cell cycle progression of human
melanoma cells: Regulation of cyclin-dependent kinases CDK2 and
CDK1. Biochem Pharmacol. 61:1205–1215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pagano M, Pepperkok R, Verde F, Ansorge W
and Draetta G: Cyclin A is required at two points in the human cell
cycle. EMBO J. 11:961–971. 1992.PubMed/NCBI
|
|
24
|
Houston A and O'Connell J: The Fas
signalling pathway and its role in the pathogenesis of cancer. Curr
Opin Pharmacol. 4:321–326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee EW, Kim JH, Ahn YH, Seo J, Ko A, Jeong
M, Kim SJ, Ro JY, Park KM, Lee HW, et al: Ubiquitination and
degradation of the FADD adaptor protein regulate death
receptor-mediated apoptosis and necroptosis. Nat Commun. 3:9782012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaufmann SH and Earnshaw WC: Induction of
apoptosis by cancer chemotherapy. Exp Cell Res. 256:42–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ashkenazi A: Targeting the extrinsic
apoptosis pathway in cancer. Cytokine Growth Factor Rev.
19:325–331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsujimoto Y: Cell death regulation by the
Bcl-2 protein family in the mitochondria. J Cell Physiol.
195:158–167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hunter AM, LaCasse EC and Korneluk RG: The
inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis.
12:1543–1568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zou H, Henzel WJ, Liu X, Lutschg A and
Wang X: Apaf-1, a human protein homologous to C. elegans
CED-4, participates in cytochrome c-dependent activation of
caspase-3. Cell. 90:405–413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fridman JS and Lowe SW: Control of
apoptosis by p53. Oncogene. 22:9030–9040. 2003. View Article : Google Scholar : PubMed/NCBI
|