Introduction
Glioma is characterized by extremely aggressive
invasive and potent malignant abilities with a high proliferation
rate (1). At present, comprehensive
treatment types (typically including surgical resection,
radiotherapy and auxiliary-assisted chemotherapy) are widely used
clinically; however, the average survival time remains only ~1 year
(2). Additionally, the majority of
chemosensitizers demonstrate negative side-effects and/or little
efficacy against glioma (3,4). Therefore, the development of novel,
more potent chemotherapy agents with fewer side-effects and higher
efficacy is a priority in glioma treatment research.
Moxidectin (MOX), derived from the actinomycete
Streptomyces cyanogriseus subsp. Noncyanogenus
(5,6), is a third generation macrocyclic
lactone with potent insecticide activity, belonging to the
milbemycin family (7,8). Previous research has revealed that
certain macrocyclic lactones, including MOX, with lower toxicity
are used widely for the treatment of internal and external
parasites in cattle, sheep, deer and horses (6,9–12). MOX
is currently being used in phase III clinical trials in the
treatment of filarial Onchocerca volvulus infection in
humans, which indicates that MOX is safe and well tolerated in
humans at doses between 3 and 36 mg (6,13).
In one previous study, some compounds that belong to
the milbemycin family including MOX were found to reverse the
multidrug resistance (MDR) of MCF-7/adr cells. Study of the
mechanisms underlying the effects of milbemycins on p-glycoprotein
(P-gp)-mediated MDR demonstrated that the milbemycins significantly
increased the intracellular accumulations of adriamycin and Rh123
via inhibiting P-gp transport function, which revealed that MOX may
function as an effective multidrug resistance agent. Additionally,
it was demonstrated that MOX was partially effective in killing
non-drug-resistant tumor cells (14). Previously, macrocyclic lactones
including avermectins (ivermectin) have been revealed to be
effective in inhibiting the proliferation of tumor cells (Hep-2 and
P388 cells) (15,16). Furthermore, ivermectin suppressed
breast cancer cell growth and induced glioblastoma cell death in
vitro and in vivo (17,18).
MOX and ivermectin, which are similar in chemical structure,
partially share certain physicochemical and pharmacological
properties. They also have broad-spectrum activity against
nematodes and arthropods (19). MOX
differs from ivermectin primarily by the lack of a sugar moiety
attached to the C13 of the macrocyclic ring (20). Previous publications have
demonstrated that both compounds have a number of similar
mechanisms of action and are part of the antiparasitic spectrum
(21–23).
To the best of our knowledge, there have been no
previous reports on the use of MOX in cancer treatment. The present
study was carried out to investigate the ability of MOX to treat
glioma, and to explore its potential molecular mechanisms in
vitro and in vivo.
Materials and methods
Reagents and antibodies
MOX was purchased from Sigma-Aldrich/Merck KGaA
(Darmstadt, Germany; European Pharmacopoeia Reference Standard),
and a purity of >95%. MOX was dissolved in dimethyl sulfoxide
(DMSO) prior to use. Cells were treated with MOX premixed cell
culture Dulbecco's modified Eagle's medium (DMEM) containing 10%
fetal bovine serum (FBS). The DMSO concentration did not exceed
0.1% of the total volume. MTT, diaminobenzidine (DAB),
polyvinylidene fluoride (PVDF) membranes, hematoxylin and eosin
(H&E) were purchased from Sigma-Aldrich; Merck KGaA. Propidium
iodide (PI)/Annexin V fluorescein isothiocyanate (FITC) were
purchased from Beyotime Institute of Biotechnology (Haimen, China).
Antibodies used in the present study were against Bcl-2-associated
× protein (Bax; cat. no. 2774; Cell Signaling Technology, Inc.,
Danvers, MA, USA), B-cell lymphoma 2 (Bcl-2; cat. no. 3498; Cell
Signaling Technology, Inc.), cyclin-dependent kinase (CDK)2 (cat.
no. 2546; Cell Signaling Technology, Inc.), CDK4 (cat no. 12790;
Cell Signaling Technology, Inc.), CDK6 (cat. no. 13331; Cell
Signaling Technology, Inc.), cleaved caspase-3 (cat. no. bs-0081R;
Bioss, Inc., Beijing, China), cleaved caspase-9 (cat. no. bs-0049R;
Bioss, Inc.), cyclin D1 (cat. no. 2922; Cell Signaling Technology,
Inc.), cyclin E (cat. no. SRP5345; Sigma-Aldrich; Merck KGaA),
Ki-67 (cat no. 9129; Cell Signaling Technology, Inc.), β-actin
(cat. no. A1978; Sigma-Aldrich; Merck KGaA), goat anti-rabbit
immunoglobulin G (IgG) (H+L)-horseradish peroxidase (HRP; cat. no.
LK2001; SunGene GmbH, Gatersleben, Germany) and goat anti-mouse IgG
(H+L)-HRP (cat. no. ZB-2305; OriGene Technologies, Inc., Rockville,
MD, USA).
Cell culture
Rat C6 glioma cells and human U251 glioma cells were
obtained from The First Affiliated Hospital of Harbin Medical
University (Harbin, China). SVG p12 was purchased from Wuhan YiPu
Biotech Co., Ltd. (Wuhan, China). Cells were cultured in DMEM
supplemented with 10% FBS and 1% penicillin and streptomycin at
37°C in a humidified incubator with 5% CO2.
Cell viability assay
Cell viability and cell number were determined using
an MTT (purity >95%) assay. C6 (2.0×103 cells/well),
U251 (1.0×104 cells/well) and normal human astrocyte
cells (SVG p12, 7.0×103 cells/well) were respectively
seeded into 96-well plates with 100 µl culture medium and treated
with the indicated concentrations (0, 10, 20, 30, 40 and 50 µmol/l)
of MOX for 48 h at 37°C. Next, 20 µl MTT (5 mg/ml) solution was
added to each well, and the cells were incubated for 4 h at 37°C.
Once the medium was carefully removed, 150 µl of DMSO was added and
agitated to dissolve the formazan crystals. The absorbance at 490
nm was measured using an enzyme-linked immunosorbent assay reader
(Nanjing Huadong Electronics Group Co., Ltd., Nanjing, China). For
relative quantification, the value of absorbance in each group was
normalized to that of the control group.
Colony formation assay
To analyze the sensitivity of the cells to MOX, an
in vitro colony formation assay was performed. Briefly, C6
(3.0×102 cells/well) and U251 (4.0×102
cells/well) cells were seeded in 6-well plates for 24 h then
treated with various concentrations of MOX (0, 10, 15 and 20
µmol/l) at 37°C. The cultures were maintained at 37°C in a 5%
CO2 incubator for 10 days, which allowed the viable
cells to grow into macroscopic colonies. Then, the medium was
removed, and the colonies were counted subsequent to being stained
with 0.1% crystal violet (Sigma-Aldrich; Merck KGaA) at room
temperature for 20 min. Quantification of colony formation was also
performed using ImageJ software (V 2.0; National Institutes of
Health, Bethesda, MD, USA).
Flow cytometry
C6 (2.5×105 cells/well) and U251
(2.8×105 cells/well) cells were seeded into 6-well
plates and treated with various concentrations of MOX (0, 10, 15
and 20 µmol/l). For cell cycle analysis, the cells were treated at
37°C for 24 and 48 h, washed with ice-cold phosphate-buffered
saline (PBS; Biotopped, Beijing, China), and collected cell
suspensions were fixed in 70% ice-cold ethanol at 4°C for 24 h.
Then, the fixed cells were washed twice with PBS and stained with
PI for 20 min at room temperature away from light. For apoptosis
analysis, cells were treated MOX at 37°C for 48 h, washed twice
with ice-cold PBS, and then cell suspensions were collected,
suspended with Annexin V binding buffer and Annexin V-FITC/PI, and
the mixture was incubated for 20 min at room temperature in the
dark. The cell cycle distribution and apoptosis ratio were measured
using a BD Biosciences FACSCalibur flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA). The DNA content of the cell cycle was
analyzed using ModFit LT v3.3 application software (Verity Software
House Inc., Topsham, ME, USA).
Transmission electron microscopy
(TEM)
C6 and U251 cells were treated with MOX (20 µmol/l)
at 37°C for 48 h, and untreated cells served as the control group.
Then, the cells were harvested, washed twice with PBS and fixed
with a solution of 2.5% glutaraldehyde in PBS at 4°C for 2 h, and
post fixed with 1% phosphate-buffered osmium tetroxide at 4°C for 1
h. Next, the cell were dehydrated using a graded series of ethanol
solutions (30, 50, 70, 90 and 100%), 8 or 10 min at a time,
transferred to propylene oxide and embedded in epon araldite
(Polybed 812; Polysciences, Inc., Warrington, PA, USA). The
ultrathin sections were observed using a JEM-100CX transmission
electron microscope (H-7650; Shanghai Yongming Automatic Equipments
Co., Ltd., Shanghai, China) and representative images were
photographed and analyzed.
Western blotting
C6 and U251 cells were treated with various
concentrations of MOX (0, 10, 15 and 20 µmol/l) for 48 h, and then
total protein from the cells was extracted using radio
immunoprecipitation assay buffer, which was subjected to sodium
dodecyl sulfate-polyacrylamide gel (12%) electrophoresis and then
transferred to PVDF membranes. Following blocking with 5% skim milk
for 1 h, the membranes were incubated at 4°C overnight with the
following antibodies: anti-CDK2 (1:1,000), anti-CDK4 (1:1,000),
anti-CDK6 (1:1,000), anti-cyclin D1 (1:1,000), anti-cyclin E
(1:1,000), anti-Bax (1:1,000), anti-Bcl-2 (1:1,000), anti-cleaved
caspase-3 (1:800), anti-cleaved caspase-9 (1:800) and anti-β-actin
(1:1,000). Subsequent to washing three times with Tris-buffered
saline Tween (TBST; Solarbio, Beijing, China), HRP-conjugated
secondary antibodies (1:5,000) were used in conjunction with
MiniChemi-Imager (Beijing Sage Creation Co., Ltd., Beijing, China)
for visualization. The intensity of the western blotting bands was
measured using ImageJ software (version 2.0; National Institutes of
Health).
Animal models
All studies were performed in accordance with the
ethical standards of the institutional and/or national research
committee (Harbin Vic Biological Technology Development Co., Ltd.,
Harbin, China) and with the 1964 Helsinki declaration and its later
amendments or comparable ethical standards. Ethical approval was
obtained for the animal experiments. Female BALB/c mice (Beijing
HFK Bioscience Co., Ltd, Beijing, China) at 5 weeks of age and an
average weight of 19–23 g were used. They were housed in plastic
cages under standard conditions and had access to rodent chow and
water ad libitum. For the glioma cancer model, U251 cells
(2.0×106) were suspended in PBS and engrafted in the
dorsal of nude mice. When the tumor volumes reached 70
mm3, mice were assigned randomly into two groups
receiving 0.1 ml saline or 20 mg/kg MOX/mouse/day by peritoneal
injection for 3 weeks. Saline or MOX were injected
intraperitoneally into mice daily. The volume of tumors were
measured every four days by using a vernier caliper and calculated
as length (mm) × width2 (mm2) × 1/2. The mice
were euthanized using CO2 for analysis following four
weeks and the maximum allowable tumor volume was no >800
mm3. The mice were sacrificed subsequent to being
anesthetized using barbiturates and tumor tissues were isolated and
frozen in liquid nitrogen or fixed in 10% formalin immediately.
Immunohistochemistry
Immunohistochemicaly staining was performed on
paraffin-embedded tissues from the harvested tumors. Sections (5
µm-thickness) were oven dried at 60°C for 1 h. Tissue sections were
then deparaffinized with xylene (absolute) and hydrated with ethyl
alcohol (100, 90, 75, 50 and 30%). The sections were labeled with
antibodies for Ki-67 (1:400), cleaved caspase-3 (1:300) and cleaved
caspase-9 (1:300) followed by HRP-conjugated secondary antibodies
(1:3,000) using DAB reagents as a substrate, and then
counterstained with hematoxylin at room temperature for 1 min. The
negative control consisted of omitting the primary antibodies. The
images were captured with digital microscope camera (E100) and
analyzed with ImageJ software (version 2.0; National Institutes of
Health).
H&E staining and terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling
(TUNEL) assay
The tumors from each mouse were immediately fixed in
formaldehyde (10%) for 24 h at 4°C and embedded in paraffin at 60°C
for 1 h. Then, the tissues were sectioned at 5 µm thickness, and
stained with H&E at room temperature for 1 min to assess the
cytopathological features.
A TUNEL assay, which detects fragmented DNA, was
performed using an In Situ Cell Death Detection kit to evaluate the
apoptotic response of tumor tissues. Briefly, subsequent to being
deparaffinized (xylene, absolute) and hydrated (95, 90, 80, 70 and
50%), slides were washed with PBS twice and incubated with
proteinase K (20 µg/ml) for 20 min at 37°C. Following a second
round of washes using PBS, slides were incubated with a TUNEL
reaction mixture prepared freshly for 1 h at 37°C in a moist
chamber. Subsequent to being washed twice with PBS, the slides were
observed under ×40 fluorescence microscopy.
Quantitative and statistical
analysis
For quantification, Ki-67, cleaved caspase-3 and
caspase-9-staining intensity was measured from the number of
positive cell nuclei in 25% fields using ImageJ software (version
2.0). All areas were chosen randomly from all sections. The
intensity of bands in western blotting was also measured by ImageJ
software. The values subsequent to normalizing to the loading
control in the control groups were set as 1.0.
All experiments were performed at least three times.
Data are presented as the mean ± standard deviation (SD).
One-factor analysis of variance (ANOVA) test was used to assess the
differences among all the experiment and control groups. The
Dunnett's test was employed for post-hoc comparison of the three
experimental groups with the control group. GraphPad Prism Package
(version 5.0; GraphPad Software, Inc., La Jolla, CA, USA) and SPSS
version 20.0 statistical software (IBM Corp., Armonk, NY, USA) were
used for statistical analysis. P<0.05 was considered to be
indicative of a statistically significant difference.
Results
MOX inhibits the viability of glioma
cells in vitro
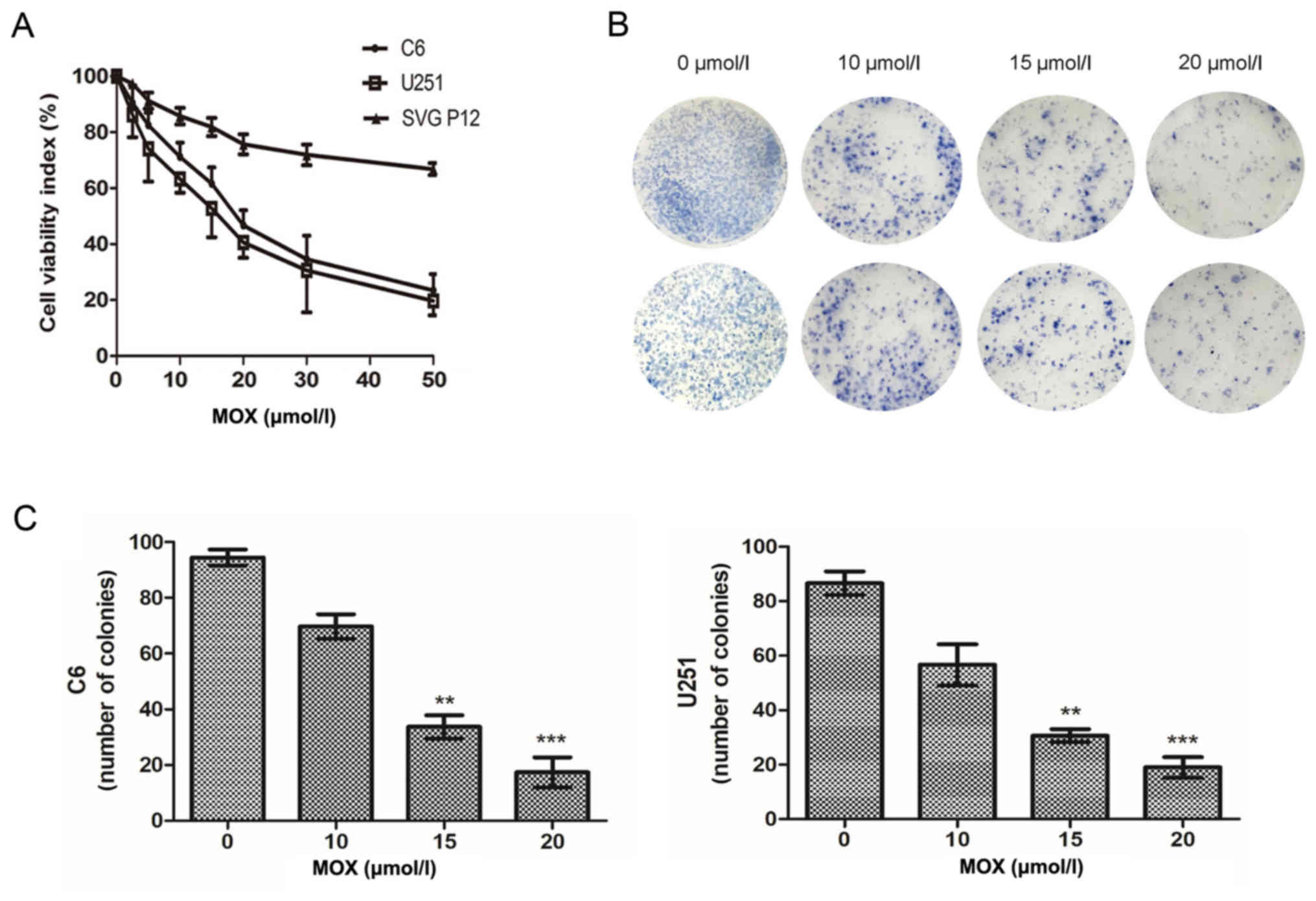
To examine the effect of MOX on glioma growth, an
MTT assay was performed to measure the cell viability rates of C6,
U251 and normal human astrocyte cells (SVG p12), respectively. As
presented in Fig. 1A, treatment
with MOX decreased the cell viability of glioma cells in a
dose-dependent manner compared with the untreated glioma cells.
Notably, MOX exerted a lesser effect on normal human astrocyte
cells. The clonogenic capacity of C6 and U251 cells was examined,
and it was revealed that MOX significantly inhibited colony
formation and induced significant decreases in the colony formation
ratio compared with the untreated cells (Fig. 1B and C). These results revealed that
MOX inhibits glioma cell viability and has little effect on normal
human astrocyte cells.
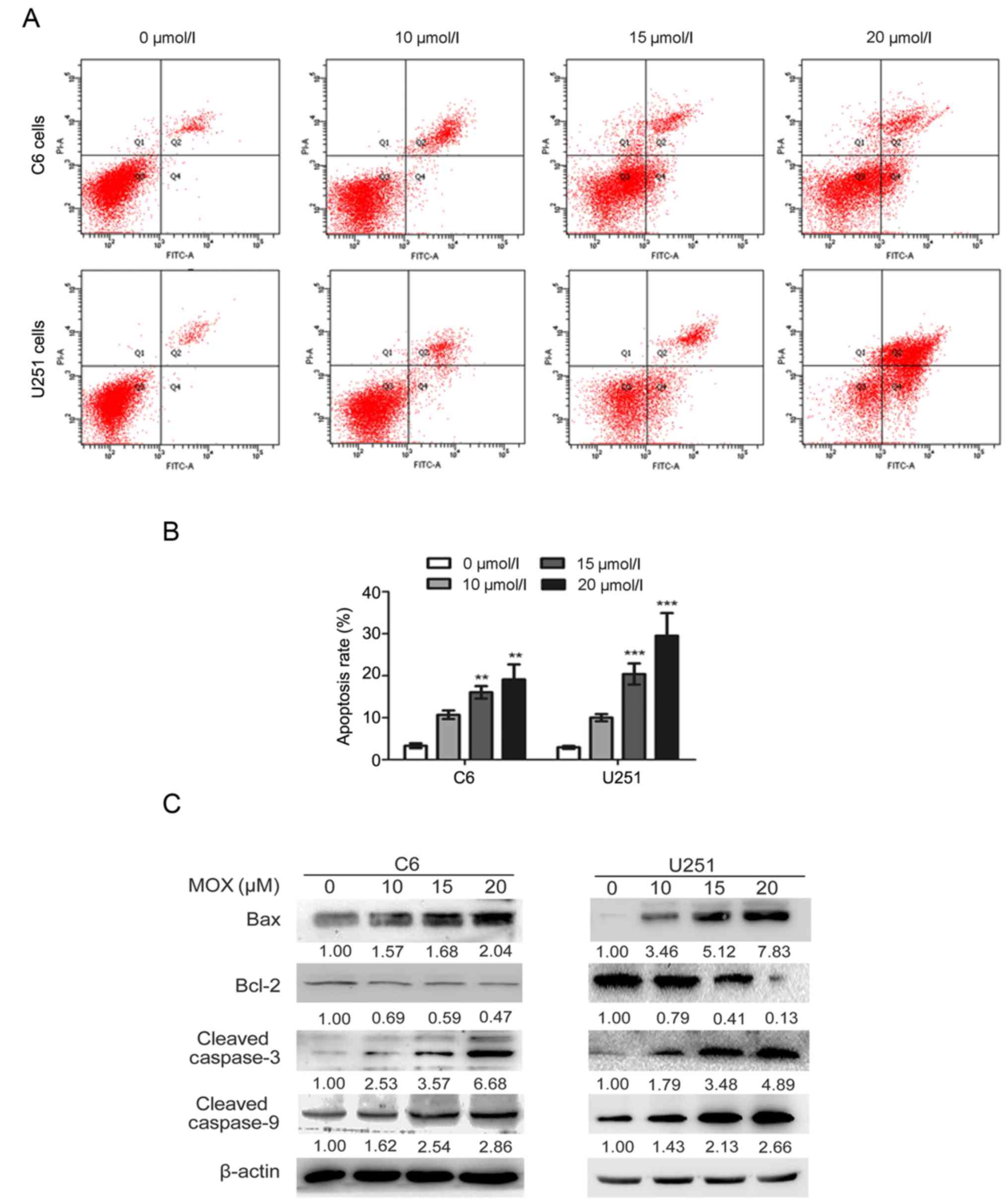
MOX induces apoptosis and the
expression of apoptosis-associated proteins in glioma cells
To analyze the effect on the induction of apoptosis
by MOX, the number of Annexin V-stained cells was determined using
flow cytometry (Fig. 2A). As
presented in Fig. 2B, C6 and U251
cells treated with MOX (20 µmol/l) had significantly induced
apoptosis compared with the control groups, and the apoptotic rate
of C6 and U251 cells was 19.10±3.59 and 29.53±5.00%, respectively.
However, the apoptotic rate of the control cells (untreated C6 and
U251 cells) was only 3.33±0.57 and 2.93±0.34%, respectively. Thus,
MOX-induced apoptosis of C6 and U251 cells occurred in a
dose-dependent manner compared with the control groups.
The mechanism of MOX-induced apoptosis was
investigated by analyzing the expression levels of mitochondrial
apoptosis proteins through western blotting. As presented in
Fig. 2C, compared with the control
group, C6 and U251 cells exposed to MOX demonstrated a
dose-dependent reduction in the protein expression levels of the
anti-apoptotic protein Bcl-2, with a concomitant increase in the
protein expression levels of the pro-apoptotic protein Bax.
Additionally, the protein expression levels of cleaved caspase-3
and cleaved caspase-9 were increased following treatment with MOX
compared with control group.
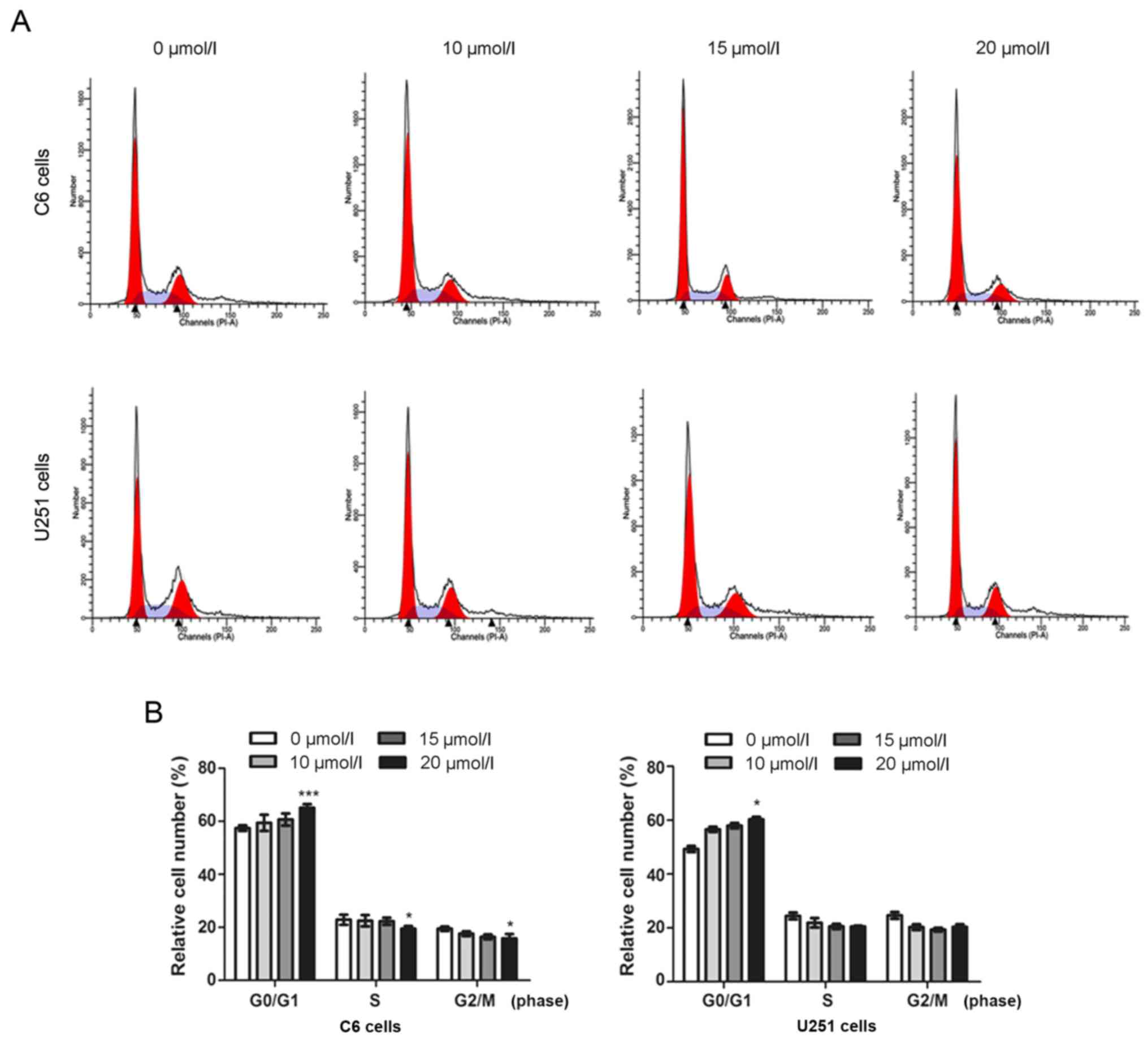
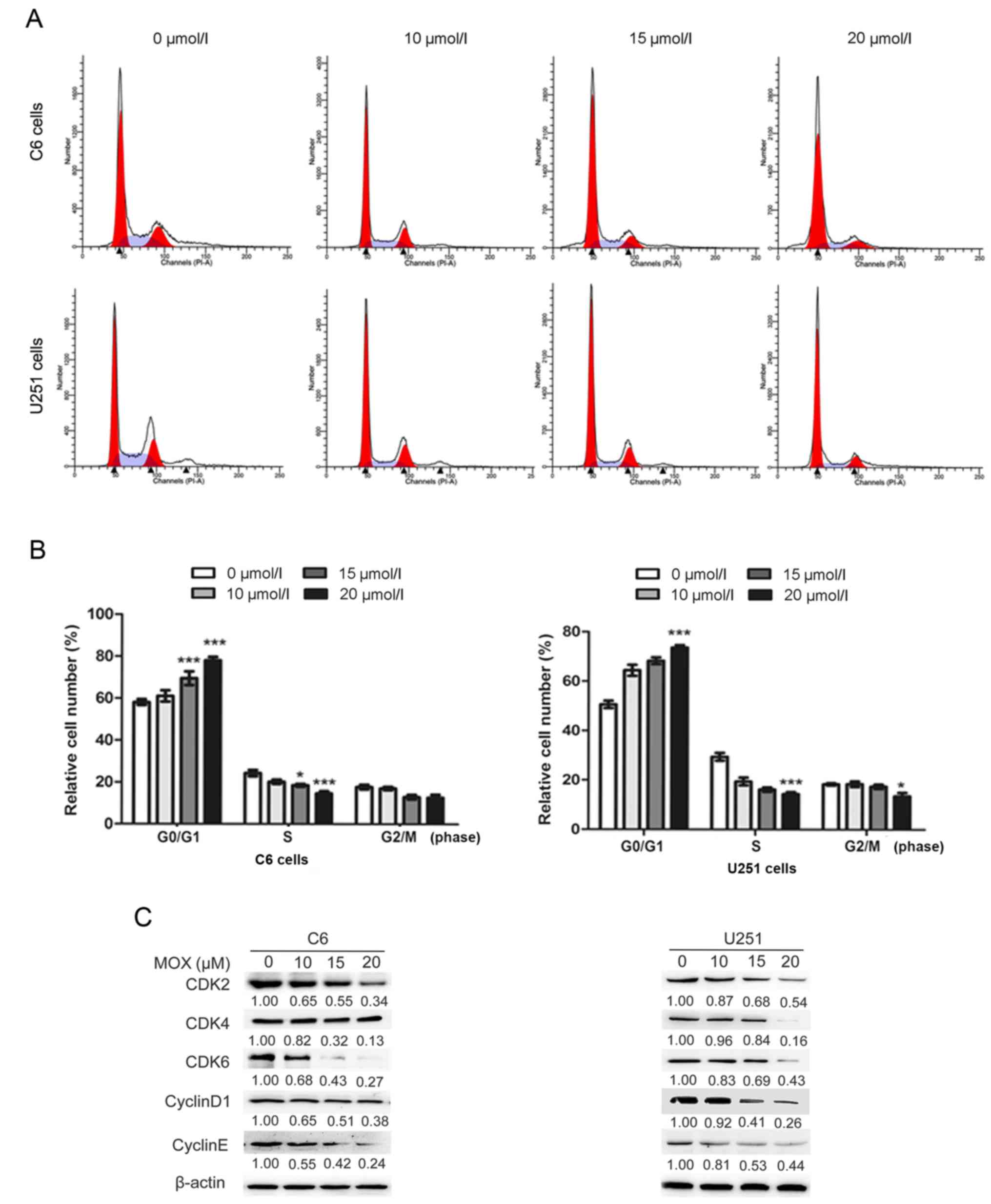
MOX induces G0/G1 arrest by modulating
cell cycle- associated proteins in glioma cells
To determine the effect of MOX on cell cycle arrest
in C6 and U251 cells, the cell cycle distribution was investigated
using flow cytometry (Figs. 3A and
4A). Interestingly, it was revealed
that the accumulation of G0/G1 phase cells, a hallmark of
apoptosis, was not notable until cells had been treated MOX for 48
h (Fig. 3B). As presented in
Fig. 4B, the results revealed that
MOX induced G0/G1 cell cycle arrest in C6 and U251 cells. The cell
population at the G0/G1 phase gradually increased from 58.08±1.40
to 78.03±1.66% for C6 cells, and from 50.58±1.50 to 73.65±1.01% for
U251 cells; and that of S phase decreased from 24.29±1.47 to
14.65±1.04% for C6 cells, and from 29.44±1.57 to 14.39±0.76% for
U251 cells. These results revealed that MOX-induced G0/G1 cell
cycle arrest of C6 and U251 cells was significantly higher compared
with the untreated cells.
To investigate the underlying mechanism of the
growth inhibitory effects of MOX, cell cycle regulators critical to
the G0/G1 phase checkpoint were evaluated, including cyclin D1,
cyclin E, CDK2, CDK4 and CDK6. The complex of cyclin D1 with CDK4
or CDK6 and the complex of cyclin E with CDK2 promote the
transition of cells from the G0/G1 phase to the S phase (24,25).
Western blot analysis confirmed that MOX downregulated the protein
expression levels of cyclin D1, cyclin E, CDK2, CDK4 and CDK6
compared with the control group (Fig.
4C).
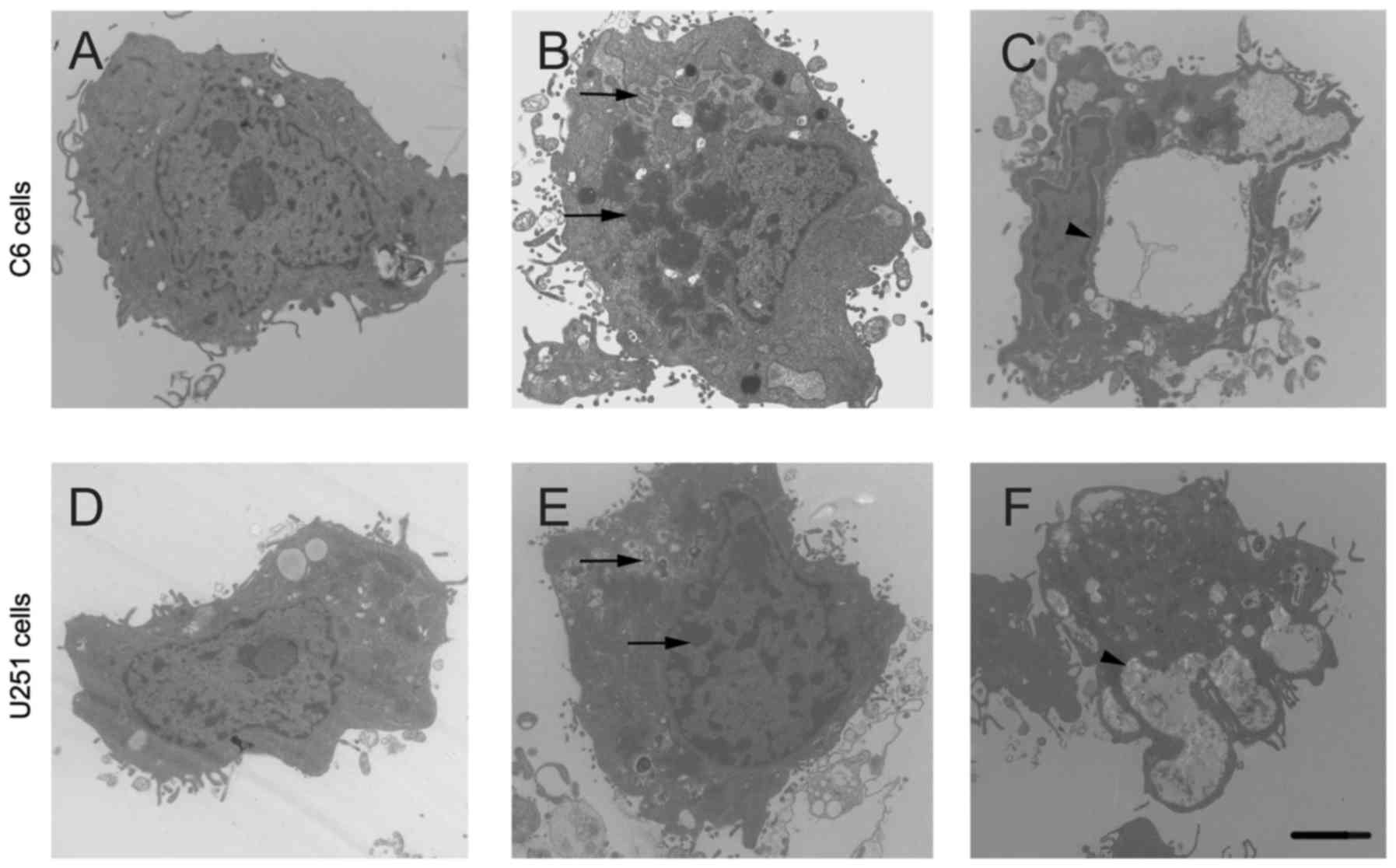
MOX induces ultrastructural
morphological changes in glioma cells
The morphological characteristics of apoptosis were
analyzed using TEM. The results revealed that untreated cells
presented no apoptotic characteristics with a complete cytoplasm
and organelles (Fig. 5A and D).
However, MOX-treated cells presented with typical apoptotic
features: including condensation and margination of nuclear
chromatin, the formation of apoptotic bodies (Fig. 5B and E) and cytoplasmic
hypervacuolization (Fig. 5C and F).
These results suggested that MOX may promote apoptosis in glioma
cells.
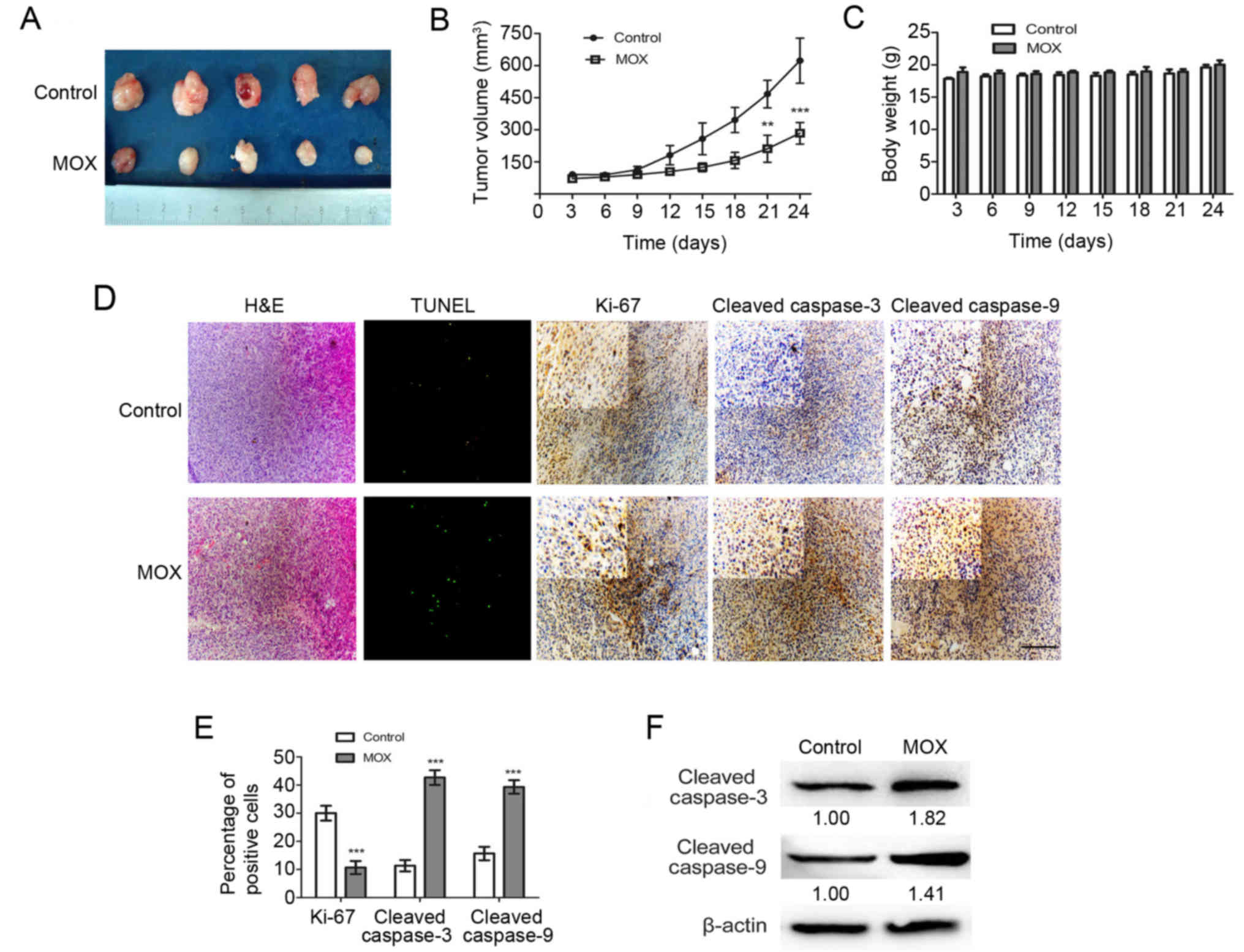
MOX suppresses U251 Xenograft growth
in vivo
Given the effect of MOX on glial cancer cell lines
in vitro, an internal engraftment model was used to examine
whether MOX may induce similar effects in vivo. BALB/c nude
mice were employed for the engraftment of human U251 cells.
Macroscopically, xenografts treated with MOX grew at a slower rate
compared with those treated with saline (Fig. 6A). Notably, tumor growth curves in
mice treated with MOX had a relatively slow trend compared with the
control group (Fig. 6B). No
significant difference in the weights of the mice was observed
between the test group and the control group on all measured days
(Fig. 6C). H&E staining and a
TUNEL assay demonstrated a greater number of dead cells and an
evident increase in the apoptotic proportion in MOX-treated tumor
tissues compared with the saline treated tissues (Fig. 6D). To confirm the change in the
proliferation status of the tumors, immunohistochemistry was
performed to label Ki-67, cleaved caspase-3 and caspase-9, which
are used clinically to assess the proliferative fraction in glioma
cells. All control xenografts displayed a stronger Ki-67 staining
compared with that of MOX-treated mice and revealed that MOX
increased the levels of cleaved caspase-3 and cleaved caspase-9
compared with saline treated tissues (Fig. 6D). As presented in Fig. 6F, the expression of cleaved
caspase-3 and cleaved caspase-9 increased substantially compared
with the control group.
Discussion
Glioma is a complex neuroglial tumor involving the
dysregulation of numerous biological pathways at multiple levels
including aberrant Akt/mTOR signaling pathway, genomic aberrations
and mitochondrial dysfunction (18,26,27).
The lack of efficacious therapeutics for glioma has necessitated
the development of novel therapeutic agents. In the present study,
the antiparasitic agent MOX was revealed to inhibit the viability
of glioma cells in a dose-dependent manner. And certain
morphological features of chromatin condensation and nuclear
membrane clustering in glioma cells were observed. These results
further demonstrated that apoptosis was induced following treatment
with MOX. Further investigation revealed that MOX significantly
induced glioma cell apoptosis by upregulating the Bax/Bcl-2 ratio
and activating the caspase-3/-9 cascade. Additionally, MOX caused
significant G0/G1 cell cycle arrest in glioma cells after 48 h of
treatment determined from the decrease in CDK2, CDK4, CDK6, cyclin
D1 and cyclin E protein expression levels. When investigated
further in vivo, MOX was able to effectively suppress U251
Xenograft growth. These results suggested that MOX likely
represents a promising agent in the treatment of glioma.
MOX is currently being used for the treatment of
onchocerciasis in humans (6).
Furthermore, in a previous study, it was demonstrated that
milbemycins including MOX displayed promising effects for
overcoming multidrug resistance in cancer therapy (14). Considering that MOX has a wide range
of biological activities, along with low neurotoxic potential,
binding affinity and intrinsic activity at relevant central nervous
system receptors (28), MOX is
considered to be an effective compound for the treatment of glioma.
Hence, the present study revealed that MOX significantly inhibited
the viability of C6 and U251 cells in a dose-dependent manner.
Furthermore, MOX exhibited a lesser effect on normal human
astrocyte (SVG p12) cells, and half-maximal inhibitory
concentration values indicated that an MOX analog is a more potent
cytotoxic reagent in breast cancer cells compared with in normal
breast cell (29). Consequently,
the present study suggested that MOX may be used to prevent the
viability of cancer cells at a low concentration and at safer
concentrations.
The balance between viability and apoptosis in
cancer cells is crucial for tumorigenesis (30,31).
Aberrant regulation of apoptotic cell death mechanisms is one of
the hallmarks of cancer development and progression and numerous
cancer cell types exhibit notable resistance to apoptosis signaling
(32,33). Apoptosis is also the main response
of cells to chemotherapeutic agent (31,34).
Previous studies have revealed that Bcl-2 family proteins,
including anti-apoptotic members (Bcl-2) and pro-apoptotic members
(Bax) serve an important role in mediating mitochondrial functions
(35–37). In the present study, it was
hypothesized that Bax and Bcl-2 were involved in the mechanism by
which MOX induces cell apoptosis. Hence, the underlying mechanism
of MOX-induced apoptosis in glioma cells was elucidated using
western blotting. Mechanistically, it was revealed that the
anticancer effect of MOX is mediated by increasing the Bax/Bcl-2
ratio in glioma cells, and that cleaved caspase-3 and cleaved
caspase-9 were also upregulated subsequent to treatment with MOX
for 48 h in C6 and U251 cells. Furthermore, the mitochondrial
apoptosis pathway is characterized by cytochrome c release
from the mitochondria to the cytosol, which activates the initiator
caspase-9, which then targets and activates the apoptotic effector
caspase-3 (38–40). The results of the present study
indicated that MOX-induced apoptosis in glioma cells upregulated
the expression of Bax and downregulated Bcl-2. However the
mitochondrial-mediated apoptotic pathway and other induction
pathways also require further in-depth study and exploration.
In addition to apoptosis, MOX also exhibited the
properties of cell cycle arrest. The flow cytometry data results
indicated that the cell cycle was arrested at the G0/G1 checkpoint
in glioma cells subsequent to MOX treatment. The underlying
mechanism of MOX on the expression of cell cycle regulation
proteins in glioma cells was determined using western blotting. The
results revealed that MOX was able to decrease the protein
expression levels of CDK2, CDK4, CDK6, cyclin D1 and cyclin E. It
has been well established that homeostasis is often disrupted by
the dysregulation of the cell cycle mechanism in cancer cells
(41,42). Cyclin-CDK heterodimers serve an
important role in regulating the progression of cells through the
G1 phase of the cell cycle and initiation of DNA replication
(43). Additionally, these results
manifested that the cell cycle was arrested at the G0/G1 phase
through downregulation of CDK4, CDK6 and cyclin D1, which induce
apoptosis, and the expression of cyclin E, which forms a complex
with CDK2, peaks in the late G1 phase, and which may have resulted
in cell cycle arrest in G0/G1.
Furthermore, experiments in vivo were
conducted according to a previous report, which indicated that the
treatment of breast cancer-bearing nude mice with MOX resulted in
significant inhibition of the tumor growth without overall gross
toxicity (26). In the present
animal study, U251 cells were selected as MOX has been demonstrated
to significantly inhibit the viability of U251 cells in
vitro, and the selected dose of the MOX injections was 20
mg/kg. The reasons are as follows: First, in our previous
experiment, treatment with various related drugs (ivermectin) was
performed in addition to MOX. It was found that the glioma cells
were treated with similar concentrations of both drugs in
vitro study and these concentrations were equal to a
physiological concentration. Then considering this reason and some
animal studies (7,17), the dose of 15 mg/kg was chosen in
our preliminary animal experiment. However, it was found that MOX
(15 mg/kg) had no obvious effect. Hence, the dose of MOX was
increased to 20 mg/kg, and it exerted an inhibitory effect on tumor
growth. Additionally, in certain studies, the bioavailability of
oral MOX compared with subcutaneous MOX in alpacas was 11%
(44); the bioavailability of MOX
in dogs co-administered with lipids was only 40% in lymph
duct-cannulated dogs (45), and the
bioavailability of MOX in rats was moderate at 19% (46). Taking this into consideration, 20
mg/kg was finally selected and was considered to be an effective
and safe concentration. Western blotting and immunohistochemistry
analysis confirmed the increase in TUNEL, Ki-67, cleaved caspase-3
and cleaved caspase-9 following treatment with MOX. These results
revealed that the subcutaneous injection of MOX may reduce the
tumor mass of U251 Xenografts. This means that MOX may be a novel
therapeutic agent for glioma.
The present study demonstrated that MOX has an
inhibitory effect on the viability of glioma cells in vitro
and in vivo by inducing mitochondrial-mediated apoptosis and
G0/G1 cell cycle arrest. Given these results and the fact that MOX
is already clinically approved or being used in clinical trials
(13), MOX may represent a potent
and promising agent to combat glioma.
Acknowledgements
The authors would like to thank Dr Matthew Pona from
the University of Ottawa (ON, Canada) for assisting in the
preparation of the manuscript.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81201723 and
81501050), the ‘Young Talents’ Project of Northeast Agricultural
University (grant no. 14QC05), the Natural Science Foundation of
Heilongjiang Province (grant no. QC2014C104) and the Heilongjiang
Postdoctoral Fund (grant no. LBH-Z13027).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
AG conceived and designed the study. DS and HL
researched the literature, performed analysis of data and drafted
the manuscript. BQ and YL were involved in the conception of the
study. JL, CC, DZ and XZ contributed to the design, execution,
support and/or manuscript review. All the authors read and approved
the final manuscript and agree to be accountable for all aspects of
the research in ensuring that the accuracy or integrity of any part
of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The study was performed in accordance with the
ethical standards of the institutional and/or national research
committee (Harbin Vic Biological Technology Development Co., Ltd.,
Harbin, China) and with the 1964 Helsinki declaration and its later
amendments or comparable ethical standards. Ethical approval was
obtained for the animal experiments.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Gao JH, Wang ZL, Liu HH, Wang LM and Huang
GH: Liposome encapsulated of temozolomide for the treatment of
glioma tumor: Preparation, characterization and evaluation. Drug
Discov Ther. 9:205–212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Liqon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gilbert MR, Wang M, Aldape KD, Stupp R,
Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT,
et al: Dose-dense temozolomide for newly diagnosed glioblastoma: A
randomized phase III clinical trial. J Clin. Oncol. 31:4085–4091.
2013.
|
|
4
|
Lan YL, Wang X, Xing JS, Lou JC, Ma XC and
Zhang B: The potential roles of dopamine in malignant glioma. Acta
Neurol Belg. 117:613–621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McKellar QA and Benchaoui HA: Avermectins
and milbemycins. J Vet Pharmacol Ther. 19:331–351. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cotreau MM, Warren S, Ryan JL,
Fleckenstein L, Vanapalli SR, Brown KR, Rock D, Chen CY and
Schwertschlag US: The antiparasitic moxidectin: Safety,
tolerability, and pharmacokinetics in humans. J Clin Pharmacol.
43:1108–1115. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prichard R, Ménez C and Lespine A:
Moxidectin and the avermectins: Consanguinity but not identity. Int
J Parasitol Drugs and Drug Resist. 14:134–153. 2012. View Article : Google Scholar
|
|
8
|
Perez M, Blazquez AG, Real R, Mendoza G,
Prieto JG, Merino G and Alvarez AI: In vitro and in vivo
interaction of moxidectin with BCRP/ABCG2. Chem Biol Interact.
180:106–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zulalian J, Stout SJ, Cunha AR, Garces T
and Miller P: Absorption, tissue distribution, metabolism, and
excretion of moxidectin in cattle. J Agric Food Chem. 42:381–387.
1994. View Article : Google Scholar
|
|
10
|
Afzal J, Stout SJ, da Cunha AR and Miller
P: Moxidectin: Absorption, tissue distribution, excretion, and
biotransformation of 14C-labeled moxidectin in sheep. J Agric Food
Chem. 42:1767–1773. 1994. View Article : Google Scholar
|
|
11
|
Afzal J, Burke AB, Batten PL, DeLay RL and
Miller P: Moxidectin: Metabolic fate and blood pharmacokinetics of
14C-labeled moxidectin in horses. J Agric Food Chem. 45:3627–3633.
1997. View Article : Google Scholar
|
|
12
|
Traversa D, Di Cesare A, Milillo P, Lohr
B, Iorio R, Pampurini F, Schaper R, Paoletti B and Heine J:
Efficacy and safety of imidacloprid 10%/moxidectin 1% spot-on
formulation in the treatment of feline aelurostrongylosis.
Parasitol Res. 105 Suppl 1:S55–S62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Verma M, Pathak M, Shahab M, Singh K,
Mitra K and Misra-Bhattacharya S: Moxidectin causes adult worm
mortality of human lymphatic filarial parasite Brugia malayi in
rodent models. Folia Parasitol. 61:561–570. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gao A, Wang X, Xiang W, Liang H, Gao J and
Yan Y: Reversal of P-glycoprotein-mediated multidrug resistance in
vitro by doramectin and nemadectin. J Pharm Pharmacol. 62:393–399.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Korystov YN, Ermakova NV, Sterlina LN,
Levitman MK, Shaposhnikova VV, Mosin VA, Drinyaev VA, Kruglyak EB,
Novik TS and Sterlina TS: Avermectins inhibit multidrug resistance
of tumor cells. Eur J Pharmacol. 493:57–64. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mani T, Bourguinat C, Keller K, Asraf S,
Blagburn B and Prichard RK: Interaction of macrocyclic lactones
with a Dirofilaria immitis P-glycoprotein. Int J Parasitol.
46:631–40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dou Q, Chen HN, Wang K, Yuan K, Lei Y, Li
K, Lan J, Chen Y, Huang Z, Xie N, et al: Ivermectin induces
cytostatic autophagy by blocking the PAK1/Akt axis in breast
cancer. Cancer Res. 76:4457–4469. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu YY, Fang SS, Sun QS and Liu B:
Anthelmintic drug ivermectin inhibits angiogenesis, growth and
survival of glioblastoma through inducing mitochondrial dysfunction
and oxidative stress. Biochem Biophys Res Commun. 480:415–421.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Farkas R, Hell E and Palfi T: The efficacy
of four anthelmintics against small strongyles in a stud farm in
Hungary. Magyar Allatorvosok Lapja. 128:291–297. 2006.
|
|
20
|
Lespine A, Martin S, Dupuy J, Roulet A,
Pineau T, Orlowski S and Alvinerie M: Interaction of macrocyclic
lactones with P-glycoprotein: Structure-affinity relationship. Eur
J Pharm Sci. 30:84–94. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mounsey KE, Bernigaud C, Chosidow O and
McCarthy JS: Prospects for moxidectin as a new oral treatment for
human scabies. PLos Negl Trop Dis. 10:e00043892016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fazzio LE, Streitenberger N, Galvan WR,
Sánchez RO, Gimeno EJ and Sanabria RE: Efficacy and productive
performance of moxidectin in feedlot calves infected with nematodes
resistant to ivermectin. Vet Parasitol. 223:26–29. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lloberas M, Alvarez L, Entrocasso C,
Virkel G, Ballent M, Mate L, Lanusse C and Lifschitz A: Comparative
tissue pharmacokinetics and efficacy of moxidectin, abamectin and
ivermectin in lambs infected with resistant nematodes: Impact of
drug treatments on parasite P-glycoprotein expression. Int. J
Parasitol Drugs Drug Resist. 3:20–27. 2013. View Article : Google Scholar
|
|
24
|
Jiang Z, Chai J, Chuang HH, Li S, Wang T,
Cheng Y, Chen W and Zhou D: Artesunate induces G0/G1 cell cycle
arrest and iron-mediated mitochondrial apoptosis in A431 human
epidermoid carcinoma cells. Anticancer Drugs. 23:606–613. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu X, Wu MY, Jiang M, Zhi QM, Bian J, Xu
MD, Gong FR, Hou J, Tao M, Shou LM, et al: TNF-α sensitizes
chemotherapy and radiotherapy against breast cancer cells. Cancer
Cell Int. 17:132017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guichet PO, Guelfi S, Ripoll C, Teigell M,
Sabourin JC, Bauchet L, Rigau V, Rothhut B and Hugnot JP:
Asymmetric distribution of GFAP in glioma multipotent Cells. PLos
One. 11:e01512742016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou XM, Sun R, Luo DH, Sun J, Zhang MY,
Wang MH, Yang Y, Wang HY and Mai SJ: Upregulated TRIM29 promotes
proliferation and metastasis of nasopharyngeal carcinoma via
PTEN/AKT/mTOR signal pathway. Oncotarget. 7:13634–13650.
2016.PubMed/NCBI
|
|
28
|
Janko C and Geyer J: Moxidectin has a
lower neurotoxic potential but comparable brain penetration in
P-glycoprotein-deficient CF-1 mice compared to ivermectin. J Vet
Pharmacol Ther. 36:275–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiang WS, Gao AL, Liang HS, Li CY, Gao JG,
Wang Q, Shuang B, Zhang J, Yan YJ and Wang XJ: Reversal of
P-glycoprotein-mediated multidrug resistance in vitro by milbemycin
compounds in adriamycin-resistant human breast carcinoma
(MCF-7/adr) cells. Toxicol In Vitro. 24:1474–1481. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mattern J and Volm M: Imbalance of cell
proliferation and apoptosis during progression of lung carcinomas.
Anticancer Res. 24:4243–4246. 2004.PubMed/NCBI
|
|
31
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Danial NN: BCL-2 family proteins: Critical
checkpoints of apoptotic cell death. Clin Cancer Res. 13:7254–7263.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiong S, Mu T, Wang G and Jiang X:
Mitochondria-mediated apoptosis in mammals. Protein Cell.
5:737–749. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ekert PG, Read SH, Silke J, Marsden VS,
Kaufmann H, Hawkins H, Hawkins CJ, Gerl R, Kumar S and Vaux DL:
Apaf-1 and caspase-9 accelerate apoptosis, but do not determine
whether factor-deprived or drug-treated cells die. J Cell Biol.
165:835–842. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang MH and Reynolds CP: Bcl-2 inhibitors:
Targeting mitochondrial apoptotic pathways in cancer therapy. Clin
Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liang BX, Liu ZY, Cao YN, Zhu C, Zuo Y,
Huang L, Wen G, Shang N, Chen Y, Yue X, et al: MC37, a new
mono-carbonyl curcumin analog, induces G2/M cell cycle arrest and
mitochondria-mediated apoptosis in human colorectal cancer cells.
Eur J Pharmacol. 796:139–148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gogvadze V, Orrenius S and Zhivotovsky B:
Multiple pathways of cytochrome c release from mitochondria in
apoptosis. Biochim Biophys Acta. 1757:639–647. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Singh PK and Kumar A: Mitochondria
mediates caspase-dependent and independent retinal cell death in
Staphylococcus aureus endophthalmitis. Cell Death Discov.
2:160342016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee JH, Jung JY, Jeong YJ, Park JH, Yang
KH, Yang KH, Choi NK, Kim SH and Kim WJ: Involvement of both
mitochondrial- and death receptor-dependent apoptotic pathways
regulated by Bcl-2 family in sodium fluoride-induced apoptosis of
the human gingival fibroblasts. Toxicology. 243:340–347. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wan D, Jiang C, Hua X, Wang T and Chai M:
Cell cycle arrest and apoptosis induced by aspidin PB through the
p53/p21 and mitochondria-dependent pathways in human osteosarcoma
cells. Anticancer Drugs. 26:931–941. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
McGinnis GR and Young ME: Circadian
regulation of metabolic homeostasis: Causes and consequences. Nat
Sci Sleep. 8:163–180. 2016.PubMed/NCBI
|
|
43
|
Lin CJ, Chang YA, Lin YL, Liu SH, Chang CK
and Chen RM: Preclinical effects of honokiol on treating
glioblastoma multiforme via G1 phase arrest and cell apoptosis.
Phytomedicine. 23:517–527. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cocquyt CM, Van Amstel S, Cox S, Rohrbach
B and Martín-Jiménez T: Pharmacokinetics of moxidectin in alpacas
following administration of an oral or subcutaneous formulation.
Res Vet Sci. 105:160–164. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lespine A, Chanoit G, Bousquet-Melou A,
Lallemand E, Bassissi FM, Alvinerie M and Toutain PL: Contribution
of lymphatic transport to the systemic exposure of orally
administered moxidectin in conscious lymph duct-cannulated dogs.
Eur J Pharm Sci. 27:37–43. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dupuy J, Larrieu G, Sutra JF, Eeckhoutte C
and Alvinerie M: Influence of verapamil on the efflux and
metabolism of 14C moxidectin in cultured rat hepatocytes. J Vet
Pharmacol Ther. 24:171–177. 2001. View Article : Google Scholar : PubMed/NCBI
|