Introduction
Pancreatic ductal adenocarcinoma (PDAC) has a 5-year
survival rate of <8% (1), and is
characterized by a highly aggressive nature and poor response to
clinical treatment. Only a small percentage of patients are able to
receive radical resection, which is the only curative treatment
option, and/or adjuvant chemotherapy with agents such as
gemcitabine and the oral fluoropyrimidine derivative S-1, or other
advanced therapeutic strategies. Among patients who undergo radical
resection, the 5-year survival rate is still only ~25% (2). At present, little is known about the
factors that contribute to the initiation and progression of PDAC,
and its specific underlying mechanisms.
Chronic psychological stress is considered to be a
powerful tumor promoter in numerous types of cancer, including
PDAC, via inducing activation of the hypothalamic pituitary adrenal
(HPA) axis and/or the sympathetic nervous system (SNS) (3,4).
Norepinephrine (NE) is a major stress hormone, which serves a vital
role in the chronic psychological stress that may induce tumor
progression. According to Lara et al (5), the concentration of NE can be ≤10 µM
in the tumor microenvironment, and NE may promote the
proliferation, invasion, migration and malignant biological
behaviors of PDAC via activation of the β2-adrenergic receptor
(β2-AR) in vitro and in vivo (6–9).
However, these studies did not elucidate the possible downstream
mechanisms; therefore, additional research is required.
The Notch-1 pathway is involved in several physical
and pathological biological processes, including cancer (10–15).
Notably, Notch is essential for embryonic development of the
pancreas and is involved in the plasticity of adult exocrine cells
(16–18); in addition, abnormal activation of
the Notch-1 pathway is correlated with the initiation and
progression of PDAC (10,11,19).
Previous studies reported that chronic stress inhibits
differentiation, and maintains the stem cell state of hematopoietic
stem cells via activating the Notch-1 pathway (20,21).
Furthermore, NE may promote angiogenesis in breast cancer via
upregulation of the Notch pathway molecule Jagged-1 (22). These studies suggest that chronic
stress, NE and the Notch-1 pathway may have an interactive
relationship in PDAC. The present study hypothesized that the
stress hormone NE may activate the Notch-1 pathway in PDAC, thus
contributing to its malignant biological behaviors.
Materials and methods
Cell culture and reagents
Human PDAC cell lines (AsPc-1, BxPc-3, Panc-1, HPAC,
Mia PaCa-2 and SW1990) were purchased from the Chinese Academy of
Sciences Cell Bank of Type Culture Collection (Shanghai, China).
Panc-1 and Mia PaCa-2 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; HyClone; GE Healthcare Life Sciences, Logan,
UT, USA), HPAC and BxPc-3 cells were cultured in RPMI-1640 (DMEM;
HyClone; GE Healthcare Life Sciences); both media were supplemented
with 10% fetal bovine serum (FBS; HyClone; GE Healthcare Life
Sciences, Logan, UT, USA) and 1% penicillin-streptomycin. AsPc-1
cells were cultured in RPMI-1640 supplemented with 20% FBS and 1%
penicillin-streptomycin. SW1990 cells were cultured in L-15
Leibovitz media (HyClone; GE Healthcare Life Sciences) supplemented
with 10% FBS and 1% penicillin-streptomycin. The cells were
cultured under standard conditions in an atmosphere containing 5%
CO2 at 37°C. To determine the optimal concentration and
duration of treatment, BxPc-3 and Panc-1 cells were treated with NE
at various concentrations (0, 0.1, 1 or 10 µM) for 24 h, or were
treated with a fixed concentration of NE (10 µM) for various
durations (0, 12, 24 or 48 h). Panc-1 and BxPc-3 cells were
separated into the following four groups for each assay: Negative
control (medium only), NE (10 µM), NE (10 µM) + DAPT (50 µM) and NE
(10 µM) + small interfering (si)RNA-Notch-1 at 37°C for 48 h.
Subsequently, these cells underwent mRNA/protein extraction and
various assays were conducted. NE and DAPT (Notch-1 pathway
inhibitor) were purchased from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany).
The following antibodies were used in the present
study: Anti-β2-AR (cat. no. sc-569, 1:200; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), anti-Notch-1 (cat. no. 3608,
1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-Jagged-1 (cat. no. ab109536, 1:1,000; Abcam, Cambridge, MA,
USA), anti-β-actin (cat. no. 3700, 1:1,000; Cell Signaling
Technology, Inc.), anti-recombination signal binding protein for
immunoglobulin κJ region (RBP-Jκ; cat. no. ab180588, 1:1,000;
Abcam), anti-Hes-1 (cat. no. ab108937, 1:1,000; Abcam), anti-matrix
metalloproteinase (MMP)-2 (cat. no. ab97779, 1:1,000; Abcam), and
anti-MMP-9 (cat. no. ab38898, 1:1,000; Abcam).
siRNA transfection
Notch-1-specific siRNA (si-Notch-1; Notch-1
siRNA-780; sense 5′-GUCCAGGAAACAACUGCAATT-3′ and antisense
5′-UUGCAGUUGUUUCCUGGACT-3′) and a negative control siRNA (sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′) were purchased from Shanghai
GenePharma Co., Ltd. (Shanghai, China). Cells (0.5×104)
were seeded in 6-well plates and were transfected at 37°C with 100
nM siRNAs using Lipofectamine RNAi MAX Reagent (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. Cells were used in the subsequent
experiments 24 h post-transfection.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using the Fastgen200 RNA
isolation system (Fastgen, Shanghai, China), according to the
manufacturer's protocol. A Prime Script RT reagent kit (Takara
Biotechnology Co., Ltd., Dalian, China) was used to reverse
transcribe total RNA into cDNA, according to the manufacturer's
protocol. qPCR was conducted using an iQ5 Multicolor Real-Time PCR
Detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and a SYBR Green PCR kit (Takara Biotechnology Co., Ltd.). The
following PCR program was used: Denaturation at 94°C for 5 min,
followed by 35 cycles consisting of denaturation at 94°C for 30
sec, annealing at 60°C for 30 sec and extension at 72°C for 45 sec;
finally, the samples were incubated at 72°C for 5 min and then
maintained at 4°C. The specificity of the amplified PCR products
was evaluated by melting curve analysis. The comparative Cq method
(23), with β-actin as the
normalization control, was used to assess the expression level of
each target gene, as previously described (24). The PCR primer sequences used were as
follows: Jagged-1, forward TTGGTTAATGGTTATCGCTGTATC, and reverse
GCAGTTCTTGCCCTCATAGTCC; Notch-1, forward GGCACTTTCTGTGAGGAGGA, and
reverse GCAGTCAGGCGTGTTGTTCT; RBP-Jκ, forward GACTCAGACAAGCGAAAGCA,
and reverse GTCGATTAAACAGAGCCACC; Hes-1, forward
TAGCTCGCGGCATTCCAAG, and reverse AAGCGGGTCACCTCGTTCA; MMP-2,
forward GATGATGCCTTTGCTCGTGC, and reverse CAAAGGGGTATCCATCGCCA;
MMP-9, forward TCCACCCTTGTGCTCTTCCCT, and reverse
CTGCCACCCGAGTGTAACCA; and β-actin forward
GACTTAGTTGCGTTACACCCTTTCT, and reverse GAACGGTGAAGGTGACAGCAGT. The
housekeeping gene β-actin was used as an internal control.
Western blot analysis
Whole-cell lysates of Panc-1 and BxPC-3 cells were
prepared using radioimmunoprecipitation assay buffer (Beyotime
Institute of Biotechnology, Haimen, China) according to the
manufacturer's protocol. The protein concentration was determined
using a bicinchoninic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). The protein lysates were resolved on a 10%
polyacrylamide gel with a 5% stacking gel. The proteins were
subsequently transferred to polyvinylidene difluoride membranes.
The membranes were blocked for 2 h in Tris-buffered saline-0.1%
(vol/vol) Tween-20 (TBST) containing 10% (wt/vol) nonfat dry milk
powder at room temperature and were then incubated with primary
antibodies overnight at 4°C. Following incubation with goat
anti-rabbit immunoglobulin G (IgG)-horseradish peroxidase (HRP)
(cat. no. sc-2004, 1:10,000) and goat anti-mouse IgG-HRP (cat. no.
sc-2005, 1:10,000) secondary antibodies (Santa Cruz Biotechnology,
Inc.), for 2 h at room temperature, the membranes were washed with
TBST, and the immunocomplexes were detected using an enhanced
chemiluminescence kit (EMD Millipore, Billerica, MA, USA) and the
Molecular Imager ChemiDoc XRS system (Bio-Rad Laboratories, Inc.).
β-actin was used as the internal loading control.
Immunofluorescence analysis
After applying the aforementioned intervention
strategies, the cancer cells were fixed in 4% formaldehyde diluted
in PBS for 20 min at room temperature. After permeabilization with
0.3% Triton X-100, the cells were treated with blocking buffer [5%
bovine serum albumin (Sigma-Aldrich; Merck KGaA) in PBS] for 1 h,
and then incubated with the primary antibodies at 4°C overnight.
The cells were then incubated with Alexa Fluor 488-conjugated goat
anti-rabbit IgG (green) secondary antibodies (cat. no. 111-545-003,
1:200; Jackson Immunoresearch Laboratories, Inc., West Grove, PA,
USA) at room temperature for 30 min, and the nuclei were stained
with DAPI. Images were pseudo-colored using a Zeiss Instruments
confocal microscope (Zeiss GmbH, Jena, Germany).
MTT assay
Cell viability was analyzed using an MTT assay
according to a previously described method (25). Cancer cells were seeded in 96-well
tissue culture plates at a density of 5,000-10,000 cells/well 24 h
prior to serum starvation. After serum starvation for 24 h, cells
were cultured in medium and were treated with the aforementioned
intervention strategies. After 12, 24 or 48 h, the medium was
removed, and MTT reagent was added to each well and incubated at
37°C for 4 h. Subsequently, 150 µl dimethyl sulfoxide was added to
each well and the cells were incubated in the dark for 10 min at
room temperature. Optical density (OD) values were measured at 490
nm using a microplate reader (BioTek Instruments, Inc., Winooski,
VT, USA). Cell viability rate was defined as follows: OD (sample
well)/OD (control well).
Apoptosis assay
Cell apoptosis was assessed by flow cytometry using
an Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide
(PI) apoptosis detection kit (BD Biosciences, San Diego, CA, USA),
according to the manufacturer's protocol, as previously described
(26). Briefly, cancer cells were
seeded into 6-well plates at a density of 2×105
cells/well, and after being serum-starved overnight, the cells were
treated with the aforementioned intervention strategies for 48 h.
Subsequently, the cells were trypsinized, washed with PBS and
stained with Annexin V and PI. The percentage of apoptotic cells
was quantified by flow cytometry using a FACSCalibur (BD
Biosciences) instrument. Samples were analyzed and the percentage
of apoptotic cells was evaluated.
Cell invasion assay
A Matrigel invasion assay was performed as
previously described (27), in
order to assess the invasive ability of PDAC cells. Briefly, the
upper chambers of the wells were coated with Matrigel (BD
Biosciences). Following treatment with the aforementioned
intervention strategies for 48 h, the cancer cells
(5×105) were suspended in serum-free medium and seeded
into the upper chamber. Cells were allowed to migrate toward media
(DMEM/RPMI-1640) supplemented with 10% FBS in the lower chamber at
37°C for 24 h. The media were aspirated from the inside of the
insert, and the non-invasive cells on the upper side were removed
using a cotton swab. The membrane of the chamber was then fixed
with 4% paraformaldehyde for 15 min at room temperature and stained
with 0.1% crystal violet for 15 min at 37°C. The number of invading
cells was quantified by counting the stained cells under a light
microscope (Nikon Corporation, Tokyo, Japan).
Statistical analysis
Each experiment was performed at least three times.
Data are presented as the means ± standard deviation. Using
GraphPad Prism version 6.0 software (GraphPad Software, Inc., La
Jolla, CA, USA), differences among groups were assessed by one-way
analysis of variance followed by Dunnett's test for multiple
comparisons. All tests were two sided, P<0.05 was considered to
indicate a statistically significant difference.
Results
Expression and location of Notch-1
pathway-associated molecules in PDAC cells
The present study detected the expression levels of
Notch-1 pathway-associated molecules in the pancreatic cell lines.
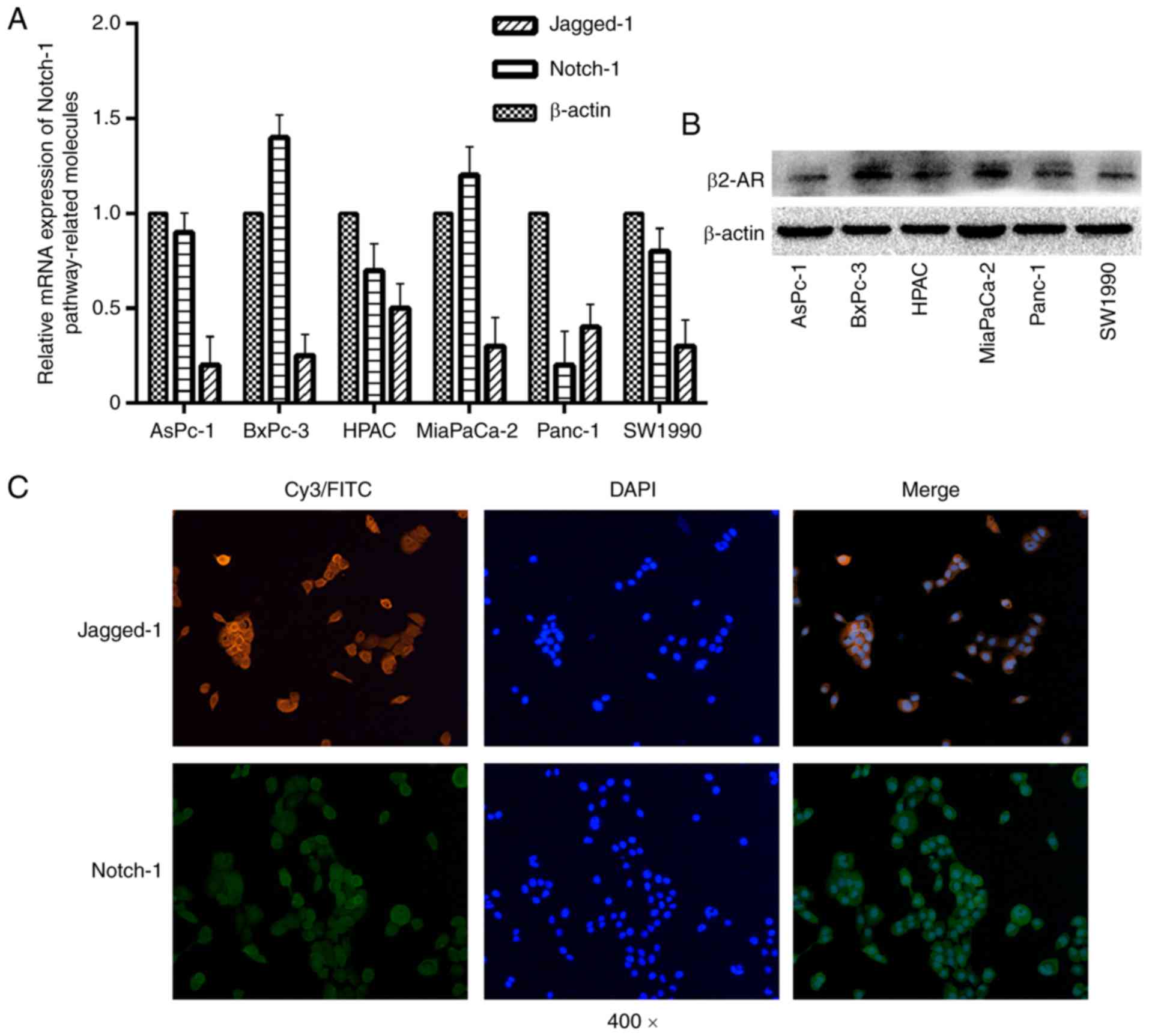
The results demonstrated that the expression levels of Notch-1 and
its ligand Jagged-1 were different in all of the cell lines tested
(Fig. 1A). β2-AR is a corresponding
receptor of NE, which has been reported to be upregulated in
pancreatic cancer tissue (28). The
present study detected the protein expression levels of β2-AR in
six pancreatic cancer cell lines; the results revealed that β2-AR
expression was different in all cell lines analyzed (Fig. 1B). The present study selected two
PDAC cell lines, BxPC-3 and Panc-1, for subsequent experiments and
used them to determine the location of Notch-1 and Jagged-1. In
BxPC-3 and Panc-1 cells (data not shown), Notch-1 and Jagged-1 were
predominantly located on the cytomembrane, rather than in the
cytoplasm or nucleus (Fig. 1C),
which is consistent with the previous findings that the Notch
pathway can be activated through various ligand-receptor
interactions, such as the interaction between receptor Notch-1 and
its ligand Jagged-1 (10). In
conclusion, the Notch-1 pathway may be involved in the development
of PDAC in vitro.
NE promotes the expression of Notch-1
pathway-associated genes in PDAC cells
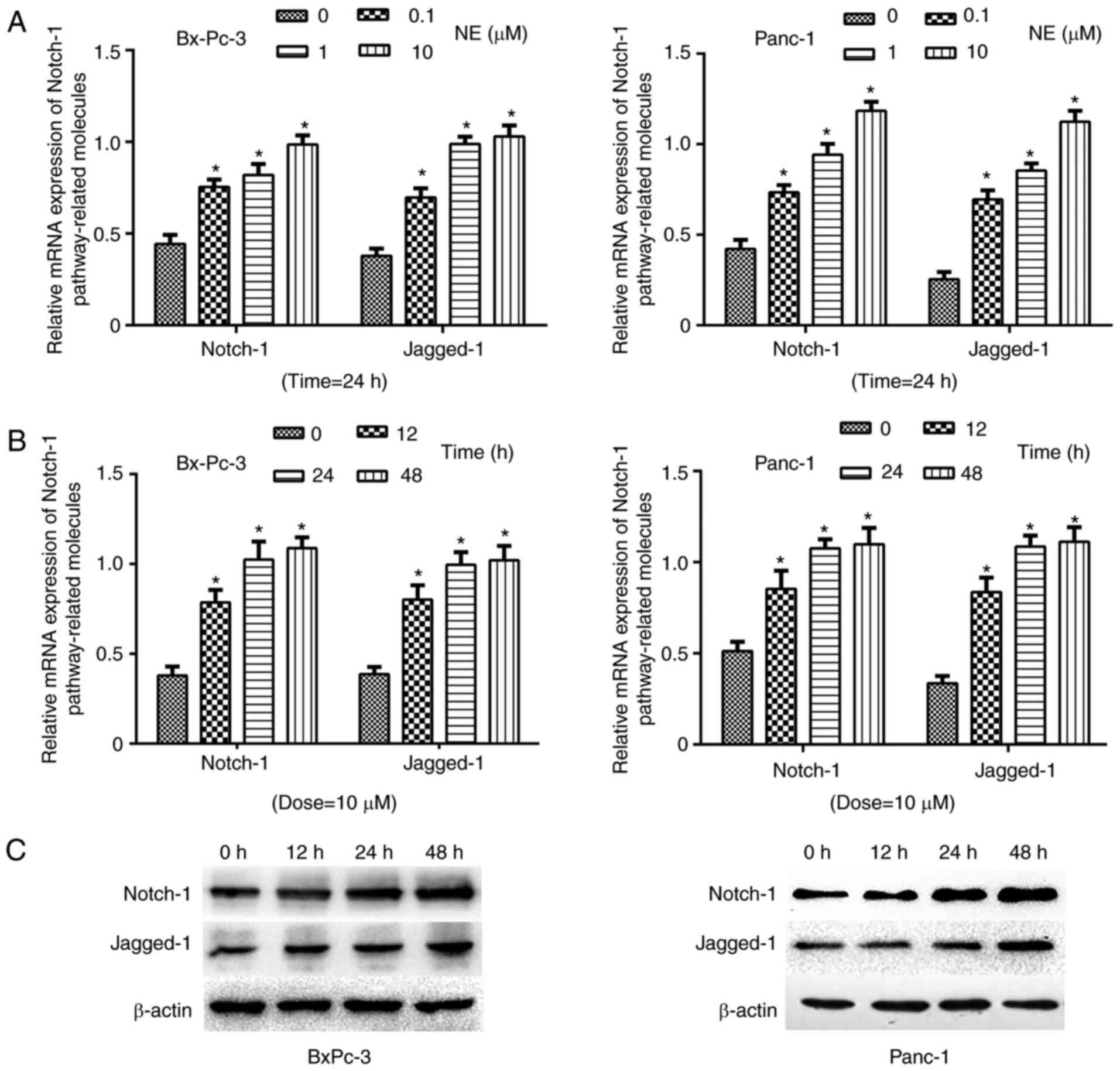
To explore the effects of NE on Notch-1
pathway-associated gene expression in PDAC cells, the cells were
treated with NE at various concentrations (0, 0.1, 1 or 10 µM).
After 24 h, Notch-1 pathway-associated gene expression was detected
by RT-qPCR. The results demonstrated that as the concentration of
NE increased, the expression levels of Notch-1 pathway-associated
genes (Notch-1 and Jagged-1) were elevated in BxPC-3 and Panc-1
cells (Fig. 2A). Furthermore, PDAC
cells were treated with 10 µM NE for various durations (0, 12, 24
or 48 h). Subsequently, RT-qPCR and western blotting indicated that
the expression levels of Notch-1 and Jagged-1 were increased as
treatment duration increased (Fig. 2B
and C). These findings indicated that NE may activate the
Notch-1 pathway in PDAC cells, which is consistent with the
hypothesis that NE may promote the progression of PDAC via
activating the Notch-1 pathway.
NE enhances cell viability and
inhibits apoptosis of PDAC cells via activation of the Notch-1
pathway
To further explore whether the Notch-1 pathway
mediated the tumor-promoting effects of NE on PDAC, si-Notch-1 and
DAPT were used to suppress the Notch-1 pathway in PDAC cells. As
shown in Fig. 3A and B, si-Notch-1
effectively inhibited Notch-1 expression at both the mRNA and
protein levels. To explore the role of Notch-1 in NE-mediated
effects on cell viability and apoptosis of PDAC cells, cells were
treated with DAPT or si-Notch-1. As shown in Fig. 3C, NE treatment alone significantly
enhanced the viability of PDAC cells; however, this effect was
abolished by si-Notch-1 and DAPT. Similarly, an apoptosis assay
indicated that NE inhibited apoptosis of PDAC cells, whereas this
effect was suppressed following inhibition of the Notch-1 pathway
(Fig. 3D and E). These findings
indicated that NE enhanced cell viability and inhibited apoptosis
of PDAC cells via activation of the Notch-1 pathway.
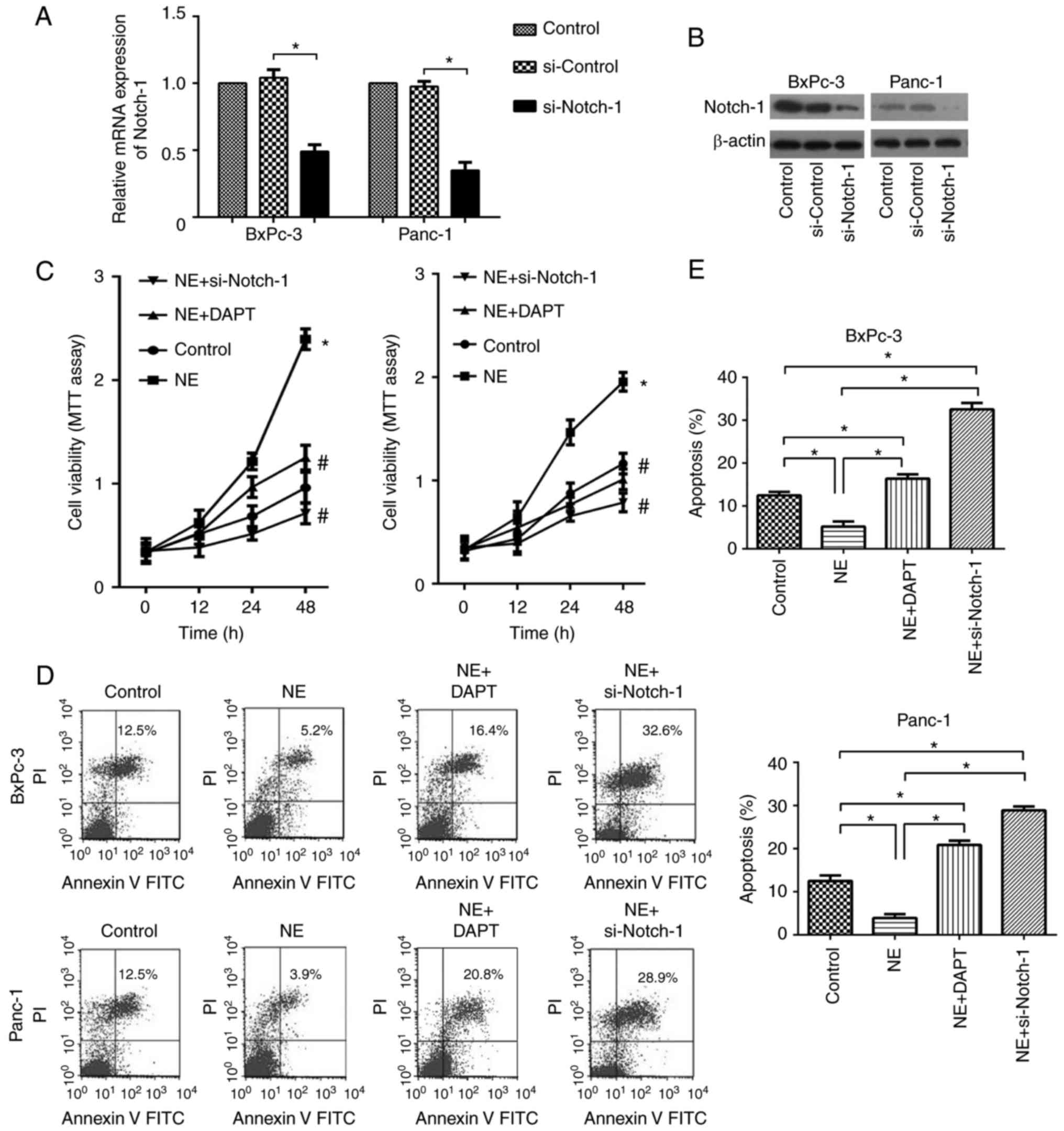 | Figure 3.NE enhances cell viability and
inhibits apoptosis of pancreatic ductal adenocarcinoma cells via
activating the Notch-1 pathway. The silencing effects of si-Notch-1
on Notch-1 (A) mRNA and (B) protein expression were confirmed by
reverse transcription-quantitative polymerase chain reaction and
western blotting, respectively. (C) Viability of BxPC-3 and Panc-1
cells in the control, NE, NE + DAPT and NE + si-Notch-1 groups, as
determined using an MTT assay. *P<0.05, vs. the control group;
#P<0.05, vs. the NE group. (D and E) Apoptosis rate
of BxPC-3 and Panc-1 cells in the control, NE, NE + DAPT and NE +
si-Notch-1 groups. *P<0.05 as indicated. NE, norepinephrine; si,
small interfering RNA. |
NE enhances the invasive ability of
PDAC cells via activation of the Notch-1 pathway
The present study confirmed that NE may promote PDAC
cell viability and inhibit apoptosis; therefore, the effects of NE
on the invasive ability of PDAC cells were subsequently
investigated using a Transwell assay. The results demonstrated that
NE promoted the invasive ability of BxPC-3 and Panc-1 cells,
whereas these effects could be blocked by si-Notch-1 and DAPT
(Fig. 4A and B). Furthermore, it
was revealed that NE upregulated the mRNA and protein expression
levels of critical Notch-1 pathway-associated and -targeted
factors, such as Notch-1, Jagged-1, RBP-Jκ (a mammalian CSL
protein) and Hes-1, and invasion-associated molecules, including
MMP-2 and MMP-9; however, these effects were reversed following
inhibition of the Notch-1 pathway (Fig.
4C and D), which means NE enhanced the invasive ability of PDAC
cells via activating the Notch-1 pathway.
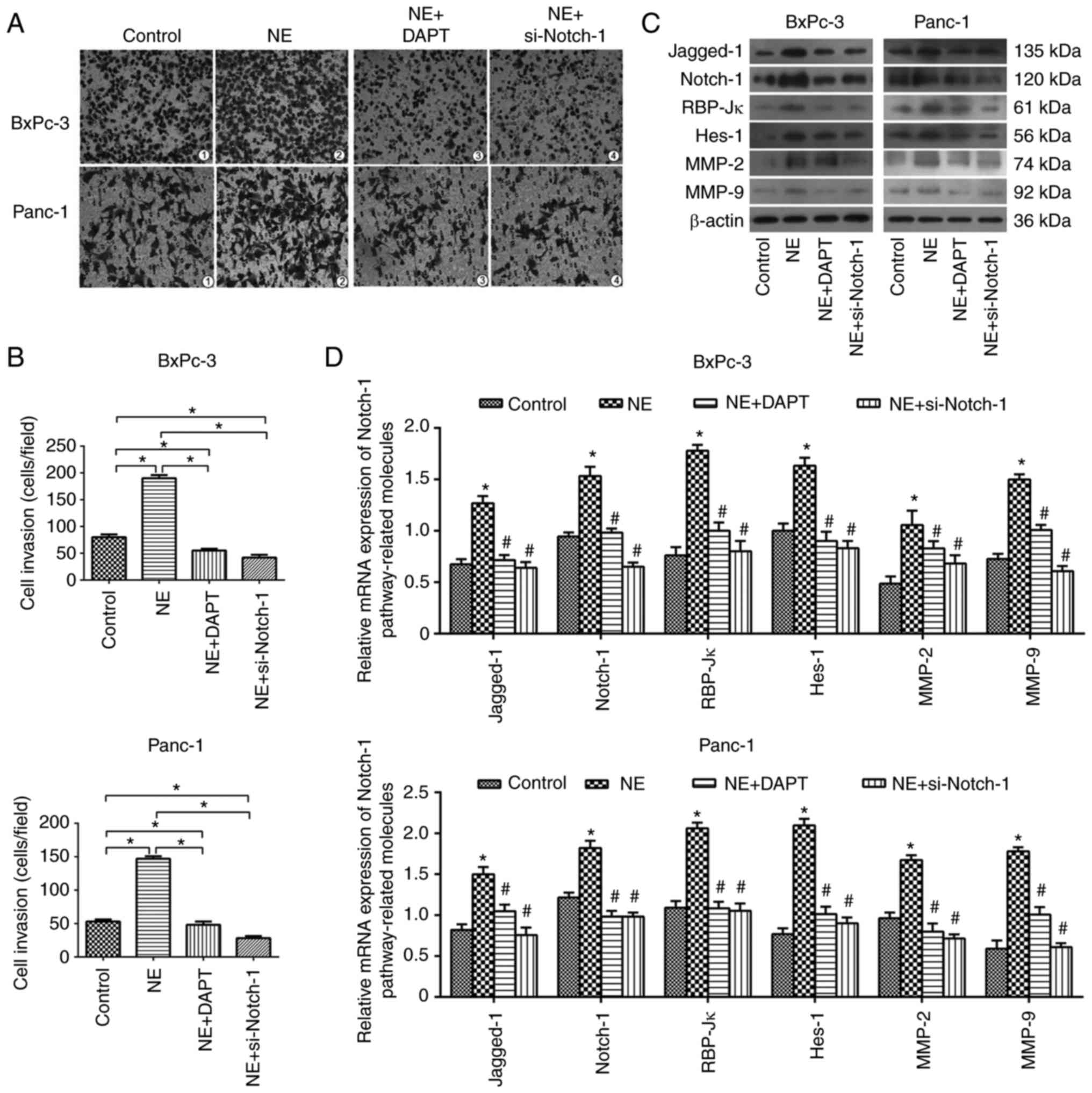 | Figure 4.NE enhances the invasive ability of
pancreatic ductal adenocarcinoma cells via activating the Notch-1
pathway. (A and B) Invasive ability of BxPC-3 and Panc-1 cells in
the control, NE, NE + DAPT and NE + si-Notch-1 groups, as
determined using a Transwell assay (magnification, ×100). Relative
expression levels of Notch-1 pathway-associated and
invasion-associated molecules at the (C) protein and (D) mRNA
levels in BxPC-3 and Panc-1 cells in the control, NE, NE + DAPT and
NE + si-Notch-1 groups. *P<0.05 vs. the control group;
#P<0.05 vs. the NE group. MMP, matrix
metalloproteinase; NE, norepinephrine; RBP-Jκ, recombination signal
binding protein for immunoglobulin κJ region; si, small interfering
RNA. |
Discussion
Complexities in the prevention, diagnosis and
treatment of PDAC may be partly due to the fact that little is
currently known regarding the factors involved in PDAC progression
and its specific mechanisms. However, the cancer-promoting effects
of chronic psychological stress, and its major downstream stress
hormone NE, are widely known (3,29,30).
Chronic psychological stress can influence various
physical and pathological biological processes, and it has been
reported to potentially initiate the progression of cancer via
activating the HPA axis and/or the SNS (3,4,31). As
a major downstream factor, NE levels are markedly elevated in the
cancer microenvironment (10 µM) compared with under normal
physiological conditions (10–1,000 pM) (5,32).
Furthermore, a clinical trial that contained a large group of
patients with cancer demonstrated that >9,000 patients were in a
state of severe psychological stress and tumor tissues from 30
patients with pancreatic cancer contained a high level of NE
(33,34). Furthermore, numerous in vitro
and in vivo studies have demonstrated that NE can induce
proliferation, invasion, migration, apoptosis inhibition,
angiogenesis and other malignant biological behaviors of PDAC via
activation of β2-AR and its downstream factors, including
p38/mitogen-activated protein kinase, cAMP response element binding
protein, nuclear factor-κB and activator protein-1; however, these
effects were blocked by β-receptor antagonists and/or inhibitory
neurotransmitter γ-aminobutyric acid (6,9,30,35).
In addition, continuous activation of the Notch-1 pathway has been
reported to affect the development of the pancreas and PDAC
(10,19,36,37).
Previous studies have used transgenic mice to indicate that
abnormal activation of the Notch pathway promotes K-RAS oncogene
mutations and mediates acinar-to-ductal metaplasia, pancreatic
intraepithelial neoplasia and PDAC (19,37–39).
In another study, a cyclin-dependent kinase inhibitor dinaciclib
(SCH727965) was revealed to suppress the growth of transplanted
tumors via inhibiting Notch-1 (40). Furthermore, in vitro studies
have reached similar conclusions (41–46).
Notably, in previous studies, the Notch-1 pathway was reported to
be associated with chronic stress and/or NE in hematopoietic stem
cells and breast cancer (20–22).
These findings indicated that chronic stress, NE and the Notch-1
pathway may have an interactive relationship in PDAC. Therefore,
the present study aimed to elucidate whether NE could promote
malignant biological behaviors in PDAC cells via activating the
Notch-1 pathway.
The present study initially measured the expression
levels of Notch-1 pathway-associated molecules in six pancreatic
cancer cell lines; after which one cell line with relatively high
Notch-1 expression, BxPC-3, and one with relatively low Notch-1
expression, Panc-1, were selected for further experimentation.
Notch-1 and Jagged-1 were revealed to be primarily located on the
cytomembrane rather than in the cytoplasm or nucleus of BxPC-3 and
Panc-1 cells. Subsequently, PDAC cells underwent a gradient
dose/time intervention strategy with NE; the results indicated that
NE activated the Notch-1 pathway by increasing the levels of two
critical molecules, Notch-1 and Jagged-1. Subsequently, in order to
explore whether NE mediated the enhancement of the malignant
biological behaviors of PDAC in a Notch-1 pathway-dependent manner,
cells were treated with si-Notch-1 or DAPT (Notch-1 pathway
inhibitor) to block the Notch-1 pathway during NE treatment.
RT-qPCR and western blotting demonstrated that NE affected the
expression of Notch-1 pathway-associated genes/proteins in BxPC-3
and Panc-1 cells, and MTT, Annexin V-FITC/PI and Transwell assays
revealed that NE enhanced cell viability and invasiveness, and
inhibited apoptosis of PDAC cells via activating the Notch-1
pathway. Notably, following inhibition of the Notch-1 pathway using
DAPT and si-Notch-1, NE-induced upregulation of Notch-1 pathway
target genes, such as Hes-1, MMP-2 and MMP-9, was inhibited.
Furthermore, the upregulation of Notch-1 pathway-associated
molecules, such as Notch-1, Jagged-1 and RBP-Jκ was also inhibited.
These findings suggested that NE may be an inducer and activator of
the Notch-1 pathway; however, the specific mechanism requires
further study.
Conversely, si-Notch-1 and DAPT could not completely
block NE-mediated progression of PDAC, thus suggesting that other
pathways or molecules may influence this process. In addition, the
Notch pathway participates in various biological processes and has
a complex network of interactions with other pathways (42,43,47).
Notably, it has been confirmed to serve dual roles in the
initiation and progression of several types of cancers or the
various stages of a single cancer type (36,38,48,49).
Furthermore, NE is just one of the chronic stress hormones, and its
use in isolation cannot completely mimic the in vivo chronic
psychological stress environment. This is a limitation of the
present study, which aimed to explore how chronic stress affects
the initiation and progression of PDAC. Therefore, additional
studies are required to prove these findings in vivo.
In conclusion, the present study demonstrated that
the stress hormone NE may activate the Notch-1 pathway in PDAC and
promote its malignant biological behaviors (Fig. 5); this may be an important factor in
its development and be associated with a specific underlying
mechanism in the progression of PDAC. These findings may provide
information regarding a novel approach for targeting NE and/or the
Notch-1 pathway in the prevention and treatment of PDAC.
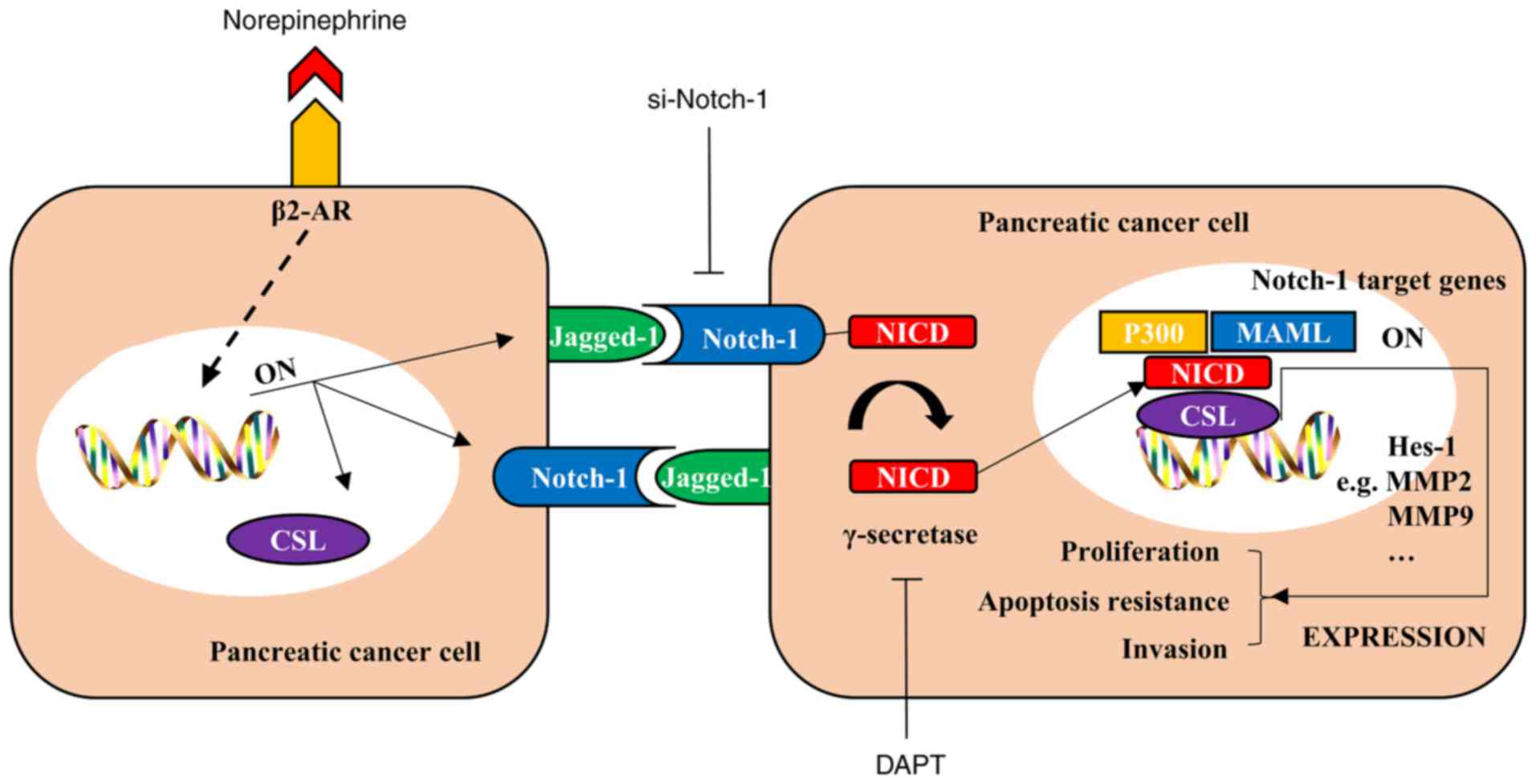 | Figure 5.NE promotes malignant biological
behaviors of pancreatic cancer cells in a Notch-1-dependent manner.
NE binds to its receptor β2-AR and activates downstream signaling,
which elevates the expression of critical Notch-1
pathway-associated molecules, such as Jagged-1, Notch-1 and CSL
(also known as recombination signal binding protein for
immunoglobulin κJ region in mammals). The Notch-1 pathway is
abnormally activated in pancreatic cancer cells and its target
genes promote several malignant biological behaviors; however,
these effects may be suppressed by a Notch-1 inhibitor (si-Notch-1)
and DAPT, an inhibitor of γ-secretase, which is a key molecule that
can release NICD from Notch 1 and activate the Notch-1 pathway.
Consequently, NE promotes malignant biological behaviors of
pancreatic cancer cells in a Notch-1-dependent manner. β2-AR,
β2-adrenergic receptor; MAML, mastermind-like; MMP, matrix
metalloproteinase; NICD, Notch intracellular cytoplasmic domain;
si, small interfering RNA. |
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81402971,
81672434 and 81472248).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
WQ, SL, QM and WD designed the experiments. JL
performed the majority of the experiments. KC, ZJ and CZ analyzed
the data. LC, BY and JC organized the figures and were involved in
the conception of the study. WQ wrote the manuscript. QM and WD
reviewed the manuscript and supervised this study. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Conti CM, Maccauro G and Fulcheri M:
Psychological stress and cancer. Int J Immunopathol Pharmacol.
24:1–5. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clarke DM and Currie KC: Depression,
anxiety and their relationship with chronic diseases: A review of
the epidemiology, risk and treatment evidence. Med J Aust.
190:S54–S60. 2009.PubMed/NCBI
|
|
5
|
Lara HE, Dorfman M, Venegas M, Luza SM,
Luna SL, Mayerhofer A, Guimaraes MA, Rosa E, Silva AA and Ramírez
VD: Changes in sympathetic nerve activity of the mammalian ovary
during a normal estrous cycle and in polycystic ovary syndrome:
Studies on norepinephrine release. Microsc Res Tech. 59:495–502.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Quốc Lu'o'ng KV and Nguyễn LT: The roles
of beta-adrenergic receptors in tumorigenesis and the possible use
of beta-adrenergic blockers for cancer treatment: Possible genetic
and cell-signaling mechanisms. Cancer Manag Res. 4:431–445.
2012.PubMed/NCBI
|
|
7
|
Chan C, Lin HJ and Lin J:
Stress-associated hormone, norepinephrine, increases proliferation
and IL-6 levels of human pancreatic duct epithelial cells and can
be inhibited by the dietary agent, sulforaphane. Int J Oncol.
33:415–419. 2008.PubMed/NCBI
|
|
8
|
Guo K, Ma Q, Li J, Wang Z, Shan T, Li W,
Xu Q and Xie K: Interaction of the sympathetic nerve with
pancreatic cancer cells promotes perineural invasion through the
activation of STAT3 signaling. Mol Cancer Ther. 12:264–273. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang XY, Wang HC, Yuan Z, Huang J and
Zheng Q: Norepinephrine stimulates pancreatic cancer cell
proliferation, migration and invasion via β-adrenergic
receptor-dependent activation of P38/MAPK pathway.
Hepatogastroenterology. 59:889–893. 2012.PubMed/NCBI
|
|
10
|
Gordon WR, Arnett KL and Blacklow SC: The
molecular logic of Notch signaling-a structural and biochemical
perspective. J Cell Sci. 121:3109–3119. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao J, Long B and Wang Z: Role of Notch
signaling pathway in pancreatic cancer. Am J Cancer Res. 7:173–186.
2017.PubMed/NCBI
|
|
12
|
Vinson KE, George DC, Fender AW, Bertrand
FE and Sigounas G: The Notch pathway in colorectal cancer. Int J
Cancer. 138:1835–1842. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Brien R and Marignol L: The Notch-1
receptor in prostate tumorigenesis. Cancer Treat Rev. 56:36–46.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brzozowa-Zasada M, Piecuch A, Dittfeld A,
Mielańczyk Ł, Michalski M, Wyrobiec G, Harabin-Słowińska M, Kurek J
and Wojnicz R: Notch signalling pathway as an oncogenic factor
involved in cancer development. Contemp Oncol. 20:267–272.
2016.
|
|
15
|
Liu J, Shen JX, Wen XF, Guo YX and Zhang
GJ: Targeting Notch degradation system provides promise for breast
cancer therapeutics. Crit Rev Oncol Hematol. 104:21–29. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jensen J: Gene regulatory factors in
pancreatic development. Dev Dyn. 229:176–200. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Siveke JT, Lubeseder-Martellato C, Lee M,
Mazur PK, Nakhai H, Radtke F and Schmid RM: Notch signaling is
required for exocrine regeneration after acute pancreatitis.
Gastroenterology. 134:544–555. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miyamoto Y, Maitra A, Ghosh B, Zechner U,
Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM,
Wolfe MS, et al: Notch mediates TGF alpha-induced changes in
epithelial differentiation during pancreatic tumorigenesis. Cancer
Cell. 3:565–576. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De La O JP, Emerson LL, Goodman JL, Froebe
SC, Illum BE, Curtis AB and Murtaugh LC: Notch and Kras reprogram
pancreatic acinar cells to ductal intraepithelial neoplasia. Proc
Natl Acad Sci USA. 105:18907–18912. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Duncan AW, Rattis FM, DiMascio LN, Congdon
KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, et
al: Integration of Notch and Wnt signaling in hematopoietic stem
cell maintenance. Nat Immunol. 6:314–322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Spiegel A, Kalinkovich A, Shivtiel S,
Kollet O and Lapidot T: Stem cell regulation via dynamic
interactions of the nervous and immune systems with the
microenvironment. Cell Stem Cell. 3:484–492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen H, Liu D, Yang Z, Sun L, Deng Q, Yang
S, Qian L, Guo L, Yu M, Hu M, et al: Adrenergic signaling promotes
angiogenesis through endothelial cell-tumor cell crosstalk. Endocr
Relat Cancer. 21:783–795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Duan W, Chen K, Jiang Z, Chen X, Sun L, Li
J, Lei J, Xu Q, Ma J, Li X, et al: Desmoplasia suppression by
metformin-mediated AMPK activation inhibits pancreatic cancer
progression. Cancer Lett. 385:225–233. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li X, Ma G, Ma Q, Li W, Liu J, Han L, Duan
W, Xu Q, Liu H, Wang Z, et al: Neurotransmitter substance P
mediates pancreatic cancer perineural invasion via NK-1R in cancer
cells. Mol Cancer Res. 11:294–302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu J, Ma J, Wu Z, Li W, Zhang D, Han L,
Wang F, Reindl KM, Wu E and Ma Q: Arginine deiminase augments the
chemosensitivity of argininosuccinate synthetase-deficient
pancreatic cancer cells to gemcitabine via inhibition of NF-κB
signaling. BMC Cancer. 14:6862014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang D, Lei J, Ma J, Chen X, Sheng L,
Jiang Z, Nan L, Xu Q, Duan W, Wang Z, et al: β2-adrenogenic
signaling regulates NNK-induced pancreatic cancer progression via
upregulation of HIF-1α. Oncotarget. 7:17760–17772. 2016.PubMed/NCBI
|
|
28
|
Weddle DL, Tithoff P, Williams M and
Schuller HM: Beta-adrenergic growth regulation of human cancer cell
lines derived from pancreatic ductal carcinomas. Carcinogenesis.
22:473–479. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vere CC, Streba CT, Streba LM, Ionescu AG
and Sima F: Psychosocial stress and liver disease status. World J
Gastroenterol. 15:2980–2986. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schuller HM, Al-Wadei HA, Ullah MF and
Plummer HK III: Regulation of pancreatic cancer by
neuropsychological stress responses: A novel target for
intervention. Carcinogenesis. 33:191–196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Herman JP, Figueiredo H, Mueller NK,
Ulrich-Lai Y, Ostrander MM, Choi DC and Cullinan WE: Central
mechanisms of stress integration: Hierarchical circuitry
controlling hypothalamo-pituitary-adrenocortical responsiveness.
Front Neuroendocrinol. 24:151–180. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schmidt C and Kraft K: Beta-endorphin and
catecholamine concentrations during chronic and acute stress in
intensive care patients. Eur J Med Res. 1:528–532. 1996.PubMed/NCBI
|
|
33
|
Zabora J, BrintzenhofeSzoc K, Curbow B,
Hooker C and Piantadosi S: The prevalence of psychological distress
by cancer site. Psychooncology. 10:19–28. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schuller HM, Al-Wadei HA and Majidi M:
GABA B receptor is a novel drug target for pancreatic cancer.
Cancer. 112:767–778. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Friedman GD, Udaltsova N and Habel LA:
Norepinephrine antagonists and cancer risk. Int J Cancer.
128:737–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dotto GP: Notch tumor suppressor function.
Oncogene. 27:5115–5123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nakhai H, Siveke JT, Klein B,
Mendoza-Torres L, Mazur PK, Algül H, Radtke F, Strobl L,
Zimber-Strobl U and Schmid RM: Conditional ablation of Notch
signaling in pancreatic development. Development. 135:2757–2765.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mazur PK, Einwächter H, Lee M, Sipos B,
Nakhai H, Rad R, Zimber-Strobl U, Strobl LJ, Radtke F, Klöppel G,
et al: Notch2 is required for progression of pancreatic
intraepithelial neoplasia and development of pancreatic ductal
adenocarcinoma. Proc Natl Acad Sci USA. 107:13438–13443. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hanlon L, Avila JL, Demarest RM, Troutman
S, Allen M, Ratti F, Rustgi AK, Stanger BZ, Radtke F, Adsay V, et
al: Notch1 functions as a tumor suppressor in a model of
K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res.
70:4280–4286. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Feldmann G, Mishra A, Bisht S, Karikari C,
Garrido-Laguna I, Rasheed Z, Ottenhof NA, Dadon T, Alvarez H,
Fendrich V, et al: Cyclin-dependent kinase inhibitor Dinaciclib
(SCH727965) inhibits pancreatic cancer growth and progression in
murine xenograft models. Cancer Biol Ther. 12:598–609. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Z, Zhang Y, Banerjee S, Li Y and
Sarkar FH: Notch-1 down-regulation by curcumin is associated with
the inhibition of cell growth and the induction of apoptosis in
pancreatic cancer cells. Cancer. 106:2503–2513. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gaspar NJ, Li L, Kapoun AM, Medicherla S,
Reddy M, Li G, O'Young G, Quon D, Henson M, Damm DL, et al:
Inhibition of transforming growth factor beta signaling reduces
pancreatic adenocarcinoma growth and invasiveness. Mol Pharmacol.
72:152–161. 2016. View Article : Google Scholar
|
|
43
|
Bao B, Wang Z, Ali S, Kong D, Li Y, Ahmad
A, Banerjee S, Azmi AS, Miele L and Sarkar FH: Notch-1 induces
epithelial-mesenchymal transition consistent with cancer stem cell
phenotype in pancreatic cancer cells. Cancer Lett. 307:26–36. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang Z, Banerjee S, Li Y, Rahman KM, Zhang
Y and Sarkar FH: Down-regulation of notch-1 inhibits invasion by
inactivation of nuclear factor-kappaB, vascular endothelial growth
factor, and matrix metalloproteinase-9 in pancreatic cancer cells.
Cancer Res. 66:2778–2784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang Z, Zhang Y, Li Y, Banerjee S, Liao J
and Sarkar FH: Down-regulation of Notch-1 contributes to cell
growth inhibition and apoptosis in pancreatic cancer cells. Mol
Cancer Ther. 5:483–493. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yabuuchi S, Pai SG, Campbell NR, de Wilde
RF, De Oliveira E, Korangath P, Streppel MM, Rasheed ZA, Hidalgo M,
Maitra A, et al: Notch signaling pathway targeted therapy
suppresses tumor progression and metastatic spread in pancreatic
cancer. Cancer Lett. 335:41–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang Z, Sengupta R, Banerjee S, Li Y,
Zhang Y, Rahman KM, Aboukameel A, Mohammad R, Majumdar AP,
Abbruzzese JL, et al: Epidermal growth factor receptor-related
protein inhibits cell growth and invasion in pancreatic cancer.
Cancer Res. 66:7653–7660. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Koch U and Radtke F: Notch and cancer: A
double-edged sword. Cell Mol Life Sci. 64:2746–2762. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
South AP, Cho RJ and Aster JC: The
double-edged sword of Notch signaling in cancer. Semin Cell Dev
Biol. 23:458–464. 2012. View Article : Google Scholar : PubMed/NCBI
|