Introduction
Arsenic disulfide (As2S2) is
an orange-red crystalline mineral and the principal effective
component of realgar, and has been used extensively to treat
various diseases in ancient China and Europe (1). In recent decades, a series of studies
have revealed the marked therapeutic potential of
As2S2 in hematopoietic tumors, particularly
acute promyelocytic leukemia (APL) (2–4). In
addition, recent evidence has revealed the potent anticancer effect
of As2S2 against various human solid cancer
cell lines, but with markedly decreased toxicity in normal somatic
cells (5–7). Previous studies have demonstrated that
As2S2 exerts potent anticancer effects in
human hepatocellular carcinoma cells, cervical cancer cells,
endometrial cancer cells, ovarian cancer cells, malignant melanoma
cells, pancreatic carcinoma cells and gastric cancer cells, whereas
human normal fibroblast cell lines and other human normal cells,
including a lung fibroblast cell line (MRC-5), dermal fibroblast
cells (HF and Hs-68), embryonic liver cells (L02) and normal breast
epithelial cells (184B5), were much less markedly affected by
As2S2 treatment (8–12).
However, relatively few studies have investigated the potential
antitumor activity of As2S2 in human breast
carcinoma and its underlying molecular mechanisms (12–14).
Breast cancer is one of the most common malignancies
among women (15,16). Although conventional therapies, such
as chemotherapy and radiotherapy, have improved the outcomes for
patients with breast cancer, drug resistance and high rates of
recurrence still hamper their efficacies in clinical application
(17). Arsenic trioxide (ATO) has
been approved by the US Food and Drug Administration in 2000 as an
agent for the treatment of APL (18) and reportedly has promising
therapeutic potential against breast cancer (19). As2S2 has a
number of benefits over ATO, including a relatively low toxicity
and safety in oral administration, while exerting a similar
antitumor effect (20,21). Exploring the antitumor effects of
As2S2 against breast carcinoma might thus
shed new light on the therapeutic potential of this arsenic
compound for the treatment of breast cancer.
Programmed cell death (PCD), which refers to any
form of cell death mediated by an intracellular death program,
serves a fundamental function in biological homeostasis (22,23).
Dysregulation of this self-destructive process leads to various
human diseases, including breast cancer. Apoptosis (type I cell
death) and autophagy (type II cell death) are the two primary forms
of PCD defined on the basis of morphological criteria (23,24).
Apoptosis, the primary and most well-researched mode of PCD, has
been regarded as the principal pathway of PCD (25). Apoptosis induction serves an
essential function in anticancer chemotherapies against various
types of cancer (26). Autophagy is
a highly regulated catabolic process that enables cells to clean up
and degrade their own cytoplasmic components (24,27).
Autophagy induction is attributed to various stresses that
ultimately lead to apoptosis, and organelle dysfunction, metabolic
stress, chemotherapies, pathogen infection and starvation are known
to induce autophagy (25). There is
a complex connection between apoptosis and autophagy; indeed,
apoptosis may begin with autophagy, and autophagy may end with
apoptosis. It has been suggested that targeting these two
self-destructive processes may be a particularly useful
chemotherapeutic strategy in the treatment of cancer (28), including breast cancer (29). Accumulating evidence has indicated
that apoptosis and autophagy can be induced by
As2S2 treatment in hematopoietic as well as
solid cancer cell lines (8,30,31).
Our previous studies revealed the inhibitory effect of
As2S2 on breast cancer cells, mediated by the
induction of apoptosis (12,13).
However, the molecular mechanism underlying the involvement of
As2S2 in apoptosis and autophagy in breast
cancer cells remains unclear, warranting further investigation.
Reactive oxygen species (ROS), as a common indicator
of oxidative stress, consist of superoxide, hydrogen peroxide and
the hydroxyl free radical (32,33).
ROS production by xenobiotics selectively kills cancer cells with
negligible effects on normal cells (34). Intriguingly, arsenic compounds
promote the generation of ROS, and this increased ROS accumulation
mediates the genotoxicity of arsenic in cancer cells, thereby
facilitating the induction of apoptosis (35–37).
ROS therefore serve a pivotal function in cancer cell death caused
by arsenic compounds, making them a tempting target for an
As2S2-based strategy of cytotoxic
intervention in breast carcinoma.
The aim of the present study was to investigate the
anticancer effects of As2S2 in human breast
cancer cells in vitro and the potential underlying molecular
mechanisms involved, particularly with respect to the induction of
PCD and the generation of ROS.
Materials and methods
Reagents
Cell Counting Kit-8 (CCK-8) was purchased from
Dojindo Molecular Technologies, Inc. (Kumamoto, Japan).
Calcein-acetoxymethyl ester (AM) and Hoechst 33342 were purchased
from Molecular Probes; Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). A Fluorescein Isothiocyanate (FITC)-Phycoerythrin Annexin V
Apoptosis Detection kit was obtained from BD Biosciences (San Jose,
CA, USA). As2S2, propidium iodide (PI), RNase
A solution and 2′,7′-dichlorofluorescin diacetate (DCF-DA) were
purchased from Sigma; Merck KGaA (Darmstadt, Germany). Chloroquine
diphosphate (CQ) was purchased from Wako Pure Chemical Industries,
Ltd. (Osaka, Japan). An Enhanced Chemiluminescence (ECL) Western
Blotting Analysis system and ECL Prime Western Blotting Detection
reagent were purchased from GE Healthcare Life Sciences (Little
Chalfont, UK). Rabbit anti-human matrix metalloproteinase-9
(MMP-9), rabbit anti-human B-cell lymphoma 2 (Bcl-2), rabbit
anti-human B-cell lymphoma extra-large (Bcl-xl), rabbit anti-human
caspase-7, mouse anti-human caspase-8, rabbit anti-human
microtubule-associated protein 1A/1B-light chain 3 (LC3A/B), mouse
anti-human cyclin B1 and rabbit anti-human cell division cycle
protein 2 (Cdc2) were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Mouse anti-human Bcl-2-associated X protein
(Bax) was purchased from Sigma; Merck KGaA.
Cell lines and cell culture
The human breast cancer MCF-7 and MDA-MB-231 cell
lines were purchased from the American Type Culture Collection
(Manassas, VA, USA). Cells were cultured in α-minimal essential
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 1%
penicillin/streptomycin and fetal bovine serum (10% for MCF-7 and
15% for MDA-MB-231; Sigma; Merck KGaA). The cells were cultured and
maintained as attached cells at 37°C in a humidified atmosphere
containing 5% CO2.
Cell culture assays and drug
treatment
MCF-7 and MDA-MB-231 cells were seeded at 10,000 and
15,000 cells/well, respectively, in 500 µl cell culture medium on
48-well plates (Iwaki microplates; Iwaki Co., Ltd., Tokyo, Japan),
followed by overnight incubation at 37°C.
As2S2 was subsequently added to the
corresponding wells to adjust the final drug concentrations to
between 0 and 16 µM. MCF-7 and MDA-MB-231 cells were allowed to
grow for 48 h in the presence of different concentrations of
As2S2, followed by a cytotoxicity assay.
Cytotoxicity assay
Cell cytotoxicity was analyzed using a CCK-8 assay.
For each cell line, ~1×104 cells/well were seeded into
48-well plates. As2S2 was subsequently added
to the corresponding wells to adjust the final drug concentrations
to between 0 and 16 µM. The plates were then incubated at 37°C in a
humidified atmosphere containing 5% CO2 for 48 h.
Following incubation, 25 µl CCK-8 reagent was added to each well,
followed by further incubation at 37°C for 3 h. The optical density
(OD) value of each well was determined using a microplate reader
(Corona MT P-32; Corona Co., Ibaraki, Japan) at 570 nm. The cell
viability rate was calculated according to the following equation:
Cell viability rate = (OD sample value - OD blank value)/(OD
control value - OD blank value) × 100%.
Morphological analysis and cell
viability assay
MCF-7 and MDA-MB-231 cells were seeded onto a
96-well plate at 5×103 cells/well in 100 µl culture
medium, followed by exposure to different concentrations of
As2S2 (0, 4, 8 and 16 µM) for 48 h. The cells
were then stained for 15 min in the dark at 37°C with the specific
live probe calcein-AM, prior to capturing images and analysis using
an Operetta CLS fluorescence microplate reader (PerkinElmer, Inc.,
Waltham, MA, USA) and the Harmony software program (version 4.5;
PerkinElmer, Inc.).
Wound healing assay
Migration was determined using a wound scratching
assay. Cells were seeded at 4×105 cells/well in 6-well
plates (Iwaki microplates) and cultured for 24 h to form a
confluent cell monolayer. A wound was then scratched onto the cells
using a sterile micropipette tip. The cells were washed with PBS
and treated with various concentrations of
As2S2 (0, 8 and 16 µM), followed by further
incubation for 48 h. Images of each scratch at the same location
were captured at 0 and 48 h using an IX70® inverted
microscope (magnification, ×100) (Olympus Corporation, Tokyo,
Japan). Cell migration was quantified by measuring the wound
opening area using the ImageJ program (version 1.50i, National
Institutes of Health, Bethesda, MD, USA).
Cell cycle analyses
MCF-7 and MDA-MB-231 cells were seeded at
4×105 cells/well in 6-well plates (Iwaki microplates),
followed by overnight incubation. Cells were treated with 0, 4, 8
and 16 µM As2S2, followed by a further 48 h
of incubation at 37°C. Cells were harvested and washed with PBS
twice. Cells were fixed in 70% ethanol overnight at −20°C and
stained with PI and RNase A solution (5 µg/ml PI and 0.5 µg/µl
RNase A). The DNA content was determined by flow cytometry (BD
Biosciences), and data were analyzed using the cell cycle analysis
software program ModFit LT (version 3.0; Verity Software House,
Inc., Topsham, ME, USA).
Morphological characteristics of
apoptosis
Hoechst 33342 staining was performed to observe
morphological characteristics of apoptotic cells. MCF-7 and
MDA-MB-231 cells were seeded onto a 96-well plate at
5×103 cells/well, followed by exposure to different
concentrations of As2S2 (0, 4, 8 and 16 µM)
for 48 h. The cells were then stained with Hoechst 33342 solution
at 37°C for 15 min in the dark. The cells were observed and
analyzed for morphological changes of the nucleus using a
fluorescence microplate reader and the Harmony software
program.
Assessment of apoptosis
MCF-7 and MDA-MB-231 cells were seeded at
2×105 cells/well in 6-well plates (2 ml/well) and
treated with serial concentrations of As2S2
(0, 4, 8 and 16 µM), followed by additional incubation for 48 h at
37°C. The apoptotic rates for the two cell lines were determined
using an FITC-Annexin V Apoptosis Detection kit. The staining
procedure was performed according to the manufacturer's protocol.
In total, ~1×104 cells were analyzed using a flow
cytometer and BD FACSDiva software (version 6.0; BD Biosciences).
The cells were subsequently assessed for the total number of
apoptotic cells, including early-apoptotic (Annexin
V+/PI−) and late-apoptotic (Annexin
V+/PI+) cells.
Autophagy inhibition in breast cancer
cells
To examine whether or not
As2S2-induced cell death was mediated through
autophagy, the autophagy inhibitor CQ (10 µM) was added to MCF-7
and MDA-MB-231 cells 1 h prior to the addition of
As2S2. Subsequently,
As2S2 was added at concentrations of 0, 4, 8
and 16 µM. After 48 h of treatment, the CCK-8 assay was performed
as aforementioned.
Western blot analyses
The standard Western blot protocol was performed in
order to evaluate the protein levels of Bcl-2, Bax, Bcl-xl,
caspase-7, caspase-8, cyclin B1, Cdc2 and LC3A/B in MCF-7 and
MDA-MB-231 cells. The total protein content was extracted from each
cell line treated by As2S2 at various final
concentrations (0, 4, 8 and 16 µM) for 48 h. In brief, cell lysates
were separated by SDS-PAGE (12.5% gel) and transferred onto a
polyvinylidene difluoride transfer membrane (Immobilon-P; Merck
KGaA). Membranes were blocked with 5% dried skimmed milk powder in
Tris-buffered saline containing 0.2% Tween-20 (TBST) for 1 h at
room temperature. The membranes were washed with TBST and incubated
overnight at 4°C with 1:1,000 anti-rabbit MMP-9 specific antibody
(cat. no. 3852; Cell Signaling Technology, Inc.), 1:1,000
anti-rabbit Bcl-2 specific antibody (cat. no. 4223; Cell Signaling
Technology, Inc.), 1:1,000 anti-rabbit Bcl-xl specific antibody
(cat. no. 2764; Cell Signaling Technology, Inc.), 1:500 anti-mouse
Bax specific antibody (cat. no. B8429; Sigma; Merck KGaA), 1:1,000
anti-rabbit caspase-7 specific antibody (cat. no. 12827; Cell
Signaling Technology, Inc.), 1:1,000 anti-mouse caspase-8 specific
antibody (cat. no. 9746; Cell Signaling Technology, Inc.), 1:1,000
anti-mouse cyclin B1 specific antibody (cat. no. 4135; Cell
Signaling Technology, Inc.), 1:1,000 anti-rabbit Cdc2 specific
antibody (cat. no. 9112; Cell Signaling Technology, Inc.) and
1:1,000 anti-rabbit LC3A/B specific antibody (cat. no. 12741; Cell
Signaling Technology, Inc.). Membranes were also probed with
anti-β-actin antibody (cat. no. ab49900; Abcam, Cambridge, UK) at
1:4,000 dilution as the internal control. The membranes were
incubated with the aforementioned primary antibodies at 4°C
overnight and then incubated with 1:1,000 anti-mouse (cat. no.
7076; Cell Signaling Technology, Inc.) or 1:1,000 anti-rabbit (cat.
no. 7074; Cell Signaling Technology, Inc.) specific polyclonal
secondary antibodies for 1 h at room temperature, followed by
washing three times with TBST. Signals were detected using an ECL
Western Blot detection kit in a luminescent image analyzer
(LAS-3000; Fujifilm Corporation, Tokyo, Japan).
Determination of ROS
MCF-7 and MDA-MB-231 cells were seeded at
4×105 cells/well in 6-well plates (Iwaki microplates),
followed by overnight incubation at 37°C. Cells were treated with
different concentrations of As2S2 (0, 4, 8
and 16 µM), followed by an additional incubation for 48 h. DCF-DA
was then added to the two cell lines to a final concentration of 10
µM and incubated at 37°C for 30 min in the dark. Subsequently,
MCF-7 and MDA-MB-231 cells were harvested, washed with PBS and
resuspended in 500 µl PBS. The intracellular ROS levels of the two
cell lines were detected and analyzed using a flow cytometer and BD
FACSDiva software.
Statistical analyses
Statistical analyses were performed using GraphPad
Prism software (version 6.0; GraphPad Software, La Jolla, CA, USA).
Results are presented as the mean ± standard error of the mean of
three or more independent experiments. A one-way analysis of
variance followed by Tukey's post hoc test was used for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
As2S2 inhibits
the cell viability of breast cancer cells
MCF-7 and MDA-MB-231 cells were cultured in the
presence of various concentrations of As2S2
ranging between 0 and 16 µM for 48 h, and a CCK-8 assay was
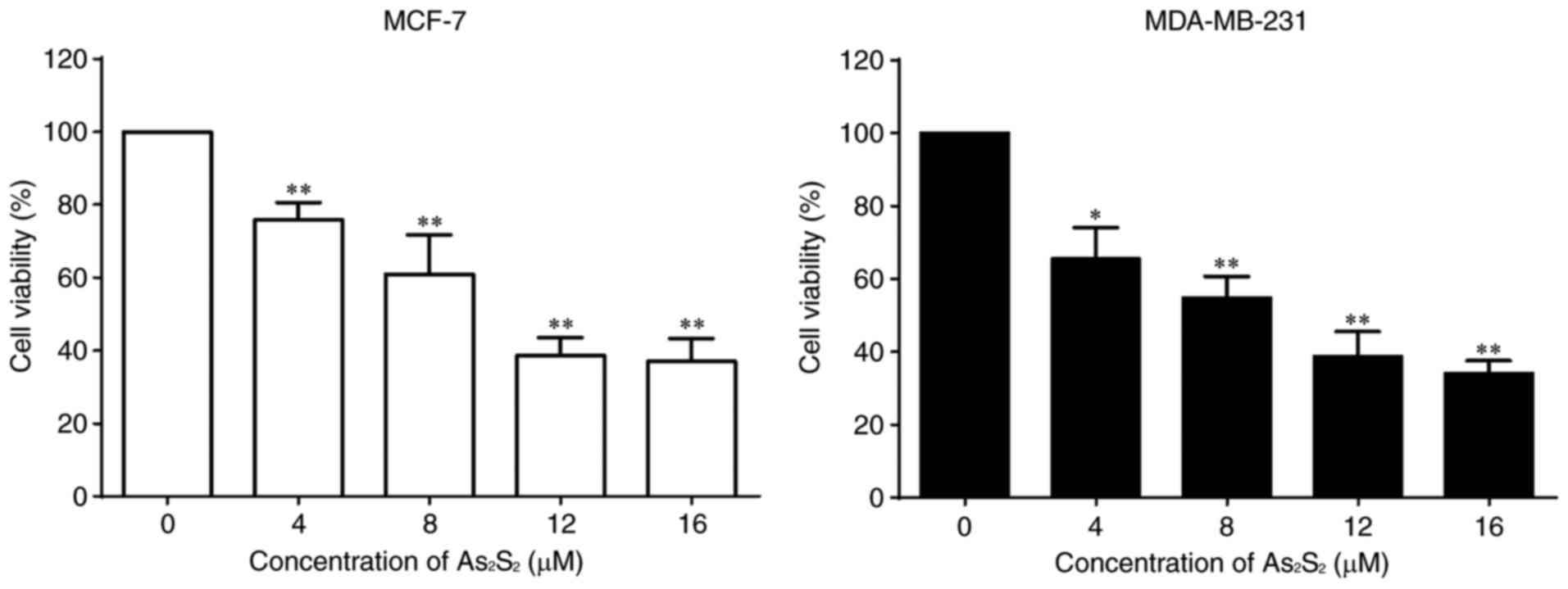
performed to determine cell viabilities. As presented in Fig. 1, As2S2
inhibited the proliferation of the breast cancer cell lines MCF-7
and MDA-MB-231 in a dose-dependent manner. The half-maximal
inhibitory concentrations (IC50 values) of
As2S2 in MCF-7 and MDA-MB-231 cells were
11.75±1.99 and 8.21±2.07 µM after 48 h of exposure,
respectively.
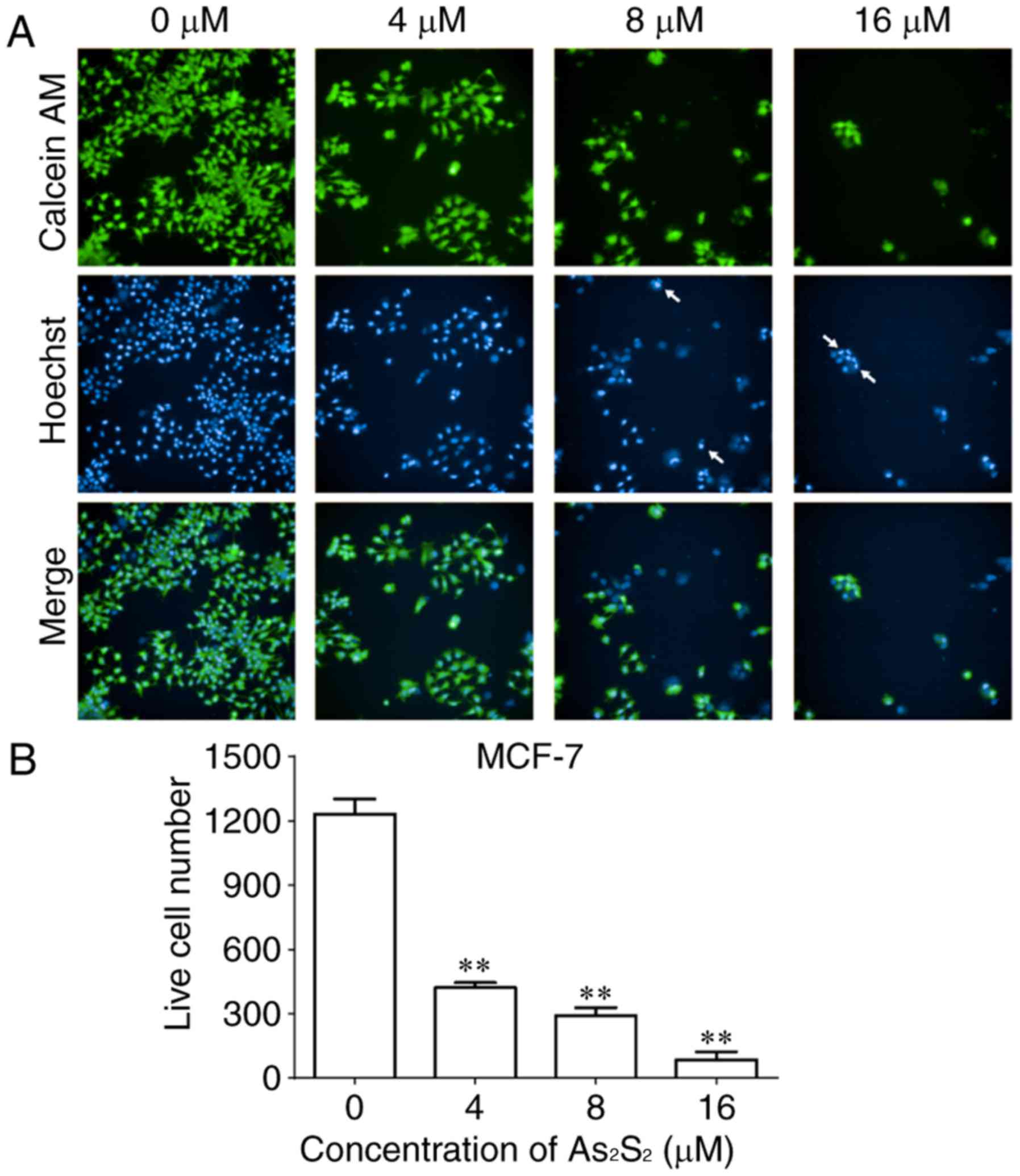
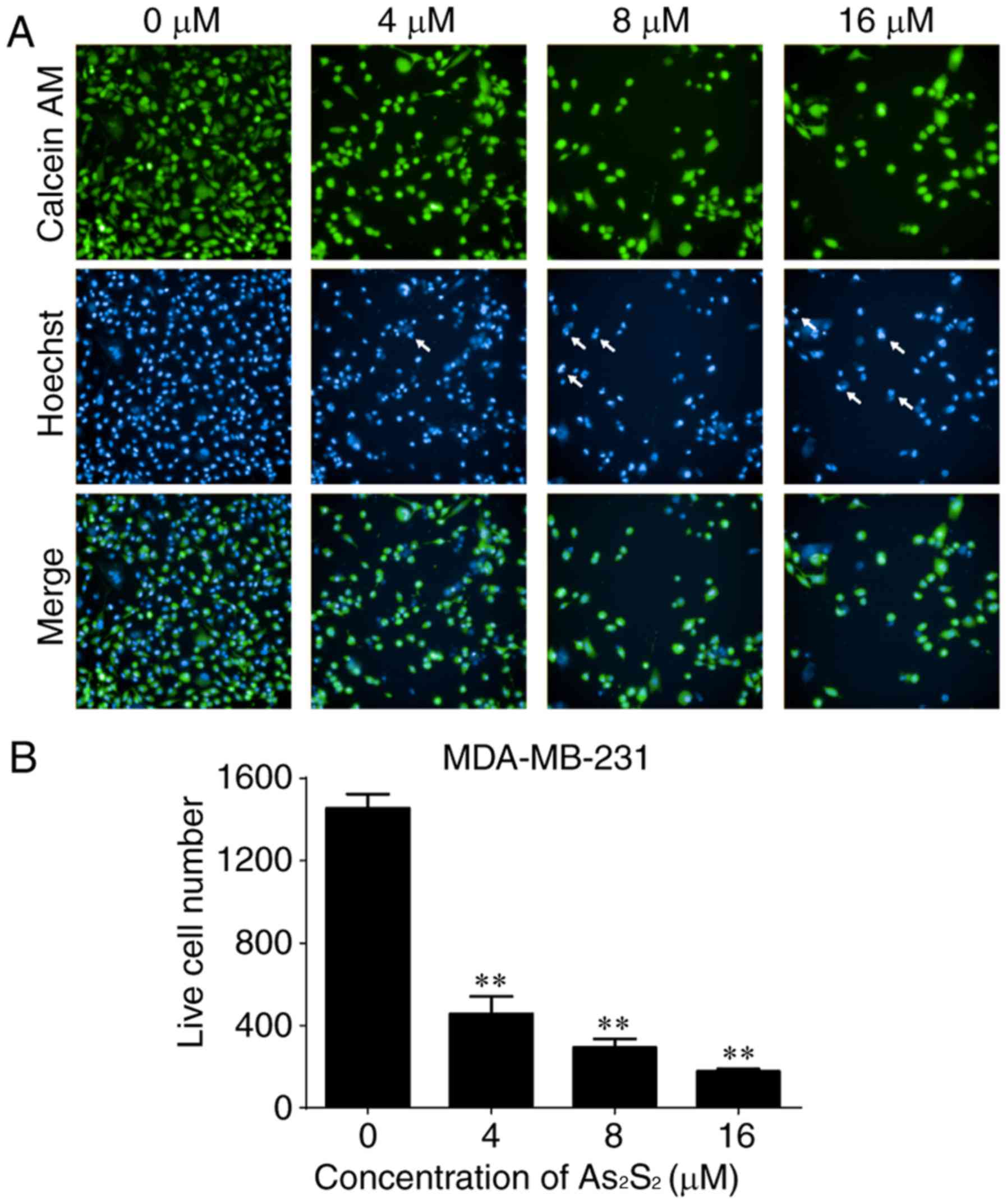
As an additional measurement to monitor cell growth
inhibition induced by As2S2 in breast cancer
cells, the fluorescent dye calcein-AM was used to identify live
cells (38). As presented in
Figs. 2 and 3, live cell numbers in the two cell lines
markedly decreased following treatment with increasing
As2S2 concentrations in a dose-dependent
manner. In MCF-7 cells, compared with the control group (0 µM
As2S2; 1,232.00±70.74 cells), the live cell
number was significantly decreased to 422.00±22.87 (P<0.0001),
291.70±37.17 (P<0.0001) and 85.00±36.76 (P<0.0001) following
exposure to 4, 8 and 16 µM As2S2 for 48 h,
respectively (Fig. 2). In
MDA-MB-231 cells, compared with the control group (0 µM
As2S2; 1,455.00±68.75 cells), the live cell
number was significantly decreased to 457.00±84.23 (P<0.0001),
292.80±42.24 (P<0.0001) and 177.00±11.92 (P<0.0001) following
exposure to 4, 8 and 16 µM As2S2 for 48 h,
respectively (Fig. 3).
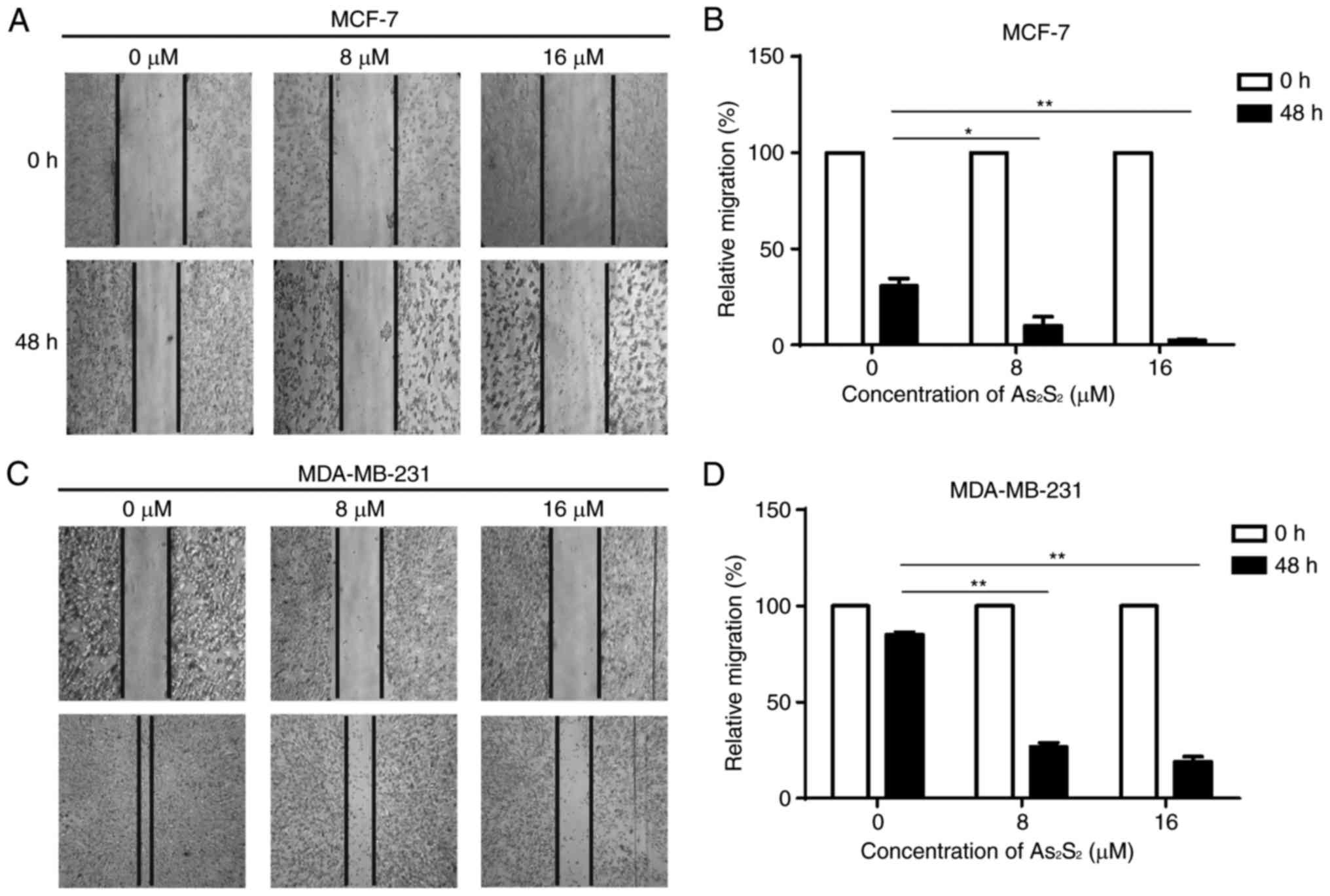
As2S2 inhibits
the motility of breast cancer cells
A scratch assay was performed to assess the effect
of As2S2 on the motility of breast cancer
cells. As presented in Fig. 4, with
the dynamic observation at 0 and 48 h after scratching,
As2S2 treatment significantly inhibited
migration of the two cell lines. In MCF-7 cells, compared with the
control group (0 µM As2S2), the relative
migration rates significantly decreased following exposure to 8 µM
(P=0.0177) and 16 µM (P=0.0042) As2S2. In
MDA-MB-231 cells, compared with the control group (0 µM
As2S2), the relative migration rates
significantly decreased following exposure to 8 µM (P<0.0001)
and 16 µM (P<0.0001) As2S2. These data
indicate that As2S2 inhibits the motility and
invasion of different types of breast cancer cell.
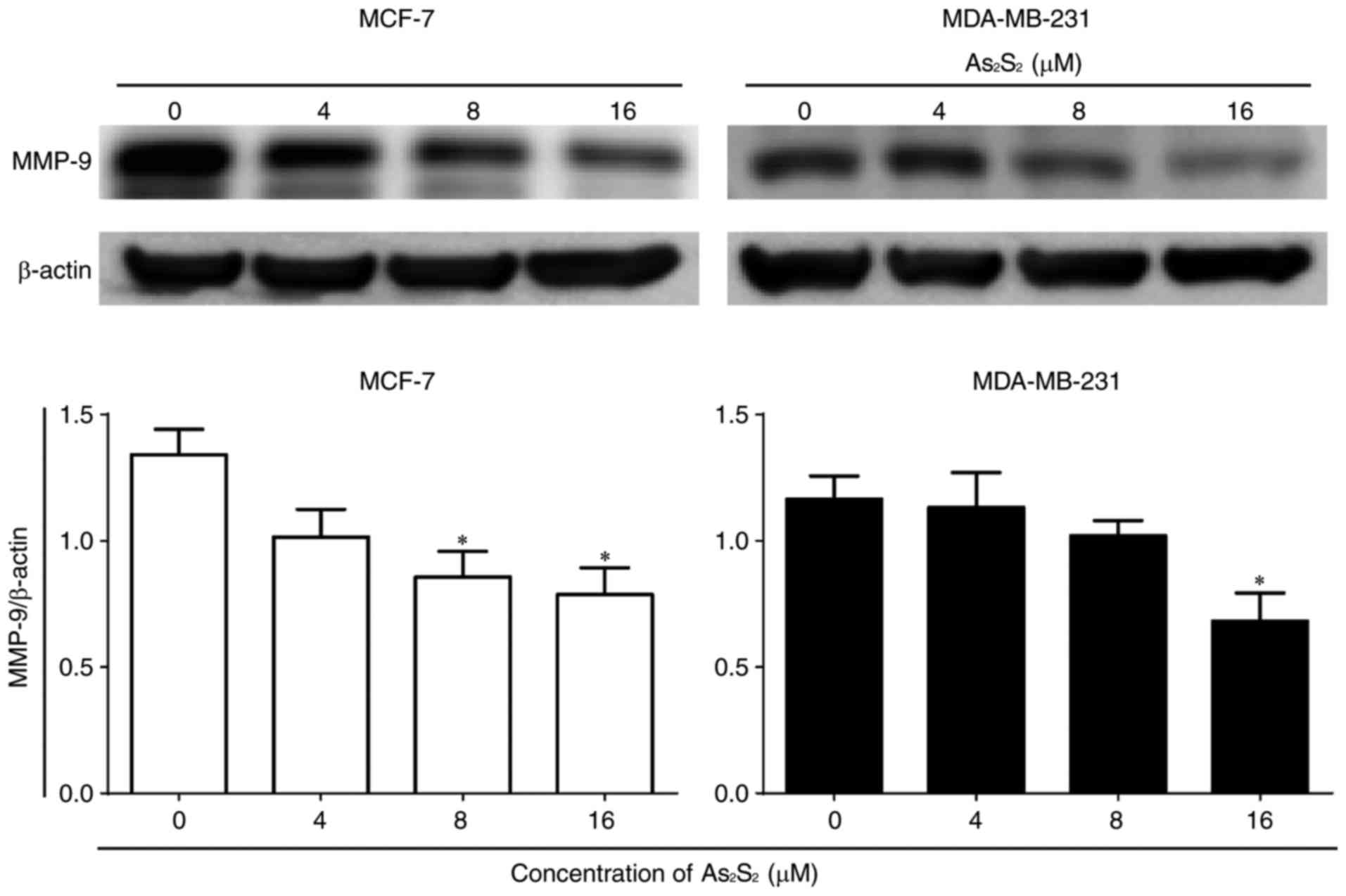
In addition, the expression of the tumor migration-
and invasion-associated protein MMP-9 was determined by western
blot analysis. As presented in Fig.
5, As2S2 treatment significantly
decreased MMP-9 expression at concentrations of 8 (P=0.0446) and 16
(P=0.0233) µM in MCF-7 cells in comparison with the control. In
contrast, in MDA-MB-231 cells, compared with the control, a
statistically significant decrease in the MMP-9 expression occurred
at 16 µM As2S2 (P=0.0444). These results
suggested that As2S2 exposure decreased the
motility of breast cancer cells due at least in part to its
downregulation of MMP-9 signals.
As2S2 triggers
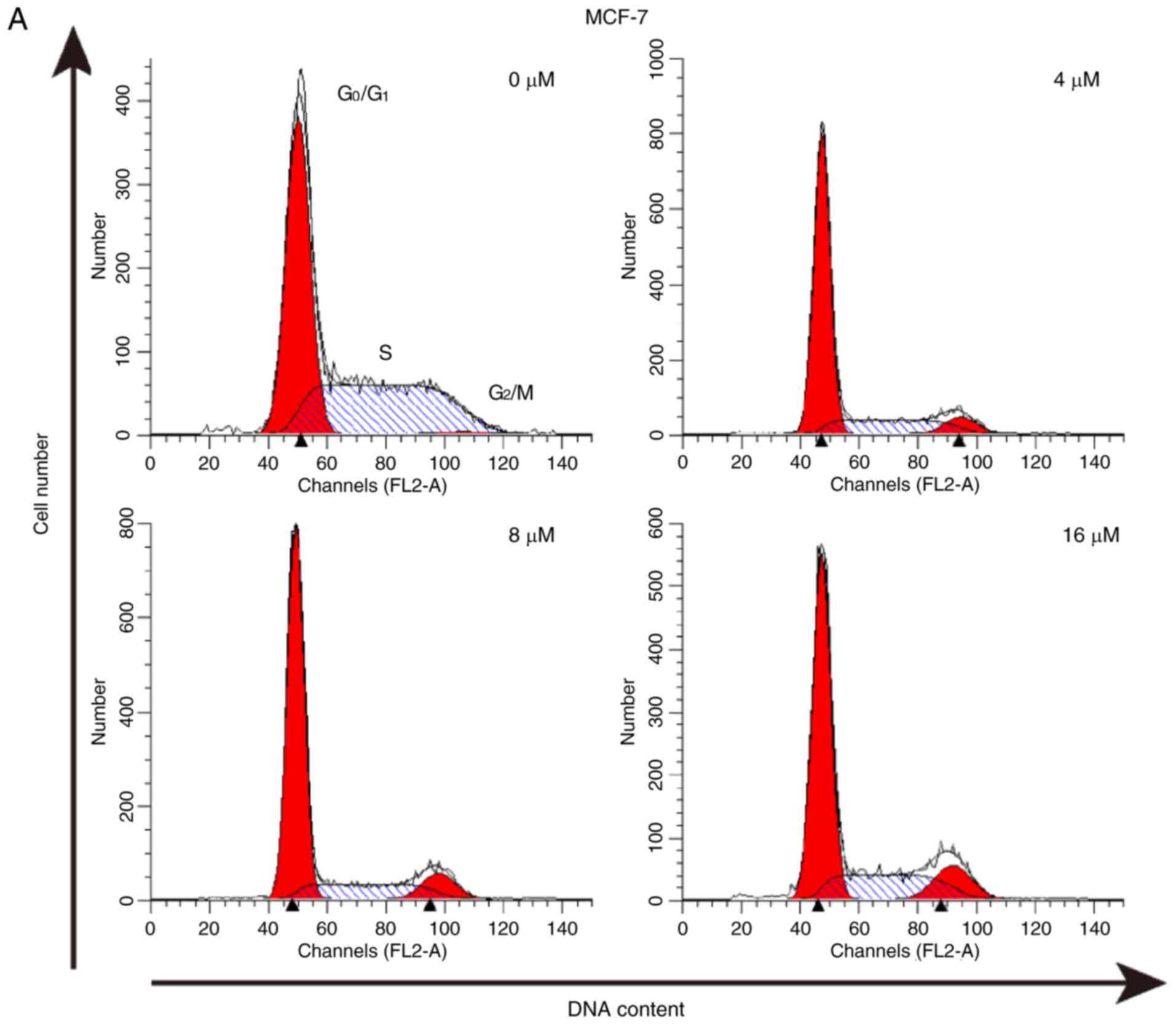
cell cycle arrest in breast cancer cells
The effect of As2S2 on the
cell cycle was assessed by evaluating the proportion of cells in
each phase compared with the control in MCF-7 and MDA-MB-231 cells
using PI staining and flow cytometry. The results indicated that
As2S2 mainly induced G2/M phase
arrest in MCF-7 cells and MDA-MB-231 cells (Fig. 6).
In MCF-7 cells, following exposure to
As2S2 at different concentrations (4, 8 and
16 µM) for 48 h, the proportion of cells in G2/M phase
significantly increased from 4.00±0.75 (0 µM) to 8.81±0.52
(P=0.0003), 9.69±0.06 (P<0.0001) and 12.05±0.31% (P<0.0001),
respectively. In MDA-MB-231 cells, As2S2
treatment at 4, 8 and 16 µM for 48 h increased the proportion of
cells in G2/M phase from 16.34±0.44 (0 µM) to 22.64±0.33
(P=0.0001), 26.11±0.30 (P<0.0001) and 43.43±1.11% (P<0.0001),
respectively, as well as increased the proportion of cells in S
phase from 20.40±0.18 (0 µM) to 23.25±0.51 (P=0.0242), 22.94±0.47
(P=0.0451) and 36.81±0.87% (P<0.0001), respectively.
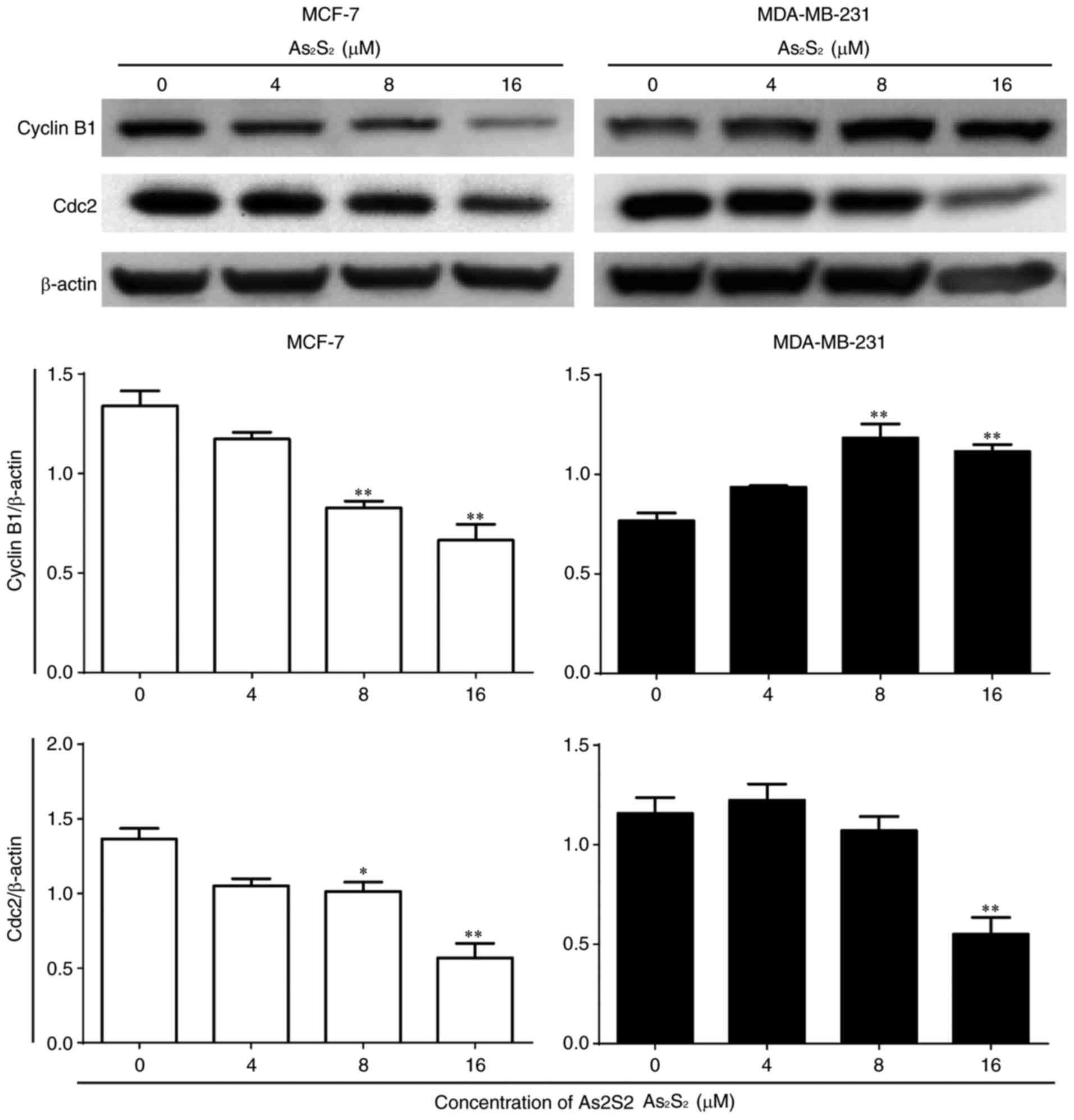
Furthermore, the expression of cell cycle-associated
proteins was determined by western blot analysis. As presented in
Fig. 7, compared with the control,
the expression of cyclin B1 significantly decreased following
As2S2 treatment at concentrations of 8
(P=0.0014) and 16 (P=0.0002) µM in MCF-7 cells, but increased with
As2S2 at concentrations of 8 (P=0.0007) and
16 (P=0.0022) µM in MDA-MB-231 cells. Following exposure to
As2S2 for 48 h, a statistically significant
decrease in the expression of Cdc2 occurred in MCF-7 cells treated
with 8 µM As2S2 (P=0.0348) and in MDA-MB-231
cells treated with 16 µM As2S2
(P=0.0028).
These results indicated that
As2S2 triggers G2/M phase arrest
in MCF-7 and MDA-MB-231 cells by regulating the expression of cell
cycle-associated proteins.
As2S2 induces
apoptosis in breast cancer cells
Apoptosis induced by As2S2 in
MCF-7 and MDA-MB-231 was validated using Hoechst 33342 staining and
a flow cytometric assay.
As presented in Figs.
2 and 3, the occurrence of
typical apoptotic characteristics, such as cell shrinkage,
chromatin condensation and nuclei fragmentation (39), was evident in MCF-7 and MDA-MB-231
cells following treatment with As2S2 (8 and
16 µM) for 48 h, whereas normal nuclei manifested with a round
shape and homogeneous staining.
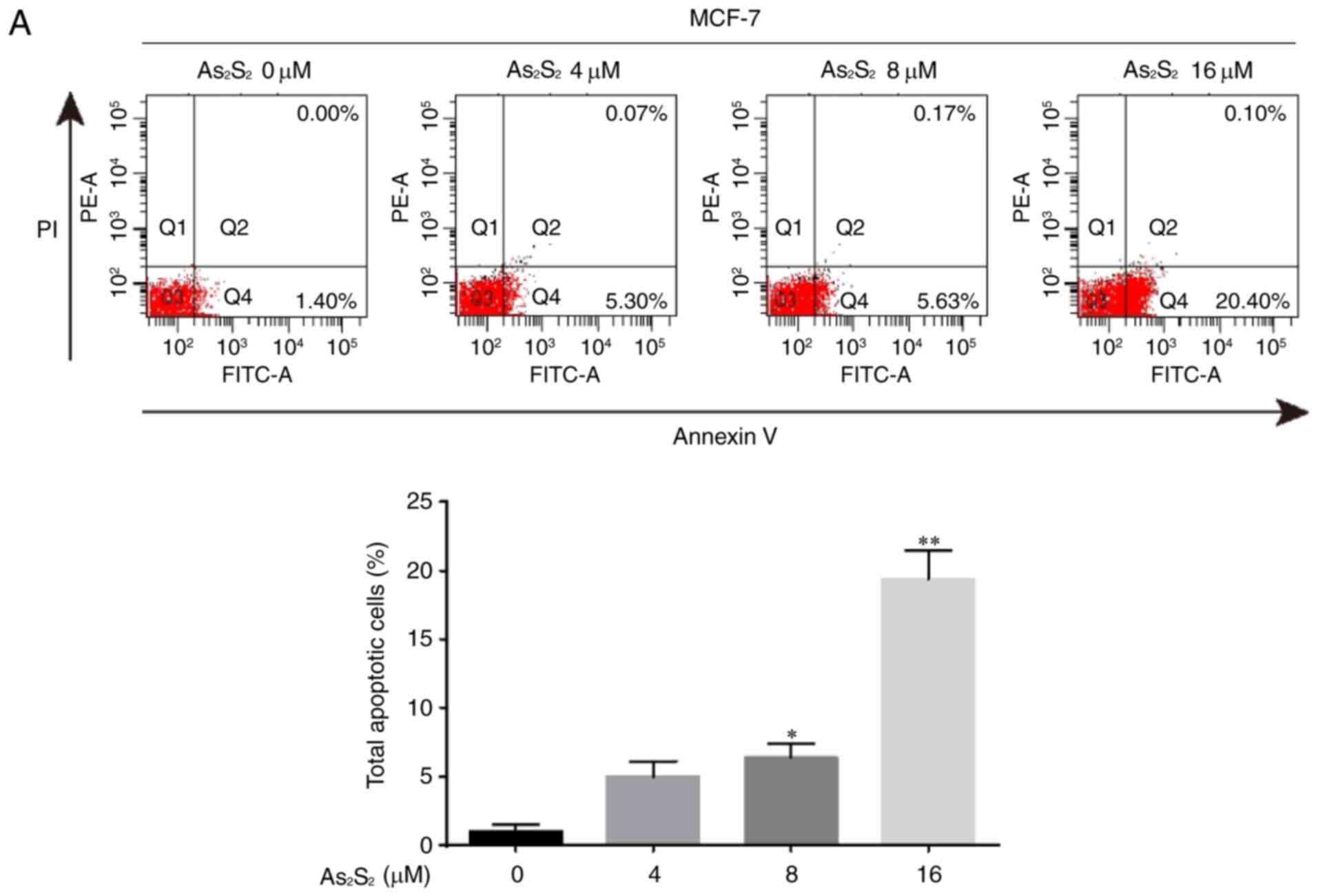
The induction of apoptosis was investigated further
using an Annexin V/PI double-staining assay followed by flow
cytometry, which was based on a probe of the total proportion of
apoptotic breast cancer cells (Annexin V-positive cells). As
presented in Fig. 8, the total
proportion of apoptotic MCF-7 cells significantly increased from
1.05±0.48 for the control (0 µM As2S2) to
6.35±1.05 (8 µM As2S2; P=0.0307) and
19.40±2.03% (16 µM As2S2; P<0.0001)
following exposure to As2S2 for 48 h. In
MDA-MB-231 cells (Fig. 7), the
proportion of apoptotic cells significantly increased from
3.73±0.32 for the control (0 µM As2S2) to
6.57±0.84 (8 µM As2S2; P=0.0345) and
15.30±1.40% (16 µM As2S2; P<0.0001)
following exposure to As2S2 for 48 h.
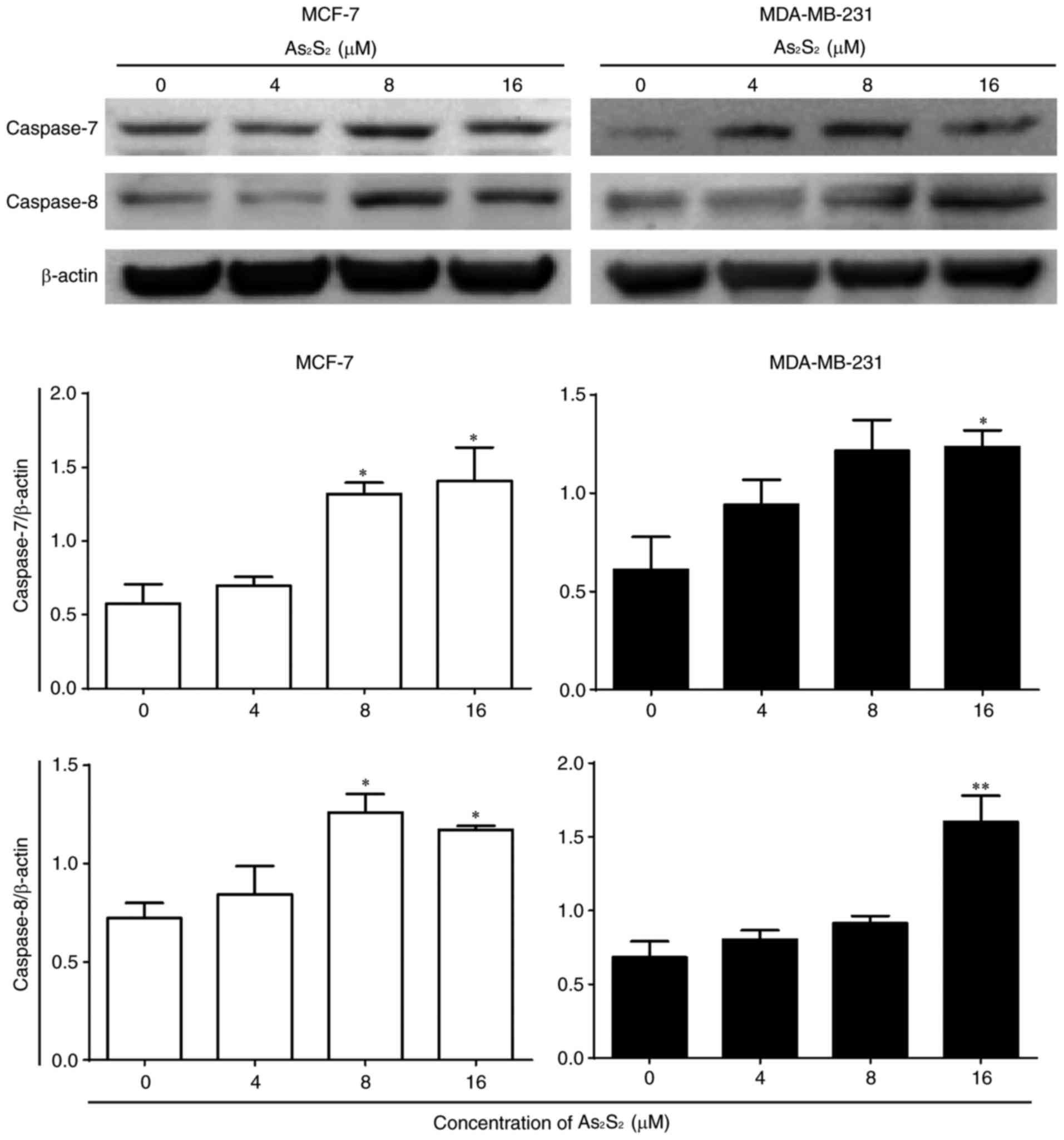
To further confirm the induction of apoptosis by
As2S2 in breast cancer cells, a western blot
analysis was performed to investigate the expression of
apoptosis-associated proteins. As presented in Fig. 9, the expression of pro-apoptotic
proteins, such as caspase-8 (apoptotic initiator) and −7 (apoptotic
executioner), was identified to be increased following treatment
with As2S2 in a dose-dependent manners in
MCF-7 and MDA-MB-231 cells. Compared with the control, the
expression of caspase-7 and −8 in MCF-7 cells was significantly
increased after 48 h of treatment with As2S2
at 8 (P=0.0234 for caspase-7; P=0.0158 for caspase-8) and 16
(P=0.0129 for caspase-7; P=0.0391 for caspase-8) µM. In MDA-MB-231
cells, the expression of caspase-7 and −8 was also significantly
increased after 48 h of treatment with As2S2
at 16 µM (P=0.0294 for caspase-7; P=0.0018 for caspase-8).
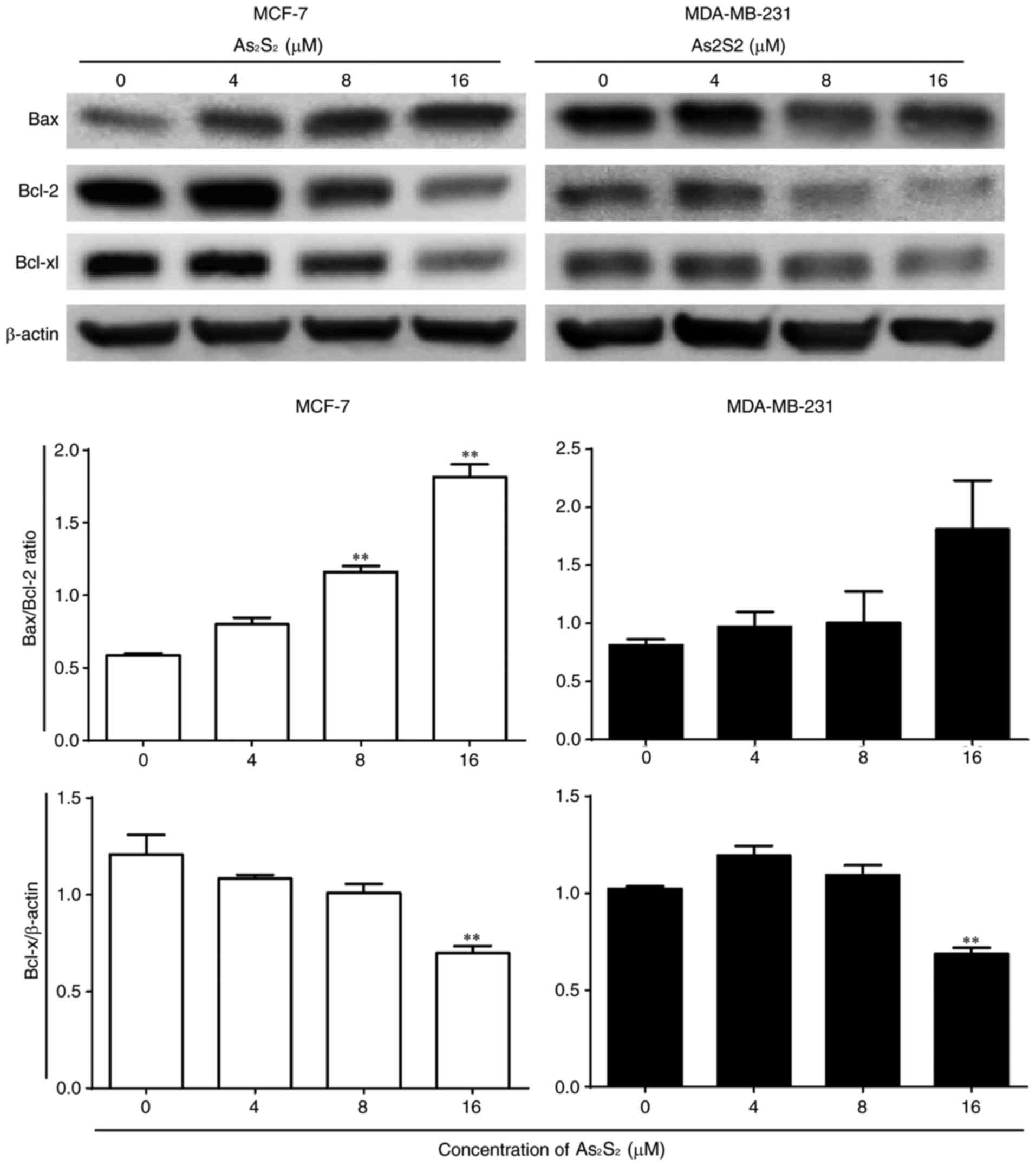
As presented in Fig.
10, the ratio of Bax expression to Bcl-2 expression was
increased by As2S2 in MCF-7 and MDA-MB-231
cells. Compared with the control, significant increases were
observed in MCF-7 cells following treatment with 8 (P=0.0003) and
16 (P<0.0001) µM As2S2 for 48 h. In
MDA-MB-231 cells, the ratio of Bax to Bcl-2 increased with
increasing doses, although the change was not statistically
significant. The anti-apoptotic protein Bcl-xl was inhibited by
As2S2 in MCF-7 and MDA-MB-231 cells. Compared
with the control, the expression of Bcl-xl was significantly
decreased by As2S2 at 16 µM in the two cell
lines (P=0.0014 in MCF-7 cells; P=0.0015 in MDA-MB-231 cells).
Taken together, these results indicate that
apoptosis was induced by As2S2 in MCF-7 and
MDA-MB-231 cells in a dose-dependent manner by regulating
apoptosis-associated proteins.
As2S2 induces
autophagy in breast cancer cells
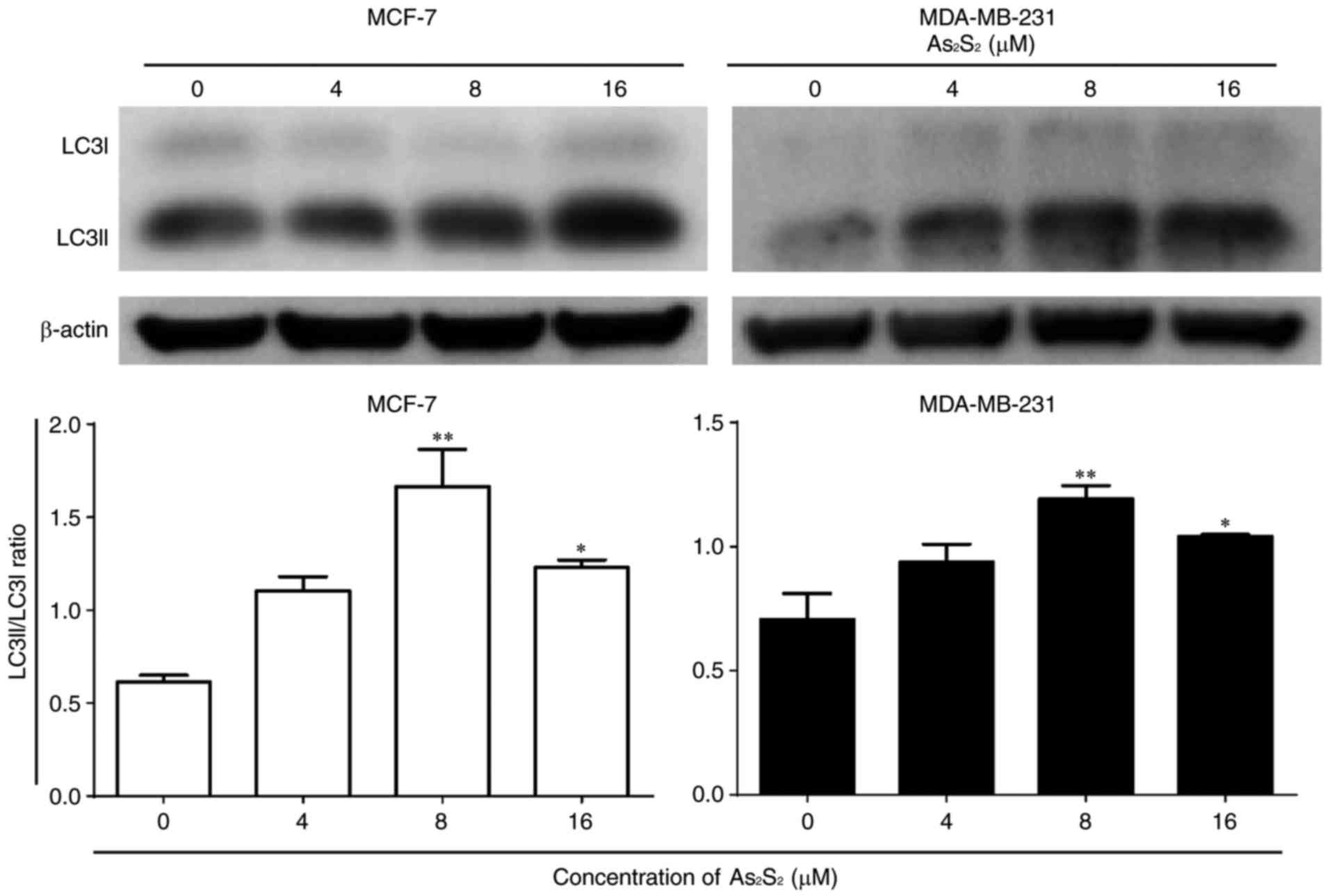
When autophagy is induced, LC3 is converted from
the cytoplasmic form LC3-I into the membrane-associated form
LC3-II. The ratio of LC3-II/LC3-I is widely considered as a primary
marker of autophagy activation (40,41).
As presented in Fig.
11, As2S2 induced an increase in the
LC3-II/LC3-I ratio in MCF-7 and MDA-MB-231 cells. Compared with the
control, significant increases were observed in MCF-7 cells
following treatment with As2S2 at 8
(P=0.0007) and 16 (P=0.0187) µM. Similarly, the ratio of
LC3-II/LC3-I was increased markedly in MDA-MB-231 cells following
treatment with As2S2 at 8 (P=0.0048) and 16
(P=0.0358) µM.
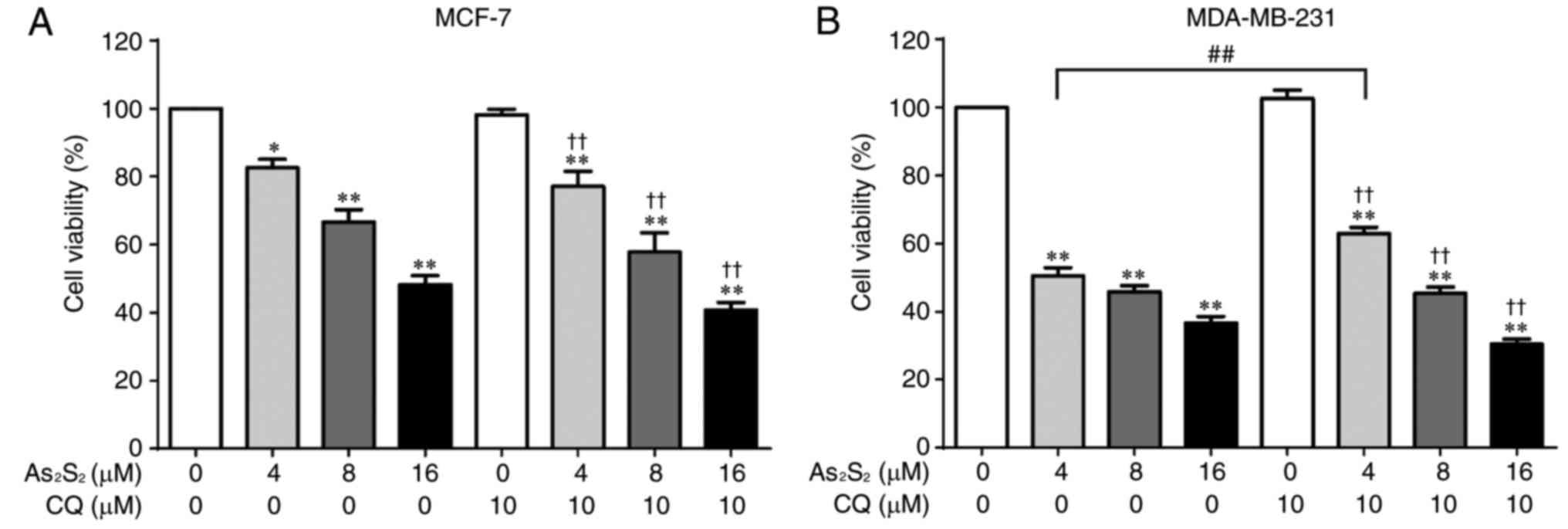
Autophagy has been identified to serve functions in
cytoprotective and cytotoxic processes. To examine the effect of
As2S2-induced autophagy on breast cancer cell
viability, MCF-7 and MDA-MB-231 cells were exposed to the autophagy
inhibitor CQ in the presence of different concentrations of
As2S2 (0, 4, 8 and 16 µM). As presented in
Fig. 12A, inhibition of autophagy
did not alter the inhibitory effect of As2S2
on MCF-7 cells. Furthermore, although pretreatment with CQ
significantly reversed the cell death induced by
As2S2 at 4 µM (P=0.0021) in MDA-MB-231 cells,
no additional effect was observed in the presence of
As2S2 treatment at 8 or 16 µM (Fig. 12B).
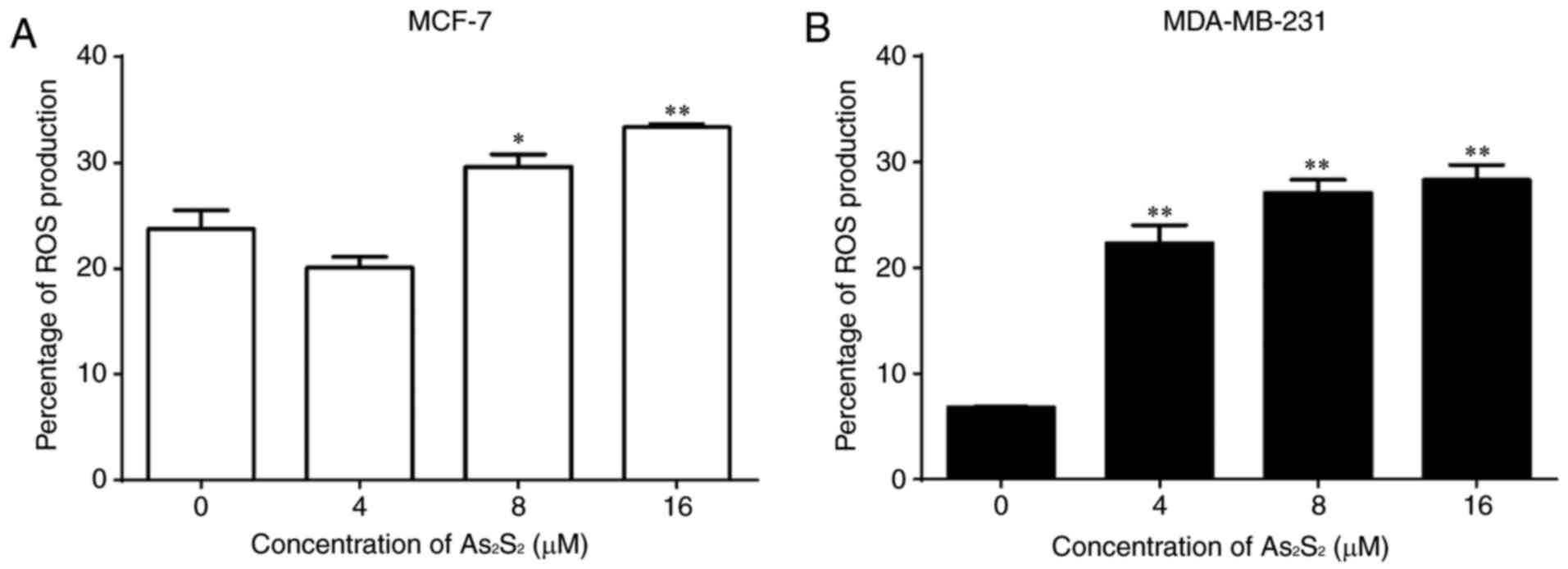
Effects of As2S2
on ROS production in breast cancer cells
The effects of As2S2 on ROS
production in breast cancer cells were assessed using an
ROS-sensitive probe, DCF-DA, and a flow cytometric assay.
As presented in Fig.
13, As2S2 induced the accumulation of ROS
in a dose-dependent manner in the two breast cancer cell lines. The
proportions of ROS production significantly increased from
23.73±1.78 in the control to 29.60±1.20 (P=0.0353) and 33.33±0.32%
(P=0.0022) by As2S2 at 8 and 16 µM in MCF-7
cells, respectively. In MDA-MB-231 cells,
As2S2 significantly increased the proportions
of ROS production from 6.80±0.06% in the control to 22.30±1.72
(P=0.0001), 27.10±1.20 (P<0.0001) and 28.30±1.43% (P<0.0001)
at As2S2 concentrations of 4, 8 and 16 µM,
respectively.
Discussion
Previous studies have identified the antitumor
effect of As2S2 against hematopoietic and
solid cancer cell lines (5–7,20,21);
however, few have investigated the effects of
As2S2 against breast cancer cell lines. In
the present study, the anticancer effect of
As2S2 on the proliferation and migration of
two distinctive subtypes of breast carcinoma cells (MCF-7 and
MDA-MB-231) and the molecular mechanisms underlying these effects
were investigated, particularly with regard to the activation of
PCD. To the best of our knowledge, the present study is the first
focusing on the two classical PCD pathways, i.e. apoptosis and
autophagy, induced by As2S2 in human breast
cancer cell lines.
Tumor metastasis, the most deadly aspect of cancer,
is a multistep aggressive process involving cell proliferation and
cell migration (42,43). In the present study, the antitumor
effects of As2S2 against the development of
breast cancer cells, including cell proliferation, survival and
migration, were investigated. Results obtained from a CCK-8 assay
indicated that As2S2 significantly inhibited
the cell viabilities in the two cell lines in a dose-dependent
manner. Consistent with these results, the data visualized and
obtained using the calcein-AM staining test showed the potent
induction of cell death by As2S2 in the two
cell lines. In addition, the results from wound healing assays
indicated a significant decrease in cell invasion by treatment with
As2S2 in a dose-dependent manner in the two
cell lines, which further suggested the tumor suppressive effects
of As2S2 by repressing the proliferative and
migratory abilities of breast carcinoma cells. MMPs are associated
with extracellular matrix degradation, which leads to cancer cell
invasion and metastasis. MMP-9 is a key factor that contributes to
the metastatic potential and cancer progression (44). The migration and invasion of cancer
cells are facilitated by MMP-9. Thus, downregulating MMP-9 may be a
possible strategy for attenuating the progression of cancer cells
(45). The results of the present
study indicated that As2S2 decreased the
protein expression of MMP-9 in the two breast cancer cell lines in
a dose-dependent manner, which may account for the decrease in
migration in the two cell lines following
As2S2 treatment.
Cell cycle dysfunction is a common feature of
proliferating and metastatic breast tumor cells (46); targeting cell cycle regulation is
therefore an important therapeutic strategy. In the present study,
it was demonstrated that As2S2 treatment
exerted pronounced cell cycle arrest at G2/M phase in
the two breast cancer cell lines in a dose-dependent manner. In
addition, As2S2 triggered
G0/G1-phase arrest in MCF-7 cells, but
S-phase arrest in MDA-MB-231 cells. The exertion of arrest at
different phases between the two cell lines is likely to be due to
characteristics specific to these cells as distinct subtypes of
breast carcinoma. Considering the observed cell cycle arrest, the
results of the present study suggest that
As2S2 treatment regulates the expression of
cell cycle-associated proteins involved in the corresponding phases
of the cell cycle in each of the breast cancer cell lines. For
example, as the main regulatory proteins in G2/M phase
(47,48), the expression of cyclin B1 and Cdc2
was regulated by As2S2 treatment in the two
cell lines, which is consistent with the blockage of the cell cycle
at G2/M by As2S2 treatment.
Intriguingly, the protein expression of cyclin B1 was regulated by
As2S2 in an opposite manner between MCF-7 and
MDA-MB-231 cells, possibly due to biological variations and
distinctions between these two cell lines. In addition to
regulating G2/M phase, Cdc2 has also been described as a
key regulator associated with G0/G1 and S
phases (38,39), which accounts for the cell cycle
arrest observed at G0/G1 phase in MCF-7 cells
and at the S phase in MDA-MB-231 cells.
Apoptosis and autophagy are two well-known PCD
mechanisms that serve essential functions in maintaining organismal
and cellular homeostasis (49).
Apoptosis, regarded as a major mechanism of chemotherapy-induced
cell death (50), is characterized
by typical morphological changes, such as nuclear condensation and
fragmentation. In the present study, apoptosis was significantly
induced by As2S2 treatment in the two breast
cancer cell lines, as demonstrated using the Annexin V staining
assay and visualized using the Hoechst 33342 staining assay. In
addition, the induction of apoptosis in the two breast cancer cell
lines was further confirmed by the activation of caspase-8 and −7,
as well as the regulation of proteins in the Bcl-2 family, which
resulted in an increase in the Bax/Bcl-2 ratio along with the
decreased expression of Bcl-xl. An essential step in triggering
apoptosis is the activation of caspases, a family of cysteine
proteases that are ubiquitously expressed as death proteases
(23,51). The caspase family has traditionally
been divided into initiator and effector caspases. Activated
initiator caspases, such as caspase-8, −9 and −10, subsequently
initiate a caspase cascade of downstream effector caspases
(52). Effector caspases, such as
caspase-3, −6 and −7, are understood to execute apoptosis following
being triggered by initiator caspases (52). Two principal signaling pathways
exist to induce cell apoptosis: The extrinsic (cell death receptor)
pathway and the intrinsic (mitochondrial) pathway (53). Of note, caspase-8 has been regarded
as a core initiating component of the extrinsic pathway,
subsequently activating the execution phase of apoptosis (e.g.
activating effector caspase-7) (54). Bcl-2 family proteins are categorized
into subgroups on the basis of their pro- or anti-apoptotic
actions: Pro-apoptotic proteins, such as Bax, and anti-apoptotic
proteins, such as Bcl-2 and Bcl-xl. These Bcl-2 family proteins
serve an important function in initiating the intrinsic apoptotic
pathway (55,56). In the present study, the expression
of caspase-8 and the Bax/Bcl-2 ratio were significantly increased,
whereas the expression of Bcl-xl was markedly decreased following
As2S2 treatment, suggesting that the
extrinsic and intrinsic pathways were involved in
As2S2-induced apoptosis in the two breast
cancer cell lines.
Apoptosis and autophagy normally occur in the same
cell, primarily in a mutually interactive manner under the same
cellular conditions (24,49). Autophagy, a catabolic process for
the degradation of unnecessary and dysfunctional cytosolic
components and organelles, has generally been regarded as the type
II (non-apoptotic) PCD and is deemed an important mechanism
involved in the tumor control process (57,58).
LC3 is a key protein involved in initiating autophagy, wherein
LC3-I is lipidated and converted into LC3-II. The ratio of
LC3-II/LC3-I is widely used as a primary marker of autophagy
activation (41). The results of
the present study indicated that As2S2
treatment induced an increase in the ratio of LC3-II/LC3-I in the
two breast cancer cell lines, suggesting that autophagy occurs in
breast cancer cells following exposure to
As2S2. Bcl-2 is an intermediary protein
shared by apoptosis and autophagy, serving an anti-apoptotic and
anti-autophagy function in the two processes (29,59).
The results of the present study indicated that
As2S2 treatment significantly decreased the
protein expression of the anti-apoptotic Bcl-2, which may in turn
potentiate the induction of autophagy. Intriguingly, autophagy is
commonly regarded as a double-edged sword (60,61)
and may positively or negatively influence cancer cell growth
(62). To improve understanding of
the function of autophagy in the present study, CQ, a
pharmacological inhibitor of autophagy, was used to clarify whether
or not As2S2-induced cell death was
associated with the induction of autophagy. The results indicated
that CQ significantly reversed the inhibitory effect of
As2S2 (4 µM) on the cell viability in
MDA-MB-231 cells, indicating that autophagy induced by
As2S2 at a relatively low concentration was
indeed involved in the death of MDA-MB-231 cells. In contrast, CQ
had little effect on the death of MCF-7 and MDA-MB-231 cells in the
presence of relatively high concentrations (8 and 16 µM) of
As2S2, suggesting that the induction of cell
death is primarily through apoptosis and cell cycle arrest, as
indicated by the results of the present study, rather than
autophagy.
ROS help to regulate a series of biological
processes, including PCD (63). It
has been identified that the accumulation of ROS is associated with
cell apoptosis, cell cycle arrest and autophagy induced by
anticancer agents, which consequently leads to negative effects on
the cell survival, proliferation and metastasis (44,64–67).
In the present study, results from flow cytometric analyses
indicated that the ROS level in MCF-7 and MDA-MB-231 cells
significantly increased following As2S2
treatment in a dose-dependent manner, which may potentiate the
induction of apoptosis, cell cycle arrest and autophagy in the two
cell lines, triggered by As2S2 treatment.
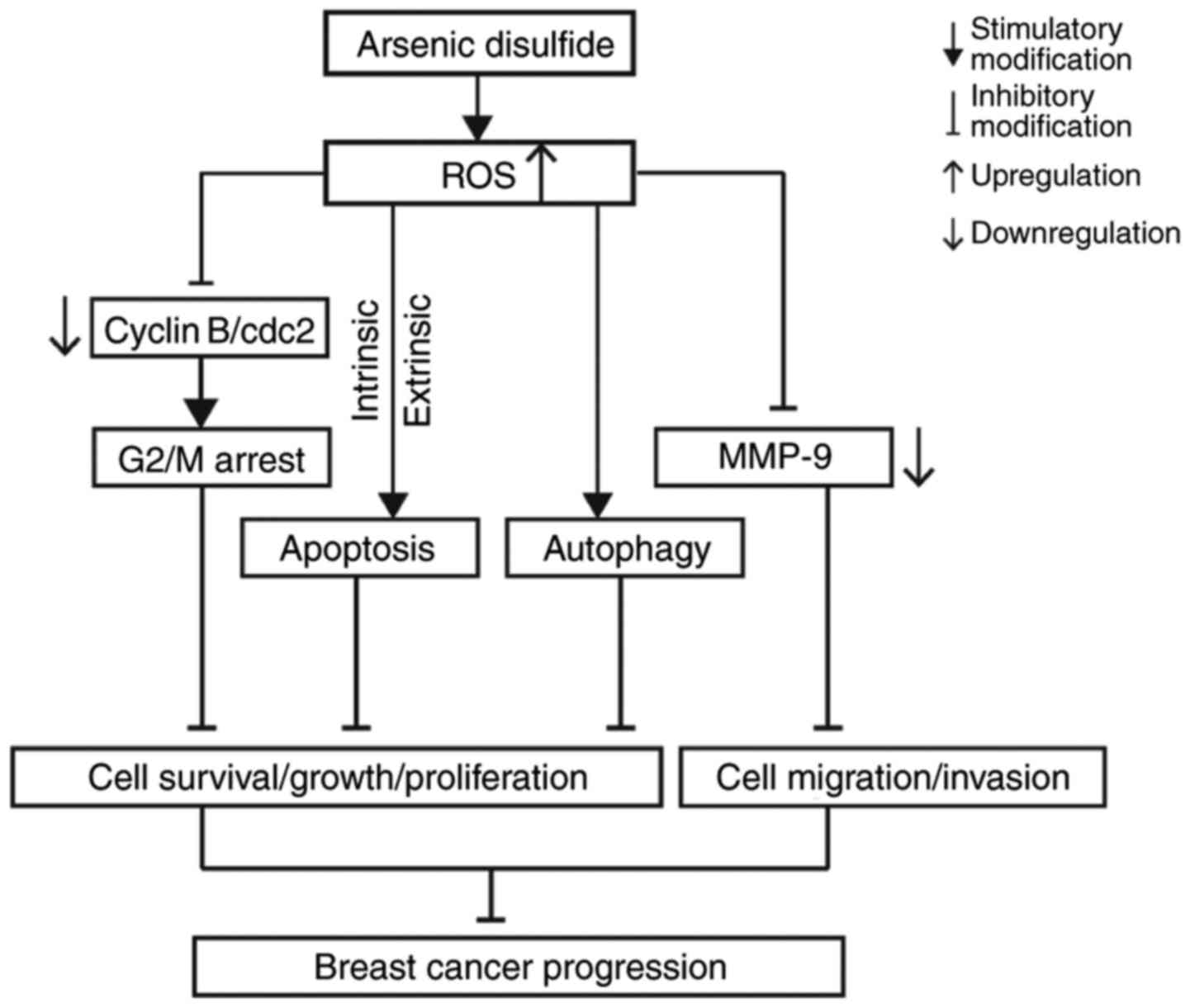
Accordingly, the oxidative stress generated by ROS production and
the activated PCD pathway markedly inhibited cell viability,
decreased the live cell number, suppressed cell viability,
attenuated cell migration and consequently decreased cell
progression in MCF-7 and MDA-MB-231 cells. The potential molecular
mechanisms underlying the inhibitory effect of
As2S2 on breast carcinoma progression are
presented in Fig. 14.
In conclusion, the results of the present study
identified the antitumor effects of As2S2
against breast carcinoma in vitro as inhibition of cell
viability, decreased cell survival and attenuated invasion of MCF-7
and MDA-MB-231 cells. These effects were associated with inhibition
of cell cycle progression, the induction of apoptosis and
autophagy, a decrease in MMP-9 expression and an increase in ROS
accumulation. In future studies, we intend to investigate the
therapeutic potential of As2S2 in vivo
in the treatment of breast cancer in animal models.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by the
China Scholarship Council (grant no. 201709110064).
Availability of data and materials
The analyzed datasets generated during the study
are available from the corresponding author on reasonable request,
while preserving the necessary confidentiality and anonymity.
Authors' contributions
TH conceived and designed the study and critically
revised the manuscript. YZ designed and performed the experiments,
analyzed the data and was a major contributor in writing the
manuscript. KO, KS, BY, ST and NT gave advice on the experiments
and contributed with reagents and technical assistance. TH and NT
supervised the study. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATO
|
arsenic trioxide
|
|
APL
|
acute promyelocytic leukemia
|
|
PCD
|
programmed cell death
|
|
CCK-8
|
Cell Counting Kit-8
|
|
PI
|
propidium iodide
|
|
CQ
|
chloroquine diphosphate
|
|
MMP-9
|
matrix metalloproteinase-9
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
Bcl-xl
|
B-cell lymphoma extra-large
|
|
Bax
|
Bcl-2-associated X protein
|
|
Cdc2
|
cell division cycle protein 2
|
|
LC3
|
microtubule-associated protein
1A/1B-light chain 3
|
|
DCF-DA
|
2′,7′-dichlorofluorescin
diacetate
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Wang H, Liu Z, Gou Y, Qin Y, Xu Y, Liu J
and Wu JZ: Apoptosis and necrosis induced by novel realgar quantum
dots in human endometrial cancer cells via endoplasmic reticulum
stress signaling pathway. Int J Nanomedicine. 10:5505–5512. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang L, Zhou GB, Liu P, Song JH, Liang Y,
Yan XJ, Xu F, Wang BS, Mao JH, Shen ZX, et al: Dissection of
mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as
an effective treatment for promyelocytic leukemia. Proc Natl Acad
Sci USA. 105:4826–4831. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang QY, Mao JH, Liu P, Huang QH, Lu J,
Xie YY, Weng L, Zhang Y, Chen Q, Chen SJ, et al: A systems biology
understanding of the synergistic effects of arsenic sulfide and
Imatinib in BCR/ABL-associated leukemia. Proc Natl Acad Sci USA.
106:3378–3383. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu Y, He P, Cheng X and Zhang M:
Long-term outcome of 31 cases of refractory acute promyelocytic
leukemia treated with compound realgar natural indigo tablets
administered alternately with chemotherapy. Oncol Lett.
10:1184–1190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang L, Kim S, Ding W, Tong Y, Zhang X,
Pan M and Chen S: Arsenic sulfide inhibits cell migration and
invasion of gastric cancer in vitro and in vivo. Drug Des Devel
Ther. 9:5579–5590. 2015.PubMed/NCBI
|
|
6
|
Wang G, Zhang T, Sun W, Wang H, Yin F,
Wang Z, Zuo D, Sun M, Zhou Z, Lin B, et al: Arsenic sulfide induces
apoptosis and autophagy through the activation of ROS/JNK and
suppression of Akt/mTOR signaling pathways in osteosarcoma. Free
Radic Biol Med. 106:24–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Song P, Chen P, Wang D, Wu Z, Gao Q, Wang
A, Zhu R, Wang Y, Wang X, Zhao L, et al: Realgar transforming
solution displays anticancer potential against human hepatocellular
carcinoma HepG2 cells by inducing ROS. Int J Oncol. 50:660–670.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qin YU, Wang H, Liu ZY, Liu J and Wu JZ:
Realgar quantum dots induce apoptosis and necrosis in HepG2 cells
through endoplasmic reticulum stress. Biomed Rep. 3:657–662. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu JZ and Ho PC: Evaluation of the in
vitro activity and in vivo bioavailability of realgar nanoparticles
prepared by cryo-grinding. Eur J Pharm Sci. 29:35–44. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ding W, Zhang L, Kim S, Tian W, Tong Y,
Liu J, Ma Y and Chen S: Arsenic sulfide as a potential anti-cancer
drug. Mol Med Rep. 11:968–974. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tse WP, Cheng CH, Che CT and Lin ZX:
Arsenic trioxide, arsenic pentoxide, and arsenic iodide inhibit
human keratinocyte proliferation through the induction of
apoptosis. J Pharmacol Exp Ther. 326:388–394. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao Y, Onda K, Yuan B, Tanaka S, Kiyomi
A, Sugiyama K, Sugiura M, Takagi N and Hirano T: Arsenic
disulfide-induced apoptosis and its potential mechanism in two- and
three-dimensionally cultured human breast cancer MCF-7 cells. Int J
Oncol. 52:1959–1971. 2018.PubMed/NCBI
|
|
13
|
Zhao Y, Yuan B, Onda K, Sugiyama K, Tanaka
S, Takagi N and Hirano T: Anticancer efficacies of arsenic
disulfide through apoptosis induction, cell cycle arrest, and
pro-survival signal inhibition in human breast cancer cells. Am J
Cancer Res. 8:366–386. 2018.PubMed/NCBI
|
|
14
|
Uematsu N, Zhao Y, Kiyomi A, Yuan BO, Onda
K, Tanaka S, Sugiyama K, Sugiura M, Takagi N, Hayakawa A and Hirano
T: Chemo-sensitivity of two-dimensional monolayer and
three-dimensional spheroid of breast cancer MCF-7 cells to
daunorubicin, docetaxel, and arsenic disulfide. Anticancer Res.
38:2101–2108. 2018.PubMed/NCBI
|
|
15
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: Defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Michailidou K, Hall P, Gonzalez-Neira A,
Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J,
Bojesen SE, Bolla MK, et al: Large-scale genotyping identifies 41
new loci associated with breast cancer risk. Nat Genet. 45:353–361,
361e1-2. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Recht A, Come SE, Henderson IC, Gelman RS,
Silver B, Hayes DF, Shulman LN and Harris JR: The sequencing of
chemotherapy and radiation therapy after conservative surgery for
early-stage breast cancer. N Engl J Med. 334:1356–1361. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cicconi L and Lo-Coco F: Current
management of newly diagnosed acute promyelocytic leukemia. Ann
Oncol. 27:1474–1481. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chow SK, Chan JY and Fung KP: Suppression
of cell proliferation and regulation of estrogen receptor alpha
signaling pathway by arsenic trioxide on human breast cancer MCF-7
cells. J Endocrinol. 182:325–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang X, Zhang X, Xu Z, Wang Z, Yue X and
Li H: Reversal effect of arsenic sensitivity in human leukemia cell
line K562 and K562/ADM using realgar transforming solution. Biol
Pharm Bull. 36:641–648. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie QJ, Cao XL, Bai L, Wu ZR, Ma YP and Li
HY: Anti-tumor effects and apoptosis induction by Realgar
bioleaching solution in Sarcoma-180 cells in vitro and transplanted
tumors in mice in vivo. Asian Pac J Cancer Prev. 15:2883–2888.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Engelberg-Kulka H, Amitai S, Kolodkin-Gal
I and Hazan R: Bacterial programmed cell death and multicellular
behavior in bacteria. PLoS Genet. 2:e1352006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fuchs Y and Steller H: Programmed cell
death in animal development and disease. Cell. 147:742–758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Booth LA, Tavallai S, Hamed HA,
Cruickshanks N and Dent P: The role of cell signalling in the
crosstalk between autophagy and apoptosis. Cell Signal. 26:549–555.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zimmermann KC, Bonzon C and Green DR: The
machinery of programmed cell death. Pharmacol Ther. 92:57–70. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levine B and Deretic V: Unveiling the
roles of autophagy in innate and adaptive immunity. Nat Rev
Immunol. 7:767–777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zambrano J and Yeh ES: Autophagy and
apoptotic crosstalk: Mechanism of therapeutic resistance in
HER2-positive breast cancer. Breast Cancer. 10:13–23.
2016.PubMed/NCBI
|
|
30
|
Shi D, Liu Y, Xi R, Zou W, Wu L, Zhang Z,
Liu Z, Qu C, Xu B and Wang X: Caveolin-1 contributes to realgar
nanoparticle therapy in human chronic myelogenous leukemia K562
cells. Int J Nanomedicine. 11:5823–5835. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pastorek M, Gronesova P, Cholujova D,
Hunakova L, Bujnakova Z, Balaz P, Duraj J, Lee TC and Sedlak J:
Realgar (As4S4) nanoparticles and arsenic trioxide (As2O3) induced
autophagy and apoptosis in human melanoma cells in vitro.
Neoplasma. 61:700–709. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shi X, Zhang Y, Zheng J and Pan J:
Reactive oxygen species in cancer stem cells. Antioxid Redox
Signal. 16:1215–1228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gupta SC, Hevia D, Patchva S, Park B, Koh
W and Aggarwal BB: Upsides and downsides of reactive oxygen species
for cancer: The roles of reactive oxygen species in tumorigenesis,
prevention, and therapy. Antioxid Redox Signal. 16:1295–1322. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jing Y, Dai J, Chalmers-Redman RM, Tatton
WG and Waxman S: Arsenic trioxide selectively induces acute
promyelocytic leukemia cell apoptosis via a hydrogen
peroxide-dependent pathway. Blood. 94:2102–2111. 1999.PubMed/NCBI
|
|
36
|
Chen YC, Lin-Shiau SY and Lin JK:
Involvement of reactive oxygen species and caspase 3 activation in
arsenite-induced apoptosis. J Cell Physiol. 177:324–333. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maeda H, Hori S, Nishitoh H, Ichijo H,
Ogawa O, Kakehi Y and Kakizuka A: Tumor growth inhibition by
arsenic trioxide (As2O3) in the orthotopic
metastasis model of androgen-independent prostate cancer. Cancer
Res. 61:5432–5440. 2001.PubMed/NCBI
|
|
38
|
Jayakumar AR, Bak LK, Rama Rao KV,
Waagepetersen HS, Schousboe A and Norenberg MD: Neuronal cell death
induced by mechanical percussion trauma in cultured neurons is not
preceded by alterations in glucose, lactate and glutamine
metabolism. Neurochem Res. 41:307–315. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Y, Chang Y, Ye N, Dai D, Chen Y, Zhang
N, Sun G and Sun Y: Advanced glycation end products inhibit the
proliferation of human umbilical vein endothelial cells by
inhibiting cathepsin D. Int J Mol Sci. 18:E4362017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aparicio IM, Martin Muñoz P, Salido GM,
Peña FJ and Tapia JA: The autophagy-related protein LC3 is
processed in stallion spermatozoa during short- and long-term
storage and the related stressful conditions. Animal. 10:1182–1191.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Singh BN, Singh HB, Singh A, Naqvi AH and
Singh BR: Dietary phytochemicals alter epigenetic events and
signaling pathways for inhibition of metastasis cascade:
Phytoblockers of metastasis cascade. Cancer Metastasis Rev.
33:41–85. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou J, Zhu YF, Chen XY, Han B, Li F, Chen
JY, Peng XL, Luo LP, Chen W and Yu XP: Black rice-derived
anthocyanins inhibit HER-2-positive breast cancer
epithelial-mesenchymal transition-mediated metastasis in vitro by
suppressing FAK signaling. Int J Mol Med. 40:1649–1656.
2017.PubMed/NCBI
|
|
44
|
Si L, Yan X, Hao W, Ma X, Ren H, Ren B, Li
D, Dong Z and Zheng Q: Licochalcone D induces apoptosis and
inhibits migration and invasion in human melanoma A375 cells. Oncol
Rep. 39:2160–2170. 2018.PubMed/NCBI
|
|
45
|
Jacob A and Prekeris R: The regulation of
MMP targeting to invadopodia during cancer metastasis. Front Cell
Dev Biol. 3:42015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Diaz-Moralli S, Tarrado-Castellarnau M,
Miranda A and Cascante M: Targeting cell cycle regulation in cancer
therapy. Pharmacol Ther. 138:255–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Stewart ZA, Westfall MD and Pietenpol JA:
Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol
Sci. 24:139–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Marconett CN, Morgenstern TJ, San Roman
AK, Sundar SN, Singhal AK and Firestone GL: BZ L101, a
phytochemical extract from the Scutellaria barbata plant, disrupts
proliferation of human breast and prostate cancer cells through
distinct mechanisms dependent on the cancer cell phenotype. Cancer
Biol Ther. 10:397–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ricci MS and Zong WX: Chemotherapeutic
approaches for targeting cell death pathways. Oncologist.
11:342–357. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Earnshaw WC, Martins LM and Kaufmann SH:
Mammalian caspases: Structure, activation, substrates, and
functions during apoptosis. Annu Rev Biochem. 68:383–424. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kadam CY and Abhang SA: Apoptosis markers
in breast cancer therapy. Adv Clin Chem. 74:143–193. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Suen DF, Norris KL and Youle RJ:
Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dickens LS, Boyd RS, Jukes-Jones R, Hughes
MA, Robinson GL, Fairall L, Schwabe JW, Cain K and Macfarlane M: A
death effector domain chain DISC model reveals a crucial role for
caspase-8 chain assembly in mediating apoptotic cell death. Mol
Cell. 47:291–305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pettersson F, Dalgleish AG, Bissonnette RP
and Colston KW: Retinoids cause apoptosis in pancreatic cancer
cells via activation of RAR-gamma and altered expression of
Bcl-2/Bax. Br J Cancer. 87:555–561. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liu F, Gao S, Yang Y, Zhao X, Fan Y, Ma W,
Yang D, Yang A and Yu Y: Curcumin induced autophagy anticancer
effects on human lung adenocarcinoma cell line A549. Oncol Lett.
14:2775–2782. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim KY, Park KI, Kim SH, Yu SN, Park SG,
Kim YW, Seo YK, Ma JY and Ahn SC: Inhibition of autophagy promotes
salinomycin-induced apoptosis via reactive oxygen species-mediated
PI3K/AKT/mTOR and ERK/p38 MAPK-dependent signaling in human
prostate cancer cells. Int J Mol Sci. 18:E10882017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tang F, Wang B, Li N, Wu Y, Jia J, Suo T,
Chen Q, Liu YJ and Tang J: RN F185, a novel mitochondrial ubiquitin
E3 ligase, regulates autophagy through interaction with BNIP1. PLoS
One. 6:e243672011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Li Y, Cui Y, Wang W, Ma M, Li M and Chen
S: Effect of the serum inhibited gene (Si1) on autophagy and
apoptosis in MCF-7 breast cancer cells. Cell Physiol Biochem.
41:2268–2278. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zeng Y, Li S, Wu J, Chen W, Sun H, Peng W,
Yu X and Yang X: Autophagy inhibitors promoted aristolochic acid I
induced renal tubular epithelial cell apoptosis via mitochondrial
pathway but alleviated nonapoptotic cell death in mouse acute
aritolochic acid nephropathy model. Apoptosis. 19:1215–1224. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Subramani R, Gonzalez E, Arumugam A, Nandy
S, Gonzalez V, Medel J, Camacho F, Ortega A, Bonkoungou S, Narayan
M, et al: Nimbolide inhibits pancreatic cancer growth and
metastasis through ROS-mediated apoptosis and inhibition of
epithelial-to-mesenchymal transition. Sci Rep. 6:198192016.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang L, Li P, Hu W, Xia Y, Hu C, Liu L and
Jiang X: CD44+CD24+ subset of PANC-1 cells
exhibits radiation resistance via decreased levels of reactive
oxygen species. Oncol Lett. 14:1341–1346. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Banerjee A, Banerjee V, Czinn S and
Blanchard T: Increased reactive oxygen species levels cause ER
stress and cytotoxicity in andrographolide treated colon cancer
cells. Oncotarget. 8:26142–26153. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li B, Zhao S, Geng R, Huo Z and Zhang H:
The sineoculis homeobox Homolog 1 (SIX1) gene regulates paclitaxel
resistance by affecting reactive oxygen species and autophagy in
human hepatocellular carcinoma cell line HepG2. Med Sci Monit.
24:2271–2279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Apel K and Hirt H: Reactive oxygen
species: Metabolism, oxidative stress, and signal transduction.
Annu Rev Plant Biol. 55:373–399. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li L, Cao W, Zheng W, Fan C and Chen T:
Ruthenium complexes containing 2,6-bis(benzimidazolyl)pyridine
derivatives induce cancer cell apoptosis by triggering DNA
damage-mediated p53 phosphorylation. Dalton Trans. 41:12766–12772.
2012. View Article : Google Scholar : PubMed/NCBI
|