Introduction
Cancer is a disease that is characterized by various
stages, including initiation, promotion and progression. It is
known that cancer cells are characterized by an increased rate of
reactive oxygen species (ROS) production, which enhances the tumor
metabolic adaptation, proliferation and survival. Excess ROS cause
oncogenic mutations through direct reaction with DNA, thus ROS are
believed to be among the initiating insults that drive
carcinogenesis (1). At the initial
stage of breast cancer, increased ROS levels may promote cell
susceptibility to DNA damage induced by environmental carcinogens,
contributing to chromosome instability and cellular carcinogenesis
(2). Thus, strategies and drugs
that are designed to decrease ROS levels would be effective in
preventing the initiation of breast cancer.
Carvedilol (CAR), a non-selective adrenergic
blocker, was primarily designed to treat cardiovascular disease by
inhibiting adrenergic receptors (3). Results from a pre-clinical study
indicated that CAR could inhibit the proliferation of cancer cells
and induce cell apoptosis in vitro (4). Our previous study identified that CAR
significantly decreased the migratory and invasive capacities of
breast cancer cells (5).
Furthermore, a population-based cohort study indicated that
long-term treatment with CAR was associated with a decreased risk
of cancer, indicating that CAR may be a potential agent in the
prevention of certain types of cancer (6). Results from a pre-clinical study
suggested that CAR may prevent malignant transformation in in
vitro and in vivo models of skin carcinogenesis
(7). These results imply that CAR
may be a novel chemopreventive agent.
MCF-10A cells are non-cancerous cells, which are
transformable and form colonies in soft agar following exposure to
tumor promoters (8).
Benzo(a)pyrene (BaP) is able to transform MCF-10A cells into
initiated breast cancer cells. This process is mediated via DNA
adduct formation and ROS serve a crucial function in the initiation
of carcinogenesis. Certain agents prevent tumorigenesis by their
antioxidant activity (9). With the
exception of the β-blocker effect, CAR exhibits antioxidant
activity, but currently there is no knowledge of whether CAR is
able to prevent breast cancer via oxidative resistance.
In the present study, non-cancerous MCF-10A cells
were used as a model to investigate the chemopreventive effect of
CAR on BaP-induced cellular carcinogenesis. The signaling pathways
that were associated with CAR during the prevention of breast
carcinogenesis were further investigated.
Materials and methods
Cells and reagents
Normal breast epithelial MCF-10A cells were
purchased from the American Type Culture Collection (Manassas, VA,
USA). BaP and CAR were purchased from Sigma; Merck KGaA (Darmstadt,
Germany). These agents were prepared as 50 µM stock solutions and
were stored at 4°C in double-distilled water. Phenol red-free
Dulbecco's modified Eagle's medium (DMEM)/Ham's F12, horse serum,
penicillin/streptomycin, antibiotic/antimycotic, epidermal growth
factor, human insulin (Novolin R), trypsin-EDTA (10X), Hanks'
balanced salt solution and PBS were purchased from Invitrogen;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Cholera
enterotoxin was obtained from Enzo Life Sciences, Inc.
(Farmingdale, NY, USA). AKT inhibitor VIII and AKT phosphorylation
activator SC79 were purchased from MedChemExpress (Monmouth
Junction, NJ, USA). The human 8-hydroxy-2-deoxyguanosine (8-OH-dG)
ELISA kit was purchased from Cusabio Technology LLC (Houston, TX,
USA). All other chemicals were purchased from Sigma-Aldrich; Merck
KGaA.
Cell culture
MCF10A cells were maintained in complete medium (1:1
mixture of DMEM and Ham's F12), supplemented with mitogenic
additives including 5% horse serum, 10 µM insulin, 0.5 ng/ml
hydrocortisol, 20 ng/ml epidermal growth factor and 100 ng/ml
cholera enterotoxin. The medium was also supplemented with 100 U/ml
penicillin and 100 U/ml streptomycin. All cultures were maintained
at 37°C under 5% CO2.
Cell treatments and harvesting
BaP and CAR were initially dissolved in
dimethylsulfoxide and subsequently in cell culture medium. All
treatments were prepared and administered under low light
conditions, prior to incubation at 37°C in a 5% CO2
humidified incubator. MCF-10A cells were divided into three groups,
namely BaP treatment, CAR/BaP co-treatment and control. The BaP
group was treated with 1 µmol/l BaP for 6, 12, 24 or 36 h. The
CAR/BaP group was treated with 1 µmol/l BaP in combination with
0.5, 1.0, 5.0 or 10 µmol/l CAR. The control group was cultured
without CAR or BaP. To determine the effect of
H2O2, MCF-10A cells were cultured with
H2O2 (200 µmol/l) and CAR (5 µmol/l) for 36
h. The cells were harvested by trypsinization at 6, 12, 24 or 36 h,
and were suspended in PBS without Mg2+ or
Ca2+. The cells were snap-frozen at −196°C in liquid
N2 until further use.
Determination of intracellular ROS
levels
The cultures were labeled with 5 µmol/l
dichlorodihydrofluorescein diacetate for 1 h. ROS levels were
detected using flow cytometry (BD Biosciences, Franklin Lakes, NJ,
USA) and fluorescence microscopy (Nikon Corporation, Tokyo, Japan).
The cells were trypsinized from the cultures and resuspended in PBS
for analysis of ROS, using a 15 mW air-cooled argon laser at an
excitation wavelength of 488 nm. Dichlorofluorescein fluorescence
emission was determined using a 529 nm band pass filter. The mean
fluorescence intensity of 2×104 cells was quantified
using Multicycle AV software (version 3.0; Phoenix Flow Systems,
San Diego, CA, USA).
Single cell gel electrophoresis (comet
assay)
MCF-10A cells were exposed to BaP/CAR for 48 h (one
cycle) and/or exposed for between 5 and 20 cycles to induce
cellular carcinogenesis (10).
Cultured cells were harvested as aforementioned. The comet assay
was performed according to a previously published protocol
(9), and tail moment was the
indicator of DNA damage. Tail moment is defined as the product of
the tail length and the fraction of total DNA in the tail, which
can be calculated using commercially available and public domain
software (9). Data analysis was
performed using Komet software (version 5.5; Andor Technology, PLC,
Belfast, Northern Ireland, UK), and Kinetic Imaging (version 5.0;
Kinetic Imaging Ltd., Wirral, UK).
ELISA detection of 8-OH-dG in MCF-10A
cells treated with BaP/CAR
The three groups of cells, namely BaP (1 µM)
treatment, CAR (5 µM)/BaP (1 µM) co-treatment and control, were
plated in 6-well dishes at a density of 1×106 cells/dish
cultured for 24 h. The cells were trypsinized and resuspended in
PBS. Subsequently, two freeze-thaw cycles were performed and the
homogenates were centrifuged at 5,000 × g for 5 min at 4°C. The
supernatant was removed and the 8-OH-dG levels were determined
using the 8-OH-dG ELISA kit, according to the manufacturer's
protocol. Absorbance was determined at 450 nm using Curve Expert
Professional software (version 2.3.0; win.cutephp.com/curveexpert_professional_1996267) to
create a standard curve. Results are expressed in ng/ml. Samples
were assayed in a blind manner.
Western blot analysis
Cell lysates were prepared by washing cells with PBS
and were incubated for 10 min at 4°C in modified
radioimmunoprecipitation assay lysis buffer [50 mmol/l Tris/HCl,
150 mmol/l NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 25
µg/ml leupeptin, 10 µg/ml aprotinin, 2 mmol/l EDTA and 1 mmol/l
sodium orthovanadate (Sigma; Merck KGaA)]. The cells were scraped
from the plates and centrifuged at 5,000 × g for 20 min at 4°C, and
the supernatants were collected. The protein concentration levels
were determined using a Bicinchoninic Acid assay kit (Pierce;
Thermo Fisher Scientific, Inc.), and 40 µg whole cell lysates were
separated by SDS-PAGE (10% gels). The samples were transferred onto
a nitrocellulose membrane by wet blotting. The membrane was blocked
with 5% fat-free dry milk in PBS for 1 h at room temperature, and
incubated with primary antibodies overnight at 4°C. The antibodies
used were anti-phospho (p)-AKT (Thr308) (1:400; cat. no.
558316; BD Biosciences), anti-murine double minute 2 (MDM2; 1:400;
cat. no. 556353; BD Biosciences), anti-p-p53
(Ser15/Ser20) (1:100; cat. no. sc-10176;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
anti-β-arrestin (1:100; cat. no. sc-21872; Santa Cruz
Biotechnology, Inc.). Following three washes with PBS, the membrane
was incubated with goat anti-mouse immunoglobulin G (IgG) (1:8,000;
cat. no. 31430; Thermo Fisher Scientific, Inc.) and goat
anti-rabbit IgG (1:8,000; cat. no. 31460; Thermo Fisher Scientific,
Inc.) secondary antibodies and visualized with SuperSignal West
Dura Extended Duration Substrate (Thermo Fisher Scientific,
Inc.).
Statistical analysis
Unless indicated otherwise, each assay was conducted
three times. Results are presented as the mean ± standard error of
the mean. Results were analyzed by one-way analysis of variance or
Student's t-test; multiple comparisons between the groups were
performed using the Student-Newman-Keuls method. P<0.05 was
considered to indicate a statistically significant difference.
Results
CAR inhibits BaP-induced ROS
production
BaP induces DNA damage through the generation of
ROS, which initiate oxidative damage to nucleic acids (2). In addition to the β-blocker effect,
CAR acts as an antioxidant and a ROS scavenger (11). MCF-10A cells were used in order to
investigate the effects of CAR on BaP-mediated ROS production. In
the absence of BaP, MCF-10A cells generated ROS spontaneously, and
CAR had the potential to decrease ROS production in cells that were
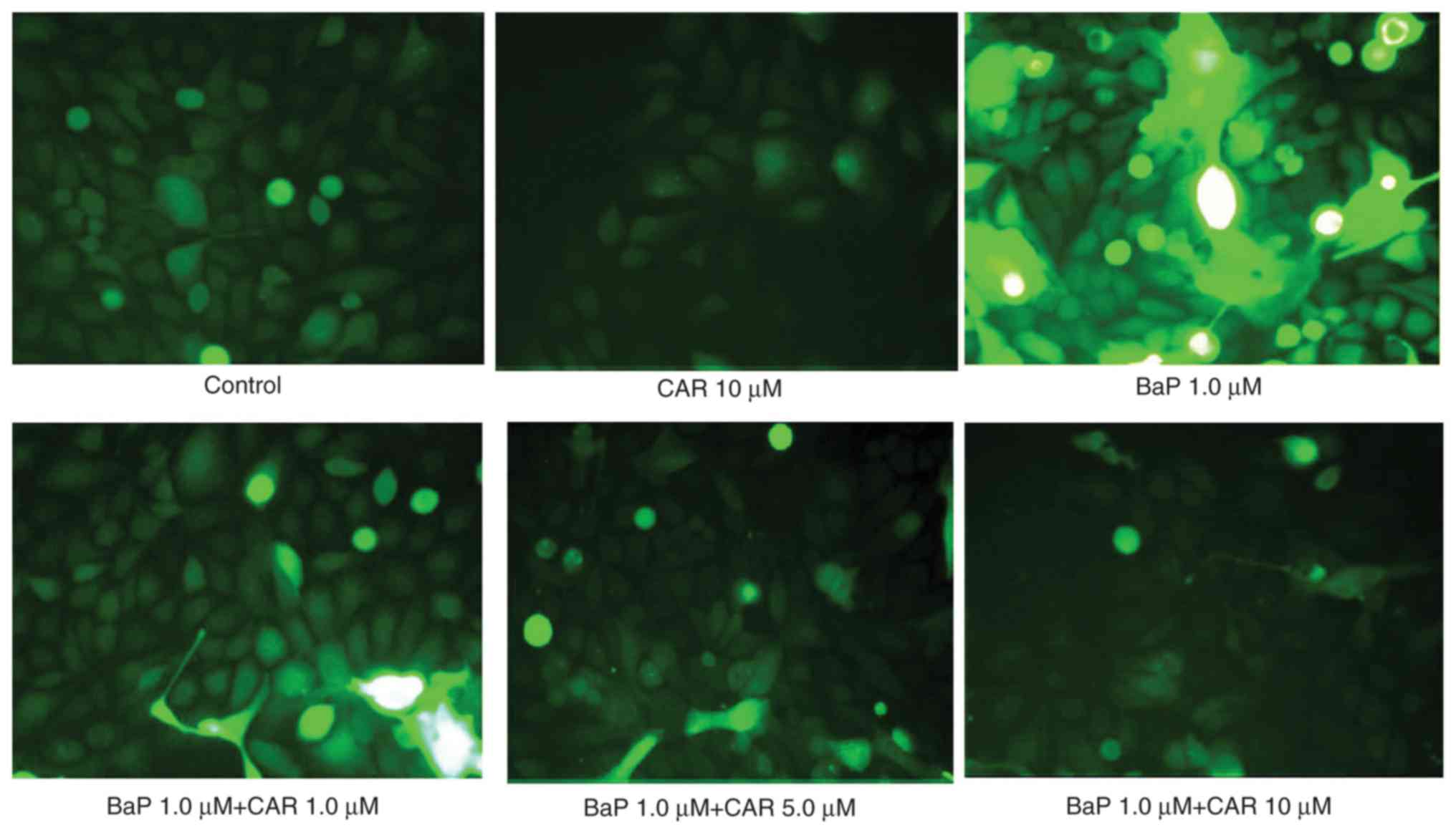
not treated with BaP (Fig. 1). BaP
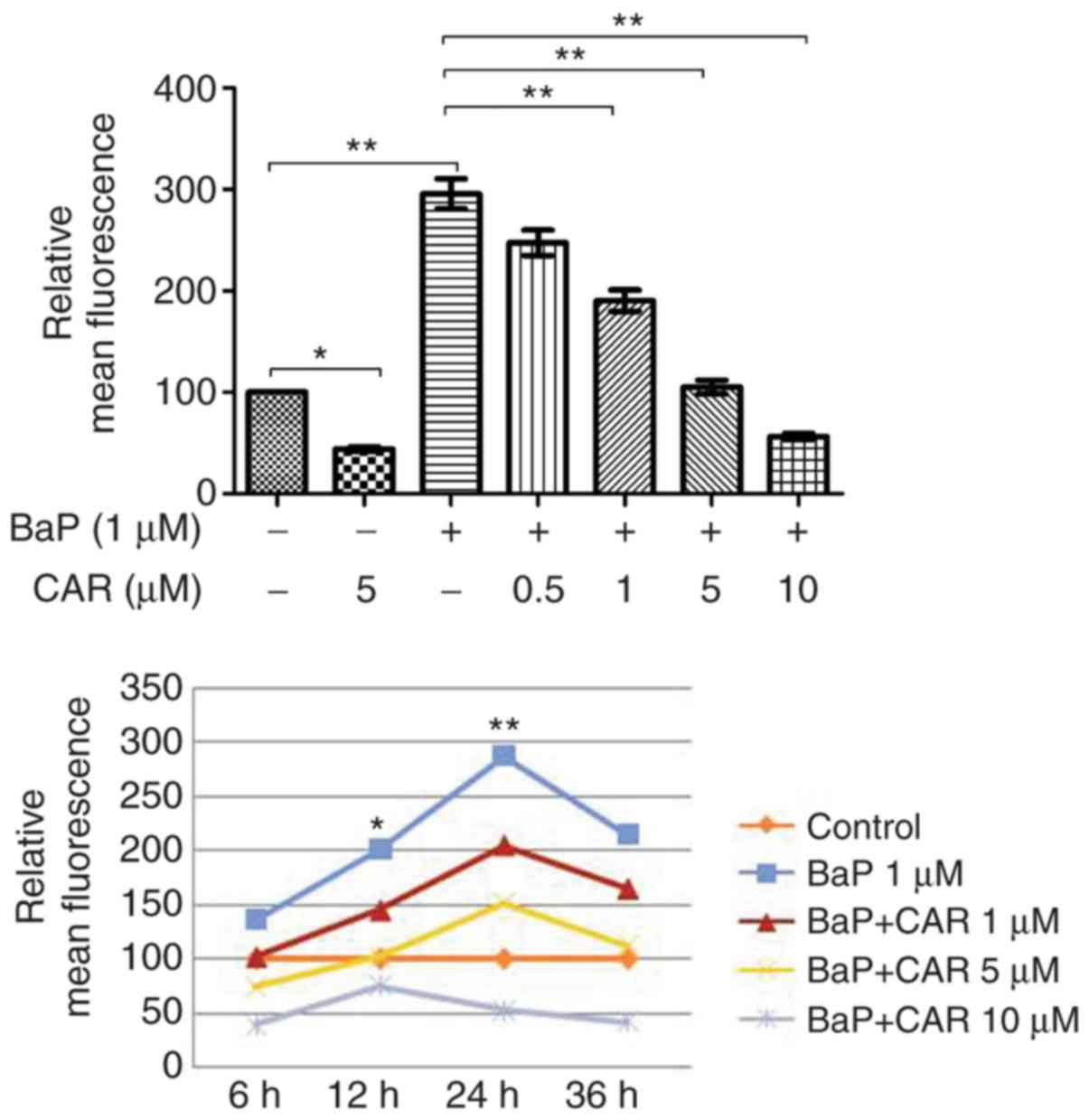
increased ROS production in MCF-10A cells; however, CAR, at
concentrations ≥1 µM, attenuated BaP-induced ROS production in a
dose-dependent manner (Fig. 2,
upper panel). Short-term exposure (6 h) of MCF-10A cells to 1 µM
BaP induced ROS, and ROS production reached a peak at 24 h
(Fig. 2, lower panel). BaP induced
a significant increase in ROS levels in a time-dependent manner
(between 6 and 24 h), although ROS levels decreased at 36 h,
indicating endogenous adjustment (Fig.
2). Co-treatment of CAR and BaP decreased BaP-induced ROS
production at all time points investigated in a dose-dependent
manner (CAR concentration ≥1 µM) (Figs.
1 and 2).
CAR eliminates BaP-induced DNA
damage
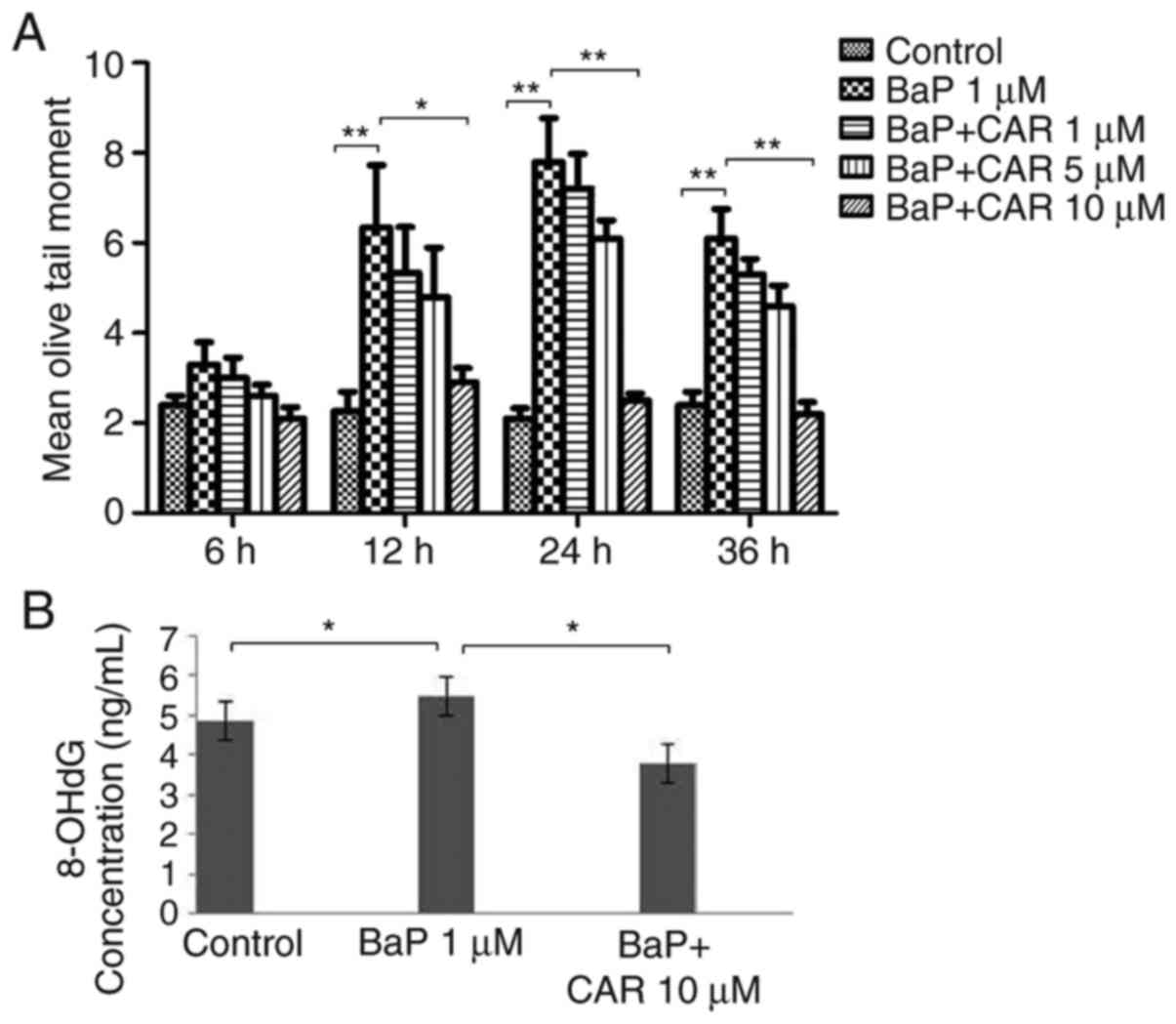
BaP induced significant DNA damage as demonstrated
using the alkaline comet assay in MCF-10A cells. Exposure of the
cells to BaP for 5 cycles was sufficient to induce DNA strand
breaks in MCF-10A cells, although no DNA damage was observed in the
15–20 exposure cycles using the alkaline comet assay. Treatment of
the cells with 1 µM BaP caused DNA damage at 12, 24 and 36 h of
culture, compared with control cells; however, CAR attenuated DNA
strand breaks that were induced by BaP (Fig. 3A). A concentration-dependent effect
was observed using CAR. Treatment of the cells with 10 µM CAR and
BaP for 24 h attenuated 68% of BaP-induced DNA damage (data not
shown), which corresponded nearly to the levels of the control
group (Fig. 3A). As a marker of DNA
oxidative damage, 8-OH-dG levels increased in MCF-10A cells treated
with BaP determined using the 8-OH-dG ELISA kit, although CAR
reversed the effect of BaP (Fig.
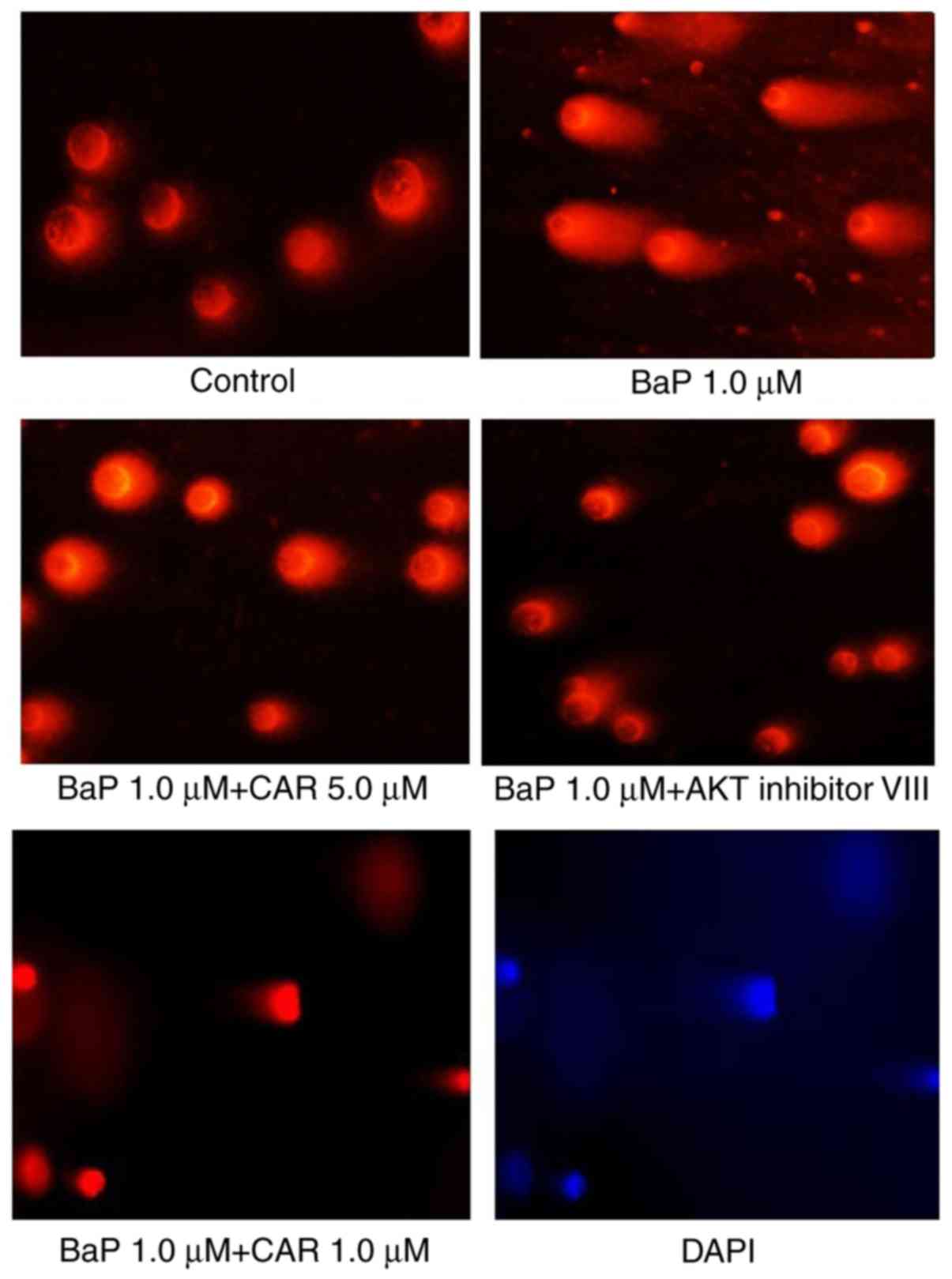
3B). Cells cultured with 1 µM BaP for 24 h exhibited apparent
DNA damage, and CAR and AKT inhibitor VIII eliminated DNA damage
caused by BaP (Fig. 4). These
results indicated that CAR has the ability to inhibit BaP-induced
DNA damage.
CAR inhibition of BaP induces the
PI3K/AKT signaling pathway via the decrease in ROS production
Certain carcinogens induce intracellular ROS
accumulation, and increase the expression of PI3K and AKT proteins.
ROS also directly activate PI3K, and activated PI3K further
phosphorylates its downstream target AKT. The alterations in the
PI3K/AKT signaling pathway have been revealed to promote cell
survival and proliferation (12,13).
In order to investigate the mechanism of CAR suppression in
BaP-induced MCF-10A cell carcinogenesis, the level of expression
and phosphorylation of target proteins associated with the PI3K/AKT
signaling pathway was determined. Western blot analysis using an
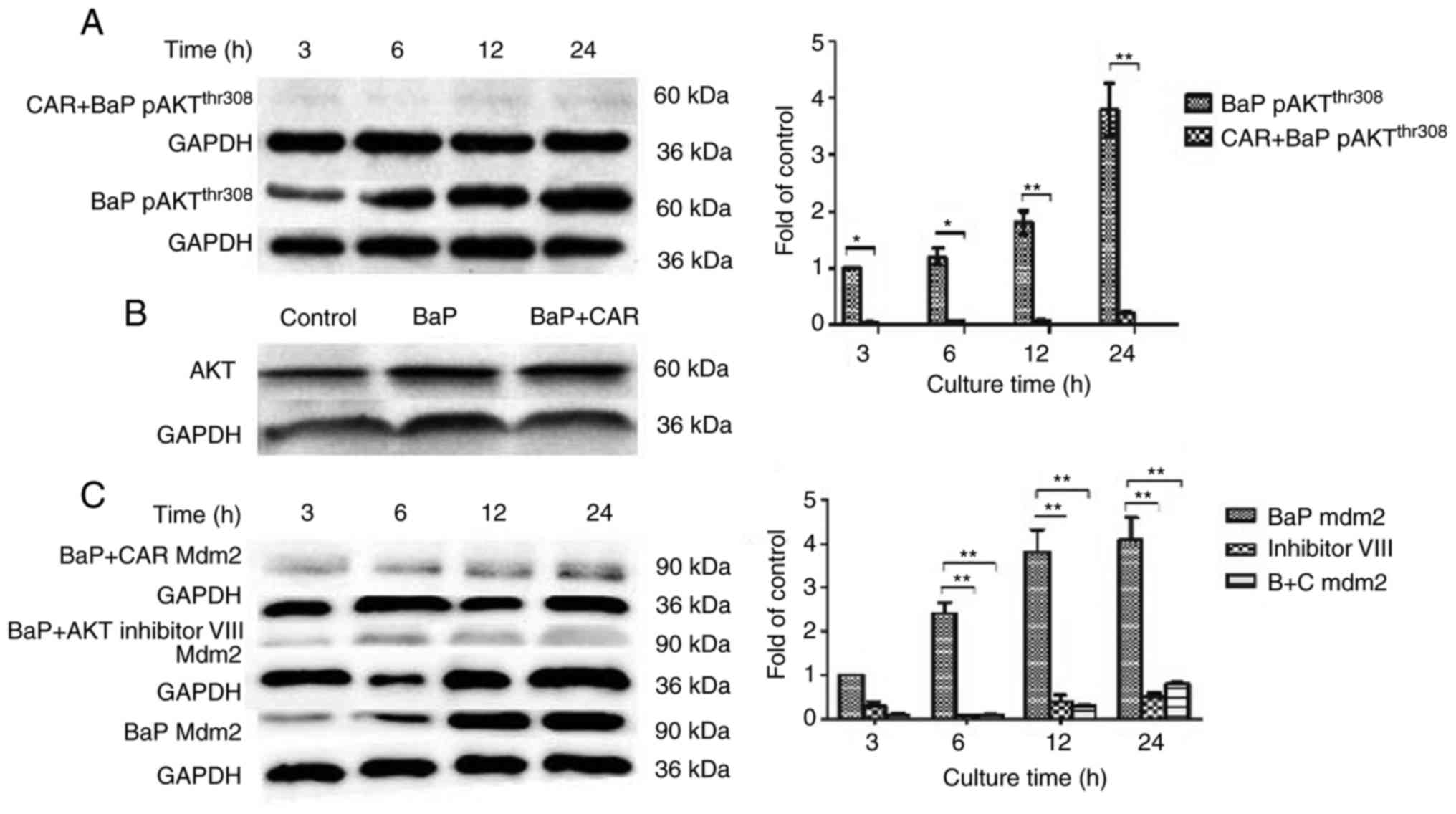
antibody against p-(activated) AKTThr308 indicated that,
compared with untreated cells, p-AKTThr308 expression
was increased significantly in MCF-10A cells that were treated with
BaP. When cells were incubated with CAR (5 µM) and BaP
simultaneously for various durations, p-AKTThr308
expression decreased in all cases (Fig.
5A). These results suggested that AKT phosphorylation induced
by BaP could be inhibited by CAR in MCF-10A cells. However, AKT
(non-phosphorylated) expression was not influenced by treatment
with BaP and CAR for 24 h (Fig.
5B). However, in the absence of BaP, MCF-10A cells were
co-cultured with H2O2 and CAR, and
p-AKTThr308 expression increased markedly (data not
shown). These results indicated that CAR inhibited BaP-induced
PI3K/AKT activation via the inhibition of ROS generation, but not
ROS scavenging.
CAR inhibition of BaP causes a change
in MDM2 and p53 expression levels
MDM2 is an important AKT substrate. Phosphorylation
of MDM2 by AKT can target the tumor suppressor p53 for degradation
through ubiquitination. p53 modulates cells that are under
oxidative stress and aids the adaptation of the cell's oxidative
mechanism to the remodeled redox balance, thus preventing oxidative
damage to DNA and proteins (14,15).
Therefore, the effect of CAR on the expression of
p53Ser15, p53Ser20 and MDM2 in BaP-induced
cells was investigated by western blotting. As CAR suppresses
BaP-induced neoplastic activity and associated protein function at
concentrations between 1 and 10 µM, 5 µM CAR was used as the
optimal concentration. It was identified that MDM2 expression was
upregulated in BaP-treated cells, although CAR was able to
eliminate the effects of BaP (Fig.
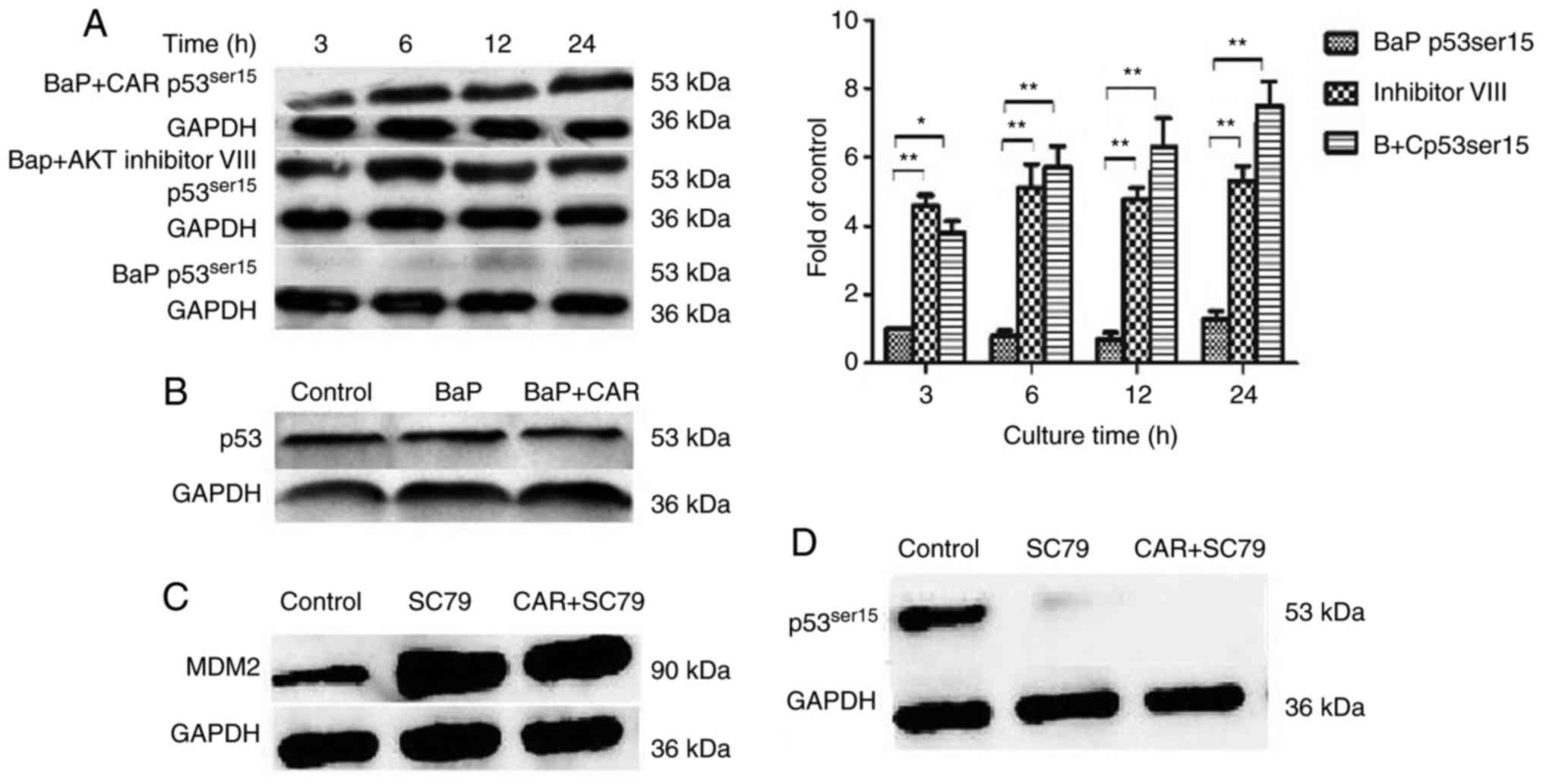
5C). In addition, the expression of p53Ser15 was
downregulated in BaP-treated cells, although CAR could prevent the
influence of BaP, and eliminate these changes caused by BaP at the
time points investigated (Fig. 6A).
However, p53Ser20 expression did not alter in
BaP-treated cells (data not shown). Furthermore, p53
(non-phosphorylated) expression was not influenced when cells were
treated with BaP and CAR for 24 h (Fig.
6B). As effectors of the PI3K/AKT signaling pathway, the MDM2
and p53Ser15 proteins serve important functions in the
effects of BaP/CAR on MCF-10A cell transformation.
When MCF-10A cells were co-cultured with BaP and the
AKT inhibitor VIII, BaP had no influence on MDM2 expression and p53
degradation (Figs. 5C and 6A). Furthermore, BaP-induced DNA damage
was counteracted by the AKT inhibitor VIII as demonstrated using
the alkaline comet assay (Fig. 4).
In contrast with the inhibition of AKT, when cells were treated
with CAR and SC79 (an AKT phosphorylation activator), enhanced MDM2
expression and p53 degradation were evident from western blot
analysis (Fig. 6C and D). CAR was
not able to inhibit the effects of SC79, which indicated that CAR
acts upstream of the PI3K/AKT signaling pathway. These data
indicated that BaP-induced ROS production activated PI3K/AKT, which
in turn upregulated MDM2, thus facilitating p53 degradation. CAR
prevents cellular carcinogenesis induced by BaP via the suppression
of PI3K/AKT and MDM2 activity, which increases p53 levels in
MCF-10A cells.
Discussion
ROS are free radicals with marked oxidative
activity. ROS oxidize proteins and DNA, and damage various cellular
organelles. This initiates the cellular injury responsible for
aging and disease (16).
ROS-mediated DNA damage can promote the malignant transformation of
cells, and facilitate cell survival and proliferation (17–20).
Increases in ROS may promote cell susceptibility to DNA damage
induced by carcinogens, contributing to cellular carcinogenesis
(21). However, the exact function
of ROS in BaP-induced carcinogenesis of breast pre-cancerous cells
is unclear.
CAR is a non-selective adrenergic blocker, primarily
designed to treat cardiovascular disease by blocking adrenergic
receptors. CAR is licensed by the US Food and Drug Administration
for the treatment of chronic heart failure partly due to its
antioxidant and ROS-scavenging properties (22). In addition, CAR has the potential of
anti-neoplastic action, including breast cancer and glioma
(4). In our previous study, we
identified that CAR could inhibit migration and invasion of several
breast cancer cell lines (5). A
recent study indicated that CAR could prevent skin carcinogenesis
induced by DMBA (7). However, there
are no studies regarding the potential prevention of breast cancer
by CAR. To address this question and to investigate the mechanisms
of CAR in eliminating the activity of BaP in cellular
carcinogenesis, CAR activity in the modulation of BaP-induced
increases in ROS, DNA damage, and associated signaling pathways in
pre-cancerous breast cells were determined.
It was identified that BaP induced a significant and
sustained increase in ROS levels in MCF-10A cells, and that CAR
attenuated BaP-induced ROS production in a concentration-dependent
manner at doses ≥1 µM. Furthermore, CAR was able to decrease ROS
production in MCF-10A cells that were not treated with BaP, since
the cells could generate ROS spontaneously. ROS are associated with
clustered DNA damage, chromosomal aberrations and induction of
carcinogenesis in normal breast cells (23). The alkaline comet assay and 8-OH-dG
ELISA kit were used to investigate the detection of DNA damage. The
data demonstrated that BaP induced significant DNA damage, and that
CAR could attenuate DNA strand breaks induced by BaP at the time
points investigated. Compared with low-dose CAR treatment, the tail
moment was lower in the high-dose CAR groups. In the present study,
exposure of MCF-10A cells to 1 µM BaP for 5 cycles was identified
to be sufficient to induce DNA damage. However, no DNA damage was
detected by the alkaline comet assay and 8-OH-dG detection, when
cells were exposed to BaP for 10–20 cycles. It was identified that
the single treatment of MCF-10A cells with a high concentration of
BaP for 48 h was sufficient to cause chromosomal aberrations
(24). Thus, we hypothesized that
MCF-10A cells treated with BaP for >10 cycles may progress to
cancerous cells, whereas BaP exhibited no effects on the cancer
cells. The investigation of the malignant transformation of the
cells requires further investigation.
Alterations in the PI3K/AKT pathway have been
observed in various types of tumor (25). Pre-cancerous cells chronically
exposed to carcinogens increase their ROS production, which
mediates the activation of PI3K, AKT and target proteins, resulting
in increased cell proliferation and anchorage-independent growth
(12). The aforementioned results
led to the investigation of whether the PI3K/AKT signaling pathway
may be involved in breast carcinogenesis induced by BaP.
Phosphorylation at Thr308 and/or Ser473
indicates the activation of AKT (26). Vincent et al (27) hypothesized that the phosphorylation
of Thr308 is a more reliable biomarker for the activity
of AKT compared with phosphorylation of Ser473. In the
present study, it was identified that chronic exposure to BaP could
induce AKTThr308 phosphorylation in MCF-10A cells,
although the function of BaP was inhibited by CAR via inhibition of
ROS production. In contrast with BaP, H2O2
treatment of cells led to AKT activation, and CAR had no influence.
This suggested that CAR inhibited BaP-induced PI3K/AKT activation
through the inhibition of ROS generation, but not through ROS
scavenging. The p53 gene may lose its repair activity and permit
passage of DNA mutations from one generation to the next. This
increases the risk of cell carcinogenesis to stress and damage.
MDM2 is a downstream substrate of PI3K/AKT and degrades p53 to
promote tumorigenesis (28).
Following DNA damage, p53 can be activated, leading to the repair
of DNA damage (29). The ability of
PI3K/AKT activation to increase DNA damage by carcinogens in
MCF-10A cells led us to hypothesize that BaP/CAR may affect the
ability of MDM2 and p53. The results of the present study indicate
that BaP could not increase the levels of p53 in MCF-10A cells. A
possible reason may be that increased ROS induced by BaP activate
AKT and subsequent MDM2, and activated MDM2 degrades p53 by
ubiquitination in MCF-10A cells. The lack of p53 response by BaP in
MCF-10A cells suggested that p53 had no protective functions on DNA
damage in these cells; this may, in part, contribute to the potent
mammary carcinogenicity of BaP. However, in the presence of CAR or
AKT inhibitor VIII, the expression of p53 in MCF-10A treated with
BaP was unknown. Generally, Ser15 and Ser20
in p53 are typically phosphorylated with DNA damage, and
Ser15 is the most common site of phosphorylation
(30–32). Therefore, antibodies against p-p53
at Ser15 and Ser20 to detect p53 activity by
western blotting. It was identified that p-p53Ser15
expression, but not p-p53Ser20, was upregulated in the
presence of CAR or AKT inhibitor VIII in MCF-10A cells treated by
BaP. This suggests that AKT activity (caused by ROS/BaP) was
inhibited by CAR or AKT inhibitor VIII, subsequent MDM2
phosphorylation by p-AKT did not occur. Therefore,
p53Ser15 protein could not be degraded by MDM2 through
ubiquitination.
Mitogen-induced activation of PI3K and its
downstream target, AKT, results in phosphorylation of MDM2 on
Ser166 and Ser186. Phosphorylation of these
residues is necessary for translocation of MDM2 from the cytoplasm
into the nucleus (28). Mutation of
the AKT phosphorylation sites in MDM2 produces a mutant protein
that is unable to enter the nucleus and increases p53 activity
(28). Zhou et al (33) reported that AKT physically
associates with MDM2 and phosphorylates it at Ser166 and
Ser186. Phosphorylation of MDM2 enhances its nuclear
localization and increases p53 degradation (33). The results of the present study
identified that MDM2 expression was upregulated, although without
detection of p-MDM2, as the downstream target of PI3K,
phosphorylation of MDM2 by activated AKT is highly likely. Thus,
increased MDM2 expression or phosphorylated MDM2 enhanced p53
degradation and may induce carcinogenesis in MCF10A cells. The
results concerning the PI3K/AKT/MDM2 signaling pathway provided
sufficient evidence for the function of CAR in the prevention of
breast tumorigenesis.
Chemoprevention has become an important approach for
decreasing breast cancer morbidity and mortality (34). Tamoxifen and raloxifene are the
classical chemopreventive drugs used to prevent estrogen receptor
(ER)-positive breast cancer that have minimal effects on
ER-negative breast cancer (34).
The results of the present study indicate that CAR may be
considered a novel chemopreventive agent, notably in the prevention
of ER-negative breast cancer, since MCF-10A cells rarely express ER
(35). The antioxidant effect of
CAR may contribute to its cancer-preventive activity. Although the
present study focused on breast cancer prevention, the results may
lay the foundation for clinical trials that can be designed to
prevent other types of cancer, as reported recently in the
prevention of hepatocarcinogenesis (36).
Acknowledgements
We are grateful to Dr Shuya Huang and Dr Chunmiao Ye
for image editing.
Funding
The present study was supported by the Science and
Technology Office of Shandong, with funding obtained from Shandong
Natural Science Funds, China (grant nos. ZR2015HM043 and
ZR2014HL074).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DG conceived and designed the experiments. XL
performed the experiments. QZ and ZY analyzed and interpreted the
data. ZM was involved in the conception of the study and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wu Q and Ni X: ROS-mediated DNA
methylation pattern alterations in carcinogenesis. Curr Drug
Targets. 16:13–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cooke MS, Evans MD, Dizdaroglu M and Lunec
J: Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB
J. 17:1195–1214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oliveira PJ, Goncalves L, Monteiro P,
Providencia LA and Moreno AJ: Are the antioxidant properties of
carvedilol important for the protection of cardiac mitochondria?
Curr Vasc Pharmacol. 3:147–158. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Erguven M, Yazihan N, Aktas E, Sabanci A,
Li CJ, Oktem G and Bilir A: Carvedilol in glioma treatment alone
and with imatinib in vitro. Int J Oncol. 36:857–866. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dezong G, Zhongbing M, Qinye F and Zhigang
Y: Carvedilol suppresses migration and invasion of malignant breast
cells by inactivating Src involving cAMP/PKA and PKCδ signaling
pathway. J Cancer Res Ther. 10:998–1003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin CS, Lin WS, Lin CL and Kao CH:
Carvedilol use is associated with reduced cancer risk: A nationwide
population-based cohort study. Int J Cardiol. 184:9–13. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang A, Yeung S, Thakkar A, Huang KM, Liu
MM, Kanassatega RS, Parsa C, Orlando R, Jackson EK, Andresen BT, et
al: Prevention of skin carcinogenesis by the β-blocker carvedilol.
Cancer Prev Res (Phila). 8:27–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kastrati I, Edirisinghe PD, Hemachandra
LP, Chandrasena ER, Choi J, Wang YT, Bolton JL and Thatcher GR:
Raloxifene and desmethylarzoxifene block estrogen-induced malignant
transformation of human breast epithelial cells. PLoS One.
6:e278762011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nkrumah-Elie YM, Reuben JS, Hudson A, Taka
E, Badisa R, Ardley T, Israel B, Sadrud-Din SY, Oriaku E and
Darling-Reed SF: Diallyl trisulfide as an inhibitor of
benzo(a)pyrene-induced precancerous carcinogenesis in MCF-10A
cells. Food Chem Toxicol. 50:2524–2530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song X, Siriwardhana N, Rathore K, Lin D
and Wang HC: Grape seed proanthocyanidin suppression of breast cell
carcinogenesis induced by chronic exposure to combined
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and
benzo[a]pyrene. Mol Carcinog. 49:450–463. 2010.PubMed/NCBI
|
|
11
|
Yue TL, McKenna PJ, Lysko PG, Gu JL, Lysko
KA, Ruffolo RR Jr and Feuerstein GZ: SB211475, a metabolite of
carvedilol, a novel antihypertensive agent, is a potent
antioxidant. Eur J Pharmacol. 251:237–243. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mohapatra P, Preet R, Das D, Satapathy SR,
Siddharth S, Choudhuri T, Wyatt MD and Kundu CN: The contribution
of heavy metals in cigarette smoke condensate to malignant
transformation of breast epithelial cells and in vivo initiation of
neoplasia through induction of a PI3K-AKT-NFκB cascade. Toxicol
Appl Pharmacol. 274:168–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Son YO, Pratheeshkumar P, Wang L, Wang X,
Fan J, Kim DH, Lee JY, Zhang Z, Lee JC and Shi X: Reactive oxygen
species mediate Cr(VI)-induced carcinogenesis through
PI3K/AKT-dependent activation of GSK-3β/β-catenin signaling.
Toxicol Appl Pharmacol. 271:239–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Astle MV, Hannan KM, Ng PY, Lee RS, George
AJ, Hsu AK, Haupt Y, Hannan RD and Pearson RB: AKT induces
senescence in human cells via mTORC1 and p53 in the absence of DNA
damage: Implications for targeting mTOR during malignancy.
Oncogene. 31:1949–1962. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He J, Zhu G, Gao L, Chen P, Long Y, Liao
S, Yi H, Yi W, Pei Z, Wu M, et al: Fra-1 is upregulated in gastric
cancer tissues and affects the PI3K/Akt and p53 signaling pathway
in gastric cancer. Int J Oncol. 47:1725–1734. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wojtovich AP and Foster TH: Optogenetic
control of ROS production. Redox Biol. 2:368–376. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gupta SC, Hevia D, Patchva S, Park B, Koh
W and Aggarwal BB: Upsides and downsides of reactive oxygen species
for cancer: The roles of reactive oxygen species in tumorigenesis,
prevention, and therapy. Antioxid Redox Signal. 16:1295–1322. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang C, Cao S, Toole BP and Xu Y: Cancer
may be a pathway to cell survival under persistent hypoxia and
elevated ROS A model for solid-cancer initiation and early
development. Int J Cancer. 136:2001–2011. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cichon MA and Radisky DC: ROS-induced
epithelial-mesenchymal transition in mammary epithelial cells is
mediated by NF-κB-dependent activation of Snail. Oncotarget.
5:2827–2838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nourazarian AR, Kangari P and Salmaninejad
A: Roles of oxidative stress in the development and progression of
breast cancer. Asian Pac J Cancer Prev. 15:4745–4751. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pluchino LA, Liu AK and Wang HC: Reactive
oxygen species-mediated breast cell carcinogenesis enhanced by
multiple carcinogens and intervened by dietary ergosterol and
mimosine. Free Radic Biol Med. 80:12–26. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yue TL, Cheng HY, Lysko PG, McKenna PJ,
Feuerstein R, Gu JL, Lysko KA, Davis LL and Feuerstein G:
Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is
an antioxidant and free radical scavenger. J Pharmacol Exp Ther.
263:92–98. 1992.PubMed/NCBI
|
|
23
|
Sigounas G, Hairr JW, Cooke CD, Owen JR,
Asch AS, Weidner DA and Wiley JE: Role of benzo[a]pha]pyrene
in generation of clustered DNA damage in human breast tissue. Free
Radic Biol Med. 49:77–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Siriwardhana N and Wang HC: Precancerous
carcinogenesis of human breast epithelial cells by chronic exposure
to benzo[a]pyrene. Mol Carcinog. 47:338–348. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Petrulea MS, Plantinga TS, Smit JW,
Georgescu CE and NeteaMaier RT: PI3K/Akt/mTOR: A promising
therapeutic target for non-medullary thyroid carcinoma. Cancer
Treat Rev. 41:707–713. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Su CH, Wang CY, Lan KH, Li CP, Chao Y, Lin
HC, Lee SD and Lee WP: Akt phosphorylation at Thr308 and Ser473 is
required for CHIP-mediated ubiquitination of the kinase. Cell
Signal. 23:1824–1830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vincent EE, Elder DJ, Thomas EC, Phillips
L, Morgan C, Pawade J, Sohail M, May MT, Hetzel MR and Tavaré JM:
Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt
protein kinase activity in human non-small cell lung cancer. Br J
Cancer. 104:1755–1761. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mayo LD and Donner DB: A
phosphatidylinositol 3-kinase/Akt pathway promotes translocation of
Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA.
98:11598–11603. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Umannová L, Machala M, Topinka J,
Schmuczerová J, Krčmář P, Neča J, Šujanová K, Kozubík A and
Vondráček J: Benzo[a]pyrene and tumor necrosis factor-α
coordinately increase genotoxic damage and the production of
proinflammatory mediators in alveolar epithelial type II cells.
Toxicol Lett. 206:121–129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cromie MM, Liu Z and Gao W:
Epigallocatechin-3-gallate augments the therapeutic effects of
benzo[a]pyrene-mediated lung carcinogenesis. Biofactors.
43:529–539. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pääjärvi G, Jernström B, Seidel A and
Stenius U: Anti-diol epoxide of benzo[a]pyrene induces
transient Mdm2 and p53 Ser15 phosphorylation, while anti-diol
epoxide of dibenzo[a,l]pyrene induces a nontransient p53 Ser15
phosphorylation. Mol Carcinog. 47:301–309. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lambert PF, Kashanchi F, Radonovich MF,
Shiekhattar R and Brady JN: Phosphorylation of p53 serine 15
increases interaction with CBP. J Biol Chem. 273:33048–33053. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou BP, Liao Y, Xia W, Zou Y, Spohn B and
Hung MC: HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2
phosphorylation. Nat Cell Biol. 3:973–982. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Freedman AN, Yu B, Gail MH, Costantino JP,
Graubard BI, Vogel VG, Anderson GL and McCaskill-Stevens W:
Benefit/risk assessment for breast cancer chemoprevention with
raloxifene or tamoxifen for women age 50 years or older. J Clin
Oncol. 29:2327–2333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hemachandra LP, Patel H, Chandrasena RE,
Choi J, Piyankarage SC, Wang S, Wang Y, Thayer EN, Scism RA,
Michalsen BT, et al: SERMs attenuate estrogen-induced malignant
transformation of human mammary epithelial cells by up-regulating
detoxification of oxidative metabolites. Cancer Prev Res (Phila).
7:505–515. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Balaha M, Kandeel S and Barakat W:
Carvedilol suppresses circulating and hepatic IL-6 responsible for
hepatocarcinogenesis of chronically damaged liver in rats. Toxicol
Appl Pharmacol. 311:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|