Introduction
Daily administered, low-dose, cytotoxic,
chemotherapeutic drugs were initially shown by Browder et al
to preferentially target the endothelium of the tumor vasculature
(1). When cyclophosphamide was
administered in low frequent doses, as opposed to the maximally
tolerated dose every three weeks, potent tumor suppression was
achieved as a result of endothelial cell apoptosis. This
anti-angiogenic, or metronomic, chemotherapeutic approach avoids
the development of tumor cell resistance by targeting the
proliferating endothelial cells required for tumor
neovascularization (2–4). Furthermore, the greater sensitivity
of endothelial cells in comparison to tumor cells allows for
significantly lower doses of the drug to be effective, thus
improving tolerability (5,6). Anti-angiogenic chemotherapy has
entered clinical trials for various vascular tumors refractory to
conventional chemotherapy (4,7–9). In
our study, 40% of children with recurrent or progressive cancer,
treated with daily low-dose oral etoposide alternating every 21
days with daily low-dose oral cyclophosphamide combined with daily
oral thalidomide and celecoxib, exhibited a prolonged or persistent
progression-free disease status (7).
Etoposide (VP16), a topoisomerase II inhibitor, is a
semisynthetic derivative of podophyllotoxin introduced in cancer
clinical trials in 1971 and FDA-approved in 1983. It is an alkaloid
cytotoxic drug that binds to and inhibits topoisomerase II-DNA
function in ligating cleaved DNA molecules, resulting in the
accumulation of single- or double-strand DNA breaks and stops the
cell cycle at the late S and G2 phases (10). Daily oral etoposide is effective
for the treatment of several tumors, including non-small cell lung
cancer, recurrent medulloblastoma and neuroblastoma, after these
tumors have developed resistance to the maximally tolerated doses
of intravenous etoposide (11,12).
Additionally, platinum-resistant epithelial ovarian cancer,
metastatic breast cancer and pediatric recurrent sarcomas have been
successfully treated with oral etoposide (13–15).
When compared to intravenous administration, treatment with oral
etoposide increased the response rate in patients with small-cell
lung and advanced breast cancers (16,17).
However, the mechanism by which low-dose oral etoposide inhibits
the growth of tumors resistant to maximally tolerated higher-dose
intravenous etoposide has not been extensively studied.
We hypothesize that tumor endothelium is a potential
target of low-dose oral etoposide, since the primary tumor and
metastatic growth are dependent on angiogenesis (18). This hypothesis is supported by
observations that etoposide inhibits the proliferation of
endothelial cells (19). In fact,
endothelial cells were found to be more sensitive to etoposide than
tumor cells in vitro (20),
suggesting that the anti-tumor effect of etoposide may, in part, be
mediated through the endothelium. Therefore, we investigated the
role of etoposide in tumor angiogenesis. We report that etoposide
inhibits primary tumor growth and metastasis through
anti-angiogenic and direct anti-tumor effects. Oral administration
of etoposide allows it to be easily incorporated into chemotherapy
regimens and supports its addition to the growing class of oral
anti-angiogenic drugs for cancer therapy.
Materials and methods
Cells and reagents
Bovine capillary endothelial (BCE) cells were
maintained on gelatinized plastic in Dulbecco's modified Eagle's
medium (DMEM) low glucose + 10% bovine calf serum. Human umbilical
vein endothelial cells (HUVECs) were maintained in EBM-2 media.
Lewis lung carcinoma (LLC), fibrosarcoma (T241), glioblastoma
(U87), breast (MDA-MB 231) and K1000 [a tumor cell line that
expresses and secretes high levels of fibroblast growth factor 2
(FGF2)] cells were cultured in DMEM + 10% heat-inactivated FBS + 1%
penicillin streptomycin glutamine. For in vitro studies,
etoposide (VP-16) (Sigma, St. Louis, MO, USA) was used and for
in vivo studies, clinical grade IV solution was
utilized.
Vascular endothelial growth factor
(VEGF) ELISA
Tumor cells that were known to secrete high levels
of VEGF (U87 glioblastoma and LLC) were plated at 15×103
cells per well (6-well plates), and 24 h later were treated with
etoposide or vehicle. Medium containing the drugs was changed on
Days 3 and 5. On Day 6, the medium was collected, and VEGF was
assayed by ELISA (R&D Systems Inc., Minneapolis, MN, USA).
Angiogenesis assays
Endothelial cell proliferation was assayed as
described (21) at
15×103 cells per well. For tumor cell proliferation,
cells were plated at 5×103 cells per well. Endothelial
cell tubes were formed by combining HUVECs (5×104
cells/well) with varying concentrations of etoposide or vehicle on
Matrigel- (Collaborative Biochemical, Bedford, MA, USA) coated
24-well plates. The animal experiments were performed in accordance
with IRB-approved protocols at Children's Hospital Boston.
For the corneal neovascularization assay, 80 ng FGF2
or 160 ng VEGF pellets were implanted into C57BL/6 mice (Jackson
Labs, Bar Harbor, ME, USA) (22).
Etoposide was administered daily over 6 days by gavage in 0.5%
methylcellulose, and control mice received vehicle (0.5%
methylcellulose).
For tumor studies, LLC was injected subcutaneously
as described (21). Glioblastoma
(U87) and T241 fibrosarcoma were injected subcutaneously
(1×106 cells in 0.1 ml PBS) into 6-week-old male severe
combined immunodeficient (MGH, Boston, MA, USA) or C57BL/6 mice,
respectively. Once tumors were 100–150 mm3, mice were
randomized into treatment and vehicle groups. Etoposide, celecoxib,
rosiglitazone and/or cyclophosphamide were administered by daily
gavage for 14–40 days. Tumors were measured every 3–7 days, and the
volume was calculated as width2 × length × 0.52.
For metastasis studies, LLC tumors were resected 15
days after implantation as described (21). After LLC resection, mice were
treated with etoposide or vehicle for 16 days when control mice
became terminally ill. On the last day of treatment, the
statistical difference between the treatment and control groups was
determined by the Student's t-test. A p-value <0.05 was accepted
as significant.
Miles vascular permeability assay
One to two days prior to the experiment, mice were
shaved to expose the skin. Mice were anesthetized with
intraperitoneally injected Avertin and injected with 1% Evan's blue
dye, either by tail vein or through the orbital plexus. VEGF (50 μl
of 1 ng/μl) and 50 μl of saline or PBS with 0.05% gelatin were
injected intradermally using a 30-gauge needle into the skin
overlying the back. Similar experiments were performed by injecting
5 μl of VEGF, saline or PBS intra-dermally into the ears. After 10
min, the animals were euthanized, and the skin was opened and
exposed to assess the intensity of Evan's blue dye extravasations.
The areas of blue skin (vascular leak) were removed and placed into
formamide for 5 days. The intensity of the reaction was quantified
by reading the samples at a wavelength of 620 nm on a SpectraMax
plate reader.
Immunohistochemistry
For PECAM1, the sections of tumors were treated with
40 μg/ml proteinase K (Roche Diagnostics Corp.) for 25 min at 37°C.
PECAM1 was amplified using tyramide signal amplification direct and
indirect kits (PerkinElmer Life Sciences, Boston, MA, USA). For
computer-enhanced imaging of tumors, histological sections were
analyzed for vessel density by computerized densitometric imaging
(Corel Photo Paint and IP Lab software). The degree of
vascularization was quantified over the entire tumor section and
expressed as a ratio of vessel area (PECAM1) to tumor area. Total
fields scored per tumor were 67–70. For control and
etoposide-treated tumors, 4 animals/group were evaluated.
Results
Etoposide has direct and indirect
anti-angiogenic and anti-tumor activity in vitro
Direct effects
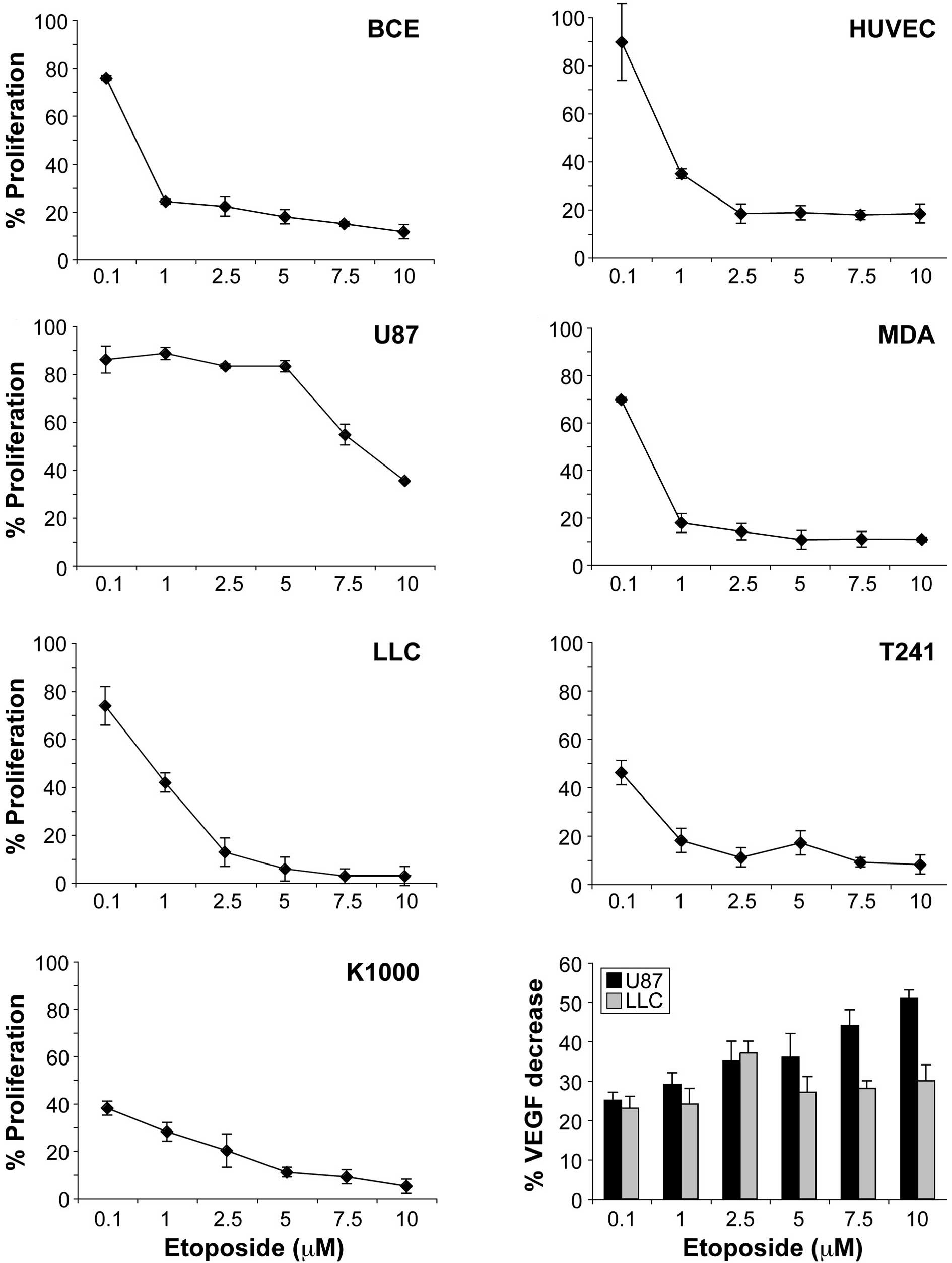
To investigate the effects of etoposide on
endothelial cell proliferation, we stimulated the proliferation of
BCE cells with FGF2, a potent mitogen for BCE cells, in a standard
proliferation assay. Etoposide inhibited FGF2-induced proliferation
of BCE cells in a dose-dependent manner, with a maximal inhibition
of 80% after a 72-h incubation period at 2.5 μM, a concentration
easily achieved orally in humans (Fig.
1A). Similarly, etoposide inhibited VEGF-induced proliferation
of HUVECs up to 80% at 2.5 μM (Fig.
1B). We next determined whether etoposide inhibits tumor cells
at similar doses as those applied to endothelial cells. Etoposide
inhibited the proliferation of human tumor cells, including
glioblastoma (U87) and breast (MDA-MB 231), differentially because
of the primary resistance of these cell lines (Fig. 1C and D). Murine tumor cell lines,
LLC and T241 fibrosarcoma demonstrated sensitivity to etoposide
(Fig. 1E–G).
Indirect effects
To determine whether etoposide inhibits angiogenesis
by down-regulating tumor-secreted growth factors, we measured VEGF
levels in tumor-conditioned media via ELISA. The tumor cell lines
glioblastoma (U87 resistant to etoposide) and LLC (sensitive to
etoposide) secreted substantial amounts of VEGF: 20,000 and 938
pg/106 cells, respectively. Etoposide inhibited VEGF
secretion in U87 cells by 51% and in LLC cells by 36% (Fig. 1H). The inhibitory effect of
etoposide on VEGF secretion in vitro suggests a potential
anti-angiogenic mechanism in vivo via decreased tumor cell
production of this angiogenic mitogen.
Etoposide inhibits endothelial cell
tube formation and FGF2- and VEGF-induced corneal
neovascularization
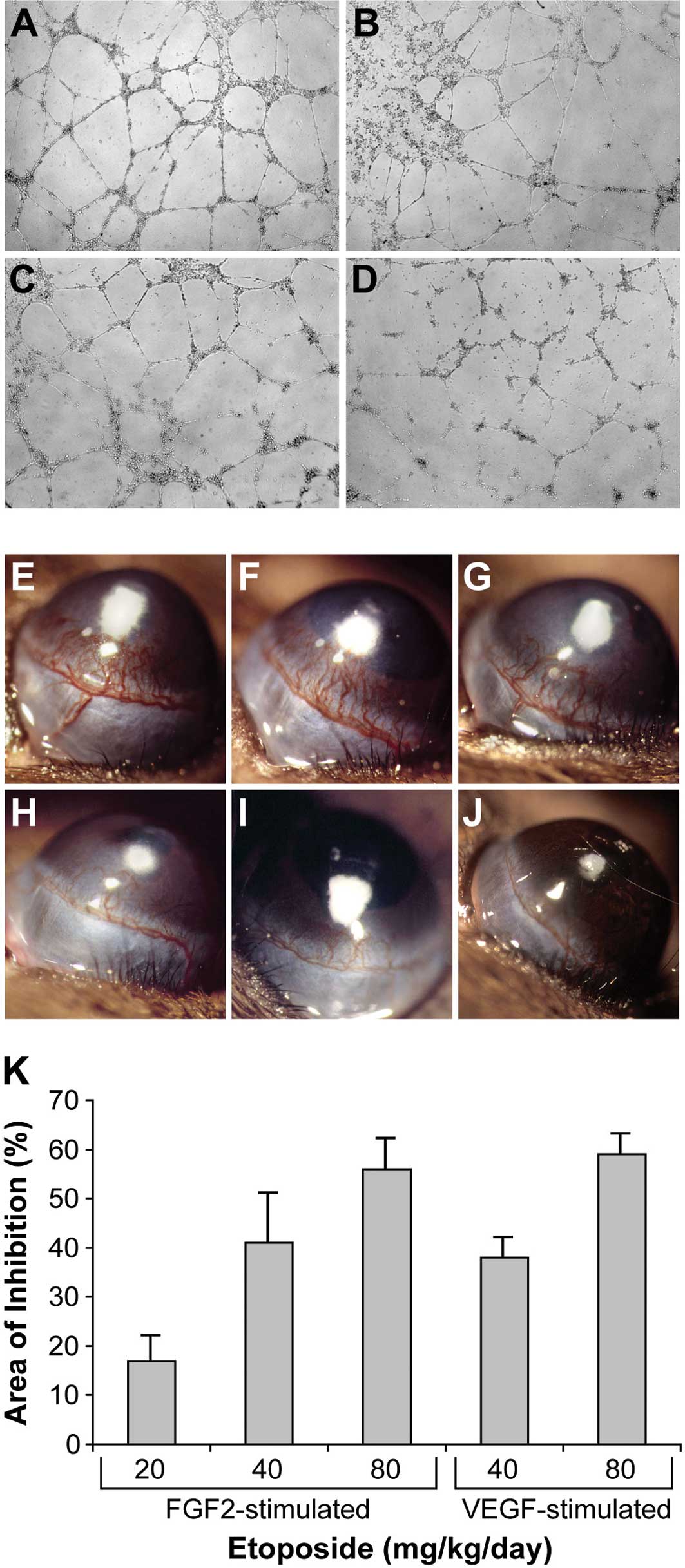
To investigate whether etoposide has an effect on
vessel morphogenesis, we seeded HUVECs on Matrigel, where they
formed branching, anastomosing tubes that mimicked capillary-like
structures (Fig. 2A). Etoposide
inhibited tube formation in a dose-dependent manner (Fig. 2B-D), consistent with previous
studies (23). To optimize the
anti-angiogenic doses of etoposide for daily administration in
mice, we implanted 80 ng FGF2 pellets into the corneas of C57BL/6
mice to stimulate neovascularization over 6 days (Fig. 2E). Systemic oral administration of
etoposide significantly inhibited FGF2-induced corneal
neovascularization in a dose-dependent fashion: 20 mg/kg/day
resulted in 17% inhibition (Fig.
2F); 40 mg/kg/day resulted in 41% inhibition (Fig. 2G); 80 mg/kg/day resulted in 56%
inhibition (Fig. 2H). To determine
the effect of etoposide on VEGF-induced corneal neovascularization,
VEGF pellets (160 ng) were implanted into the corneas of C57BL/6
mice. Systemic oral administration of etoposide (40 and 80
mg/kg/day) inhibited VEGF-induced corneal neovascularization by 38
and 59%, respectively (Fig. 2I and
J). In summary, daily administration of etoposide exhibited
dose-dependent inhibition of both FGF2- and VEGF-stimulated corneal
neovascularization (Fig. 2K).
Etoposide inhibits VEGF-induced
vascular permeability and raises endostatin levels in vivo
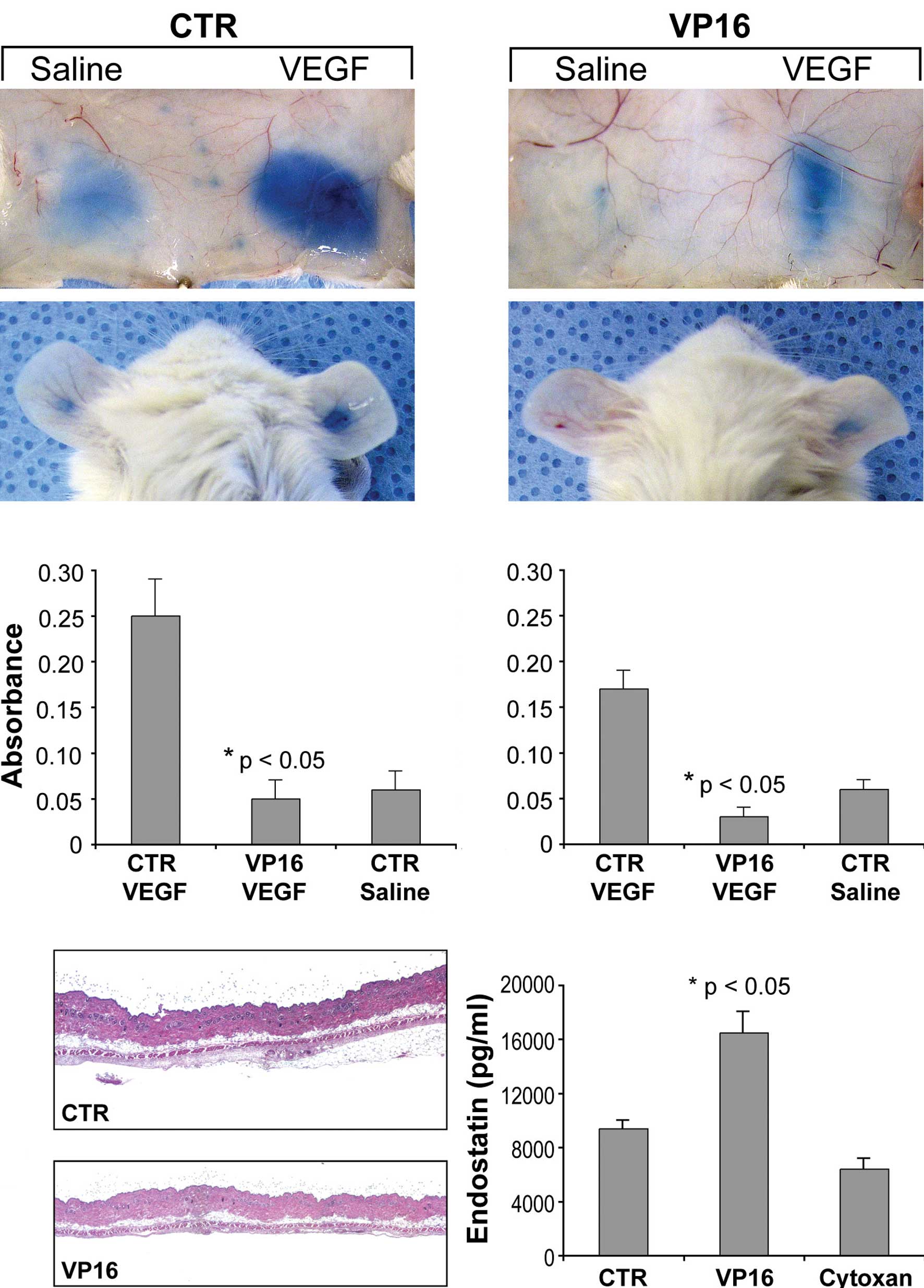
We next determined whether etoposide (VP16) affects
VEGF-induced vascular permeability, a standard test of in
vivo VEGF activity (24). In
response to VEGF, control mice displayed Evan's blue extravasation
into the subcutaneous skin and ears (Fig. 3A) 80–82% greater than that of
etoposide-treated mice (Fig. 3B).
There was also a decrease in vascular leakage between the two
saline groups in the etoposide-treated mice, presumably
representing the inhibition of basal circulating VEGF.
Spectrophotometric analysis of extravasated Evan's blue in both the
skin and ear of etoposide-treated mice exhibited a dramatic
reduction in VEGF-induced vascular permeability (Fig. 3C and D). Immunohistochemical
analysis (H&E staining) revealed that the area of skin edema
was greatly reduced in the etoposide-treated mice when compared to
the vehicle-treated mice (Fig. 3E and
F). Together, these results indicate that daily low-dose oral
etoposide is a potent inhibitor of VEGF-dependent signaling.
Since etoposide raises biologically active
endostatin levels in vitro (25), we examined whether the
administration of etoposide raises endostatin levels in
vivo. Mice treated with etoposide exhibited a 75% increase in
plasma levels of endostatin (Fig.
3G). Another oral chemotherapeutic agent, cyclophosphamide, had
no effect on endostatin levels in the plasma (Fig. 3G).
Systemic therapy with etoposide
inhibits primary tumor growth and metastasis
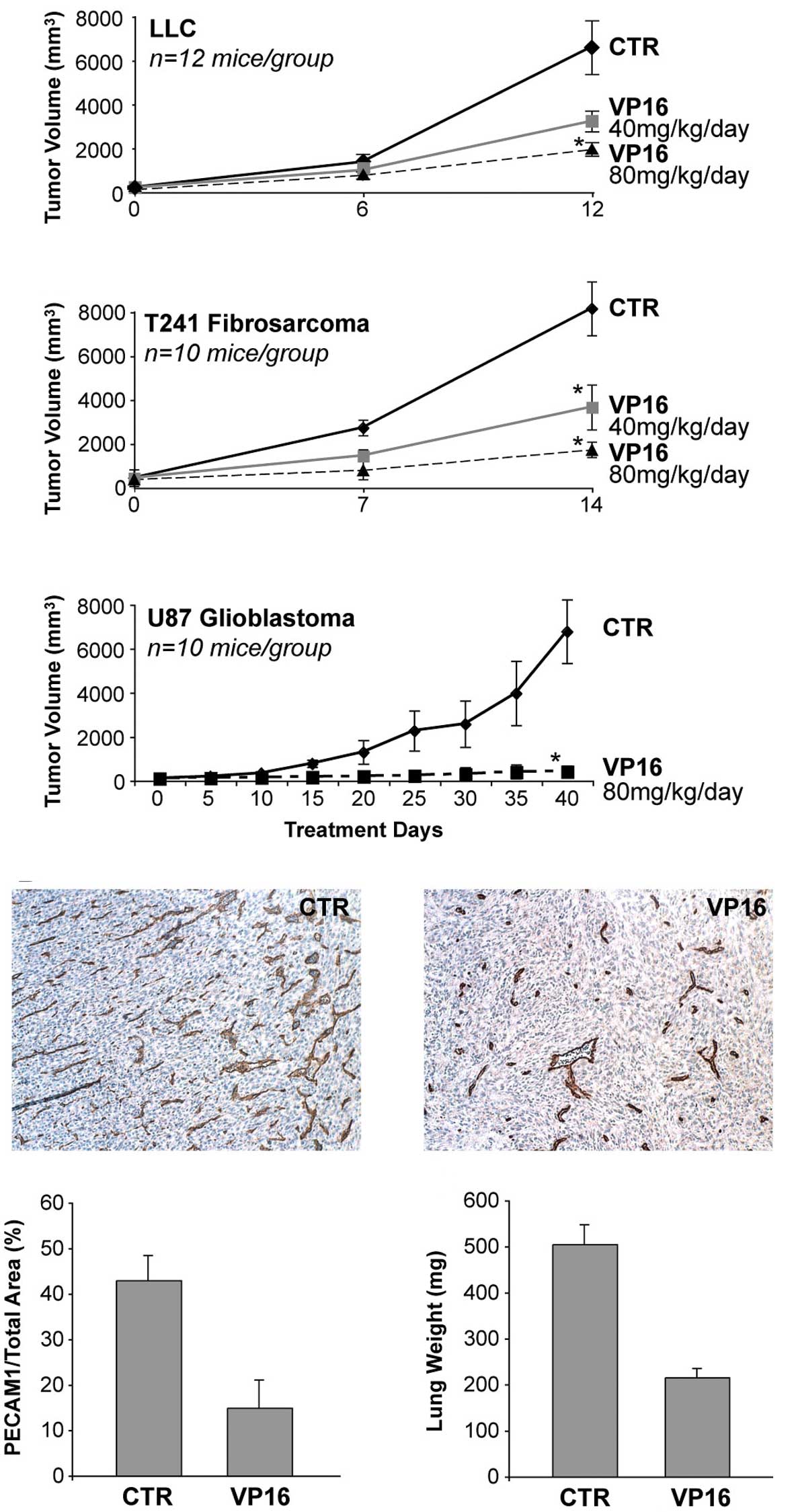
In order to examine the anti-angiogenic effect of
daily, low-dose, oral etoposide (VP16) on the growth of primary
tumors, we treated established subcutaneous tumors of 100–150
mm3 volume grown in mice. We utilized the optimal doses
of etoposide identified in the corneal neovascularization assay.
Oral etoposide at 40 and 80 mg/kg/day inhibited the growth of LLC
by 29 and 56%, respectively (Fig.
4A), and T241 fibrosarcoma by 55 and 79%, respectively
(Fig. 4B); 80 mg/kg of etoposide
inhibited glioblastoma (U87) by 95% (Fig. 4C). There was no evidence of
significant weight loss or other drug-related toxicity in any of
the mice. To determine whether etoposide inhibited primary tumor
growth by inhibiting angiogenesis, we measured the microvessel
density in the treated and control tumors. A decrease in the
microvessel density during treatment with an angiogenesis inhibitor
suggests an anti-angiogenic effect on tumor growth (18). Etoposide treatment reduced
microvessel density relative to that in the control tumors, thus
indicating the presence of its anti-angiogenic efficacy (Fig. 4D–F).
Etoposide, when administered in mice via liposomes,
was found to inhibit the formation of lung nodules in a metastatic
tail vein model (26). The tail
vein model illustrates only the homing step of tumor cells from
circulation to an organ. By contrast, the LLC metastasis model is a
model of spontaneous lung metastasis with all of the steps involved
in metastasis including the invasion of tumor cells from the
primary tumor to the circulation. Removal of the primary LLC was
found to decrease the circulating angiogenesis inhibitor
angiostatin, resulting in rapid growth of lung metastasis (21). In the present study, mice were
treated for 15 days with oral daily etoposide (80 mg/kg/day) or
vehicle after removal of the primary LLC. In mice treated with
vehicle, growing invasive metastasis almost entirely replaced the
normal lung tissue, leading to lung weights in these mice of 505±42
mg. In marked contrast, mice treated with oral etoposide (80
mg/kg/day) had a lung weight of 216±19 mg vs. normal lung weights
of 152±10 mg (Fig. 4G).
Etoposide has synergistic anti-tumor
activity with oral anti-angiogenic drugs, including celecoxib and
rosiglitazone
To determine whether combining other classes of
drugs improves the anti-tumor efficacy of etoposide, we utilized
the cyclooxygenase-2 (COX2) inhibitor, celecoxib and
peroxisome-proliferator activated receptor (PPAR)γ ligand
rosiglitazone, which are both orally administered and target
endothelial and tumor cells (22,27).
We administered celecoxib, rosiglitazone and either etoposide or
cyclophosphamide at the lowest doses necessary for minimal
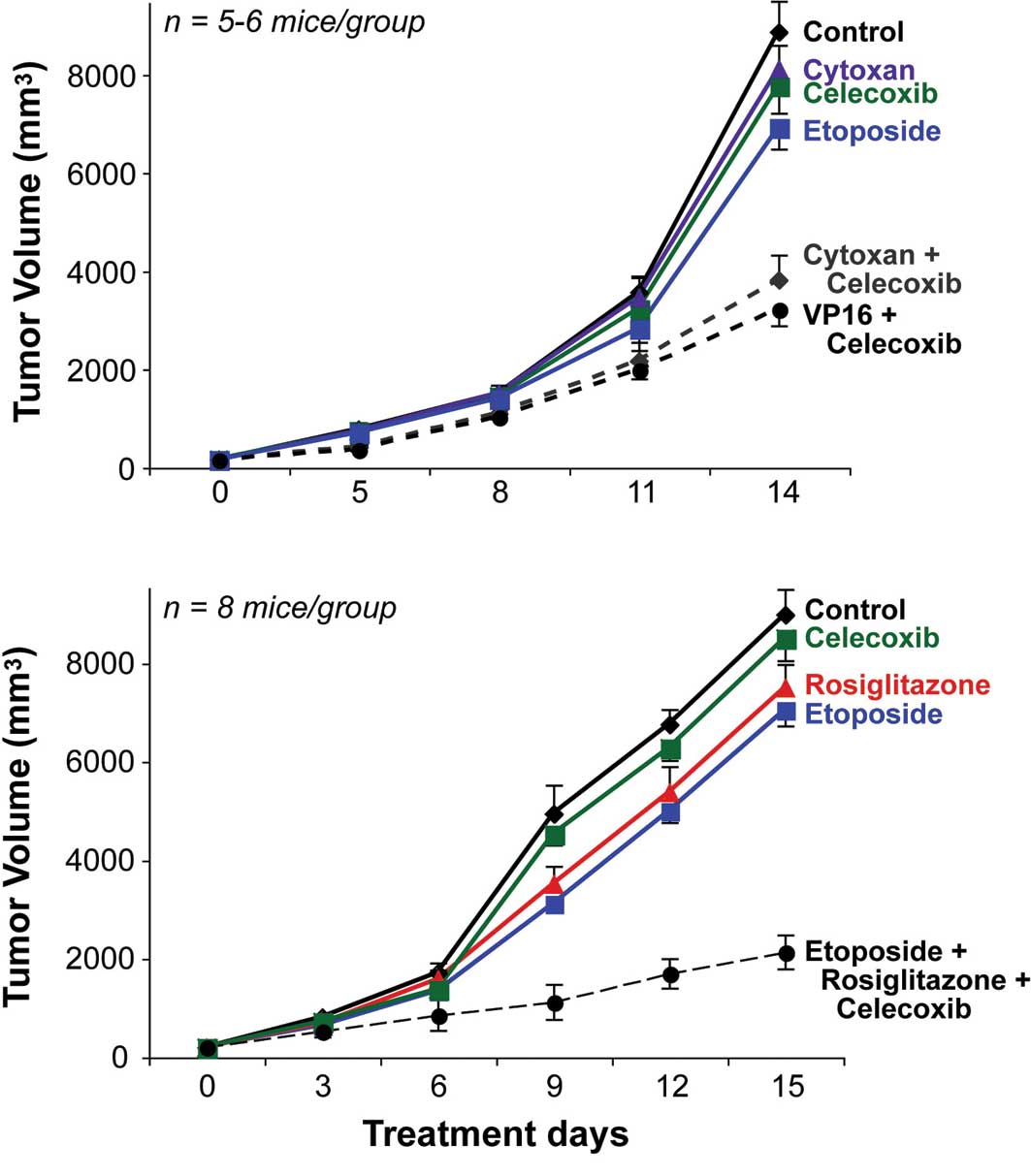
anti-tumor effect. Oral celecoxib (30 mg/kg/day) significantly
enhanced the anti-tumor activity of low-dose oral etoposide (10
mg/kg/day) by 42% (Fig. 5A). When
combined, PPARγ agonist rosiglitazone (50 mg/kg/day) and celecoxib
(30 mg/kg/day) enhanced the anti-tumor activity of low-dose oral
etoposide (10 mg/kg/day) by 69% (Fig.
5B), with no evidence of drug-related toxicity.
Discussion
Most chemotherapy regimens are associated with
significant toxicity when administered at maximum tolerated doses.
There is now increasing evidence that multi-drug-resistant tumors
are effectively targeted by anti-angiogenic chemotherapy (1,2), in
which low doses of cytotoxic drugs are given at close, regular
intervals, with minimal toxic side effects (4). Therefore, standard chemotherapeutic
agents, when modified by frequency and dose, target tumor
angiogenesis. The mechanism by which cytotoxic chemotherapy affects
the tumor vasculature may include selective killing of endothelial
cells, suppression of circulating endothelial precursor cells
and/or increasing levels of the endogenous angiogenesis inhibitors,
such as thrombospondin-1 (4,28–31),
and decreasing levels of angiogenesis stimulators, such as
VEGF.
Oral etoposide, a chemotherapeutic drug, is an
active agent in the treatment of various malignancies, including
recurrent brain tumors, leukemia, lymphoma, hepatocellular
carcinoma, Kaposi's sarcoma, ovarian and testicular cancer
(13,32–34).
Patients with small-cell lung cancer treated with a prolonged
maintenance of low serum etoposide concentrations (>1 μg/ml)
were found to have a high response rate (35), while tumoricidal doses usually
require >10 μg/ml (36).
Multiple pre-clinical and clinical studies have shown that the
anti-tumor activity of etoposide is schedule-dependent, as smaller
doses over several days or small daily doses result in higher
response rates than single large doses (12,14,32,37).
In addition to its effect on tumor cells, etoposide
has been reported to reduce tumor angiogenesis in one of two renal
cell carcinoma cell lines (38).
Our studies support the role of etoposide in inhibiting
angiogenesis in vitro and in vivo by decreasing VEGF
production by tumor cells and microvessel density and increasing
endostatin levels in vivo, consistent with other studies
showing that etoposide increases the expression of biologically
active endostatin in vitro (25). This increase in endostatin may
explain in part the anti-tumor efficacy of etoposide (5). Results from our studies suggest that
etoposide is an addition to the growing class of drugs that
increase systemic endostatin levels, including tamoxifen, celecoxib
and prednisolone plus salazosulphapyridine (in joint fluid)
(39–41).
Tumor angiogenesis involves various pathways,
thereby providing multiple molecular targets for anti-angiogenic
drugs. Despite the potential efficacy of anti-angiogenic drugs,
when used as single agents, resistance occurs by various mechanisms
(6,42). Therefore, there is an urgent need
for multi-drug regimens in treating drug-resistant cancer in the
clinic. Anti-angiogenic ‘metronomic’ chemotherapy with
cyclophosphamide was shown to be synergistic with the
thrombospondin peptide ABT-510 in suppressing tumor growth
(43). Recent studies show synergy
between PPARγ ligands, such as rosiglitazone, and platinum-based
chemotherapeutic agents in inhibiting tumor growth (44). The use of oral etoposide in a
number of combinations, such as with other angiogenesis inhibitors,
chemotherapy and/or radiation, has demonstrated activity in mouse
tumor models and patients (19,25,45,46).
Our results show that etoposide has synergistic anti-tumor activity
with COX2 inhibitors and PPARγ ligands. COX2 inhibitors, such as
celecoxib, have both anti-angiogenic and anti-tumor activities
(27); we previously demonstrated
that the PPARγ ligand rosiglitazone inhibits primary tumor growth
and metastasis by targeting the tumor endothelium (22). The mechanism by which etoposide
inhibits tumor angiogenesis may complement the anti-angiogenic
effects of COX2 and PPARγ ligands resulting in greater inhibition
of endothelial proliferation and a decrease in VEGF secretion.
Already, several human studies support the clinical
relevance of oral etoposide. We recently incorporated etoposide as
part of a four-drug anti-angiogenic chemotherapy regimen
(thalidomide, celecoxib, etoposide and cyclophosphamide), which
showed prolonged disease-free status in pediatric patients with
recurrent or progressive cancer (7). Similarly, etoposide was part of a
four-drug regimen named COMBAT (combined oral maintenance
biodifferentiating and anti-angiogenic therapy), which was
effective in solid tumors in children which had relapsed (9). This regimen included celecoxib,
cis-retinoic acid, metronomic temozolomide and low-dose etoposide.
Anti-angiogenic ‘metronomic’ chemotherapy is significantly
cost-effective in the treatment of metastatic breast cancer
(47). Therefore, oral etoposide,
which is very well tolerated, may result in increased patient
compliance; it can also be administered on an outpatient basis,
thereby reducing costs, which is becoming an important issue
(48,49).
Our studies suggest that etoposide may be beneficial
in treating angiogenic diseases, such as cancer, because of its
effect on the endothelium and on angiogenesis pathways. Moreover,
the endothelium is also an important target in the treatment of
non-neoplastic diseases, such as arthritis, psoriasis and
endometriosis. In fact, suboptimal doses of etoposide were found to
improve collagen II-induced arthritis without monocyte depletion
(50). As an orally administered
FDA-approved drug, etoposide is ideally suited for use in
combination with other anti-angiogenesis regimes and can complement
conventional cancer treatment modalities.
Acknowledgements
We dedicate this research study to the
memory of Dr Judah Folkman. The excellent technical assistance of
Ricky Sanchez is acknowledged. We thank Kristin Johnson for
photography. We thank Jessica Barnes for the helpful discussion.
This study was supported by the Stop and Shop Family Pediatric
Brain Tumor Fund and the C.J. Buckley Pediatric Brain Tumor
Research Fund (M.W.K., D.P. and A.L.) and by the Department of
Defense Innovator Award #W81XWH-04-1-0316 (J.F.) and private
philanthropic funds.
References
|
1.
|
Browder T, Butterfield CE, Kraling BM, et
al: Antiangiogenic scheduling of chemotherapy improves efficacy
against experimental drug-resistant cancer. Cancer Res.
60:1878–1886. 2000.PubMed/NCBI
|
|
2.
|
Klement G, Baruchel S, Rak J, et al:
Continuous low-dose therapy with vinblastine and VEGF receptor-2
antibody induces sustained tumor regression without overt toxicity.
J Clin Invest. 105:15–24. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Hanahan D, Bergers G and Bergsland E: Less
is more, regularly: metronomic dosing of cytotoxic drugs can target
tumor angiogenesis in mice. J Clin Invest. 105:1045–1047. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kerbel RS and Kamen BA: The
anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer.
4:423–436. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Folkman J: Angiogenesis: an organizing
principle for drug discovery? Nat Rev Drug Discov. 6:273–286. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Man S, Bocci G, Francia G, et al:
Antitumor effects in mice of low-dose (metronomic) cyclophosphamide
administered continuously through the drinking water. Cancer Res.
62:2731–2735. 2002.
|
|
7.
|
Kieran MW, Turner CD, Rubin J, et al: A
feasibility trial of antiangiogenic (metronomic) chemotherapy in
pediatric patients with recurrent or progressive cancer. J Pediatr
Hematol Oncol. 27:573–581. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Vogt T, Hafner C, Bross K, et al:
Antiangiogenetic therapy with pioglitazone, rofecoxib, and
metronomic trofosfamide in patients with advanced malignant
vascular tumors. Cancer. 98:2251–2256. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Sterba J, Valik D, Mudry P, et al:
Combined biodifferentiating and antiangiogenic oral metronomic
therapy is feasible and effective in relapsed solid tumors in
children: single-center pilot study. Onkologie. 29:308–313. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Aisner J and Lee EJ: Etoposide. Current
and future status. Cancer. 67:215–219. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Kakolyris S, Samonis G, Koukourakis M, et
al: Treatment of non-small cell lung cancer with prolonged oral
etoposide. Am J Clin Oncol. 21:505–508. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Ashley DM, Meier L, Kerby T, et al:
Response of recurrent medulloblastoma to low-dose oral etoposide. J
Clin Oncol. 14:1922–1927. 1996.PubMed/NCBI
|
|
13.
|
Alici S, Saip P, Eralp Y, Aydiner A and
Topuz E: Oral etoposide (VP16) in platinum-resistant epithelial
ovarian cancer (EOC). Am J Clin Oncol. 26:358–362. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Martin M, Lluch A, Casado A, et al:
Clinical activity of chronic oral etoposide in previously treated
metastatic breast cancer. J Clin Oncol. 12:986–991. 1994.PubMed/NCBI
|
|
15.
|
Kebudi R, Gorgun O and Ayan I: Oral
etoposide for recurrent/progressive sarcomas of childhood. Pediatr
Blood Cancer. 42:320–324. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Cavalli F, Sonntag RW, Jungi F, Senn HJ
and Brunner KW: VP-16-213 monotherapy for remission induction of
small cell lung cancer: a randomized trial using three dosage
schedules. Cancer Treat Rep. 62:473–475. 1978.PubMed/NCBI
|
|
17.
|
Bontenbal M, Planting AS, Verweij J, et
al: Second-line chemotherapy with long-term low-dose oral etoposide
in patients with advanced breast cancer. Breast Cancer Res Treat.
34:185–189. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Folkman J: Tumor angiogenesis. Cancer
Medicine. Holland JF, Frei EI, Bast RCJ, Kufe DW, Pollock RE and
Weichselbaum RR: 5th edition. BC Decker Inc; Ontario: pp. 132–152.
2000
|
|
19.
|
Ma G, Masuzawa M, Hamada Y, et al:
Treatment of murine angiosarcoma with etoposide, TNP-470 and
prednisolone. J Dermatol Sci. 24:126–133. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Drevs J, Fakler J, Eisele S, et al:
Antiangiogenic potency of various chemotherapeutic drugs for
metronomic chemotherapy. Anticancer Res. 24:1759–1763.
2004.PubMed/NCBI
|
|
21.
|
O'Reilly MS, Holmgren L, Shing Y, et al:
Angiostatin: a novel angiogenesis inhibitor that mediates the
suppression of metastases by a Lewis lung carcinoma. Cell.
79:315–328. 1994.PubMed/NCBI
|
|
22.
|
Panigrahy D, Singer S, Shen LQ, et al:
PPARγ ligands inhibit primary tumor growth and metastasis by
inhibiting angiogenesis. J Clin Invest. 110:923–932. 2002.
|
|
23.
|
Kamiyama H, Takano S, Tsuboi K and
Matsumura A: Anti-angiogenic effects of SN38 (active metabolite of
irinotecan): inhibition of hypoxia-inducible factor 1 alpha
(HIF-1alpha)/vascular endothelial growth factor (VEGF) expression
of glioma and growth of endothelial cells. J Cancer Res Clin Oncol.
131:205–213. 2005. View Article : Google Scholar
|
|
24.
|
Dvorak HF: Vascular permeability
factor/vascular endothelial growth factor: a critical cytokine in
tumor angiogenesis and a potential target for diagnosis and
therapy. J Clin Oncol. 20:4368–4380. 2002. View Article : Google Scholar
|
|
25.
|
Hong SY, Lee MH, Kim KS, et al:
Adeno-associated virus mediated endostatin gene therapy in
combination with topoisomerase inhibitor effectively controls liver
tumor in mouse model. World J Gastroenterol. 10:1191–1197.
2004.
|
|
26.
|
Sant VP, Nagarsenker MS, Rao SG and Gude
RP: Sterically stabilized etoposide liposomes: evaluation of
antimetastatic activity and its potentiation by combination with
sterically stabilized pentoxifylline liposomes in mice. Cancer
Biother Radiopharm. 18:811–817. 2003. View Article : Google Scholar
|
|
27.
|
Masferrer JL, Leahy KM, Koki A, et al:
Antiangiogenic and antitumor activities of cyclooxgenase-2
inhibitors. Cancer Res. 60:1306–1311. 2000.PubMed/NCBI
|
|
28.
|
Kerbel RS: Antiangiogenic therapy: a
universal chemosensitization strategy for cancer? Science.
312:1171–1175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Shaked Y, Emmenegger U, Francia G, et al:
Low-dose metronomic combined with intermittent bolus-dose
cyclophosphamide is an effective long-term chemotherapy treatment
strategy. Cancer Res. 65:7045–7051. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Bocci G, Nicolaou KC and Kerbel RS:
Protracted low-dose effects on human endothelial cell proliferation
and survival in vitro reveal a selective antiangiogenic window for
various chemotherapeutic drugs. Cancer Res. 62:6938–6943. 2002.
|
|
31.
|
Hamano Y, Sugimoto H, Soubasakos MA, et
al: Thrombospondin-1 associated with tumor microenvironment
contributes to low-dose cyclophosphamide-mediated endothelial cell
apoptosis and tumor growth suppression. Cancer Res. 64:1570–1574.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Needle MN, Molloy PT, Geyer JR, et al:
Phase II study of daily oral etoposide in children with recurrent
brain tumors and other solid tumors. Med Pediatr Oncol. 29:28–32.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Doll DC, Kasper LM, Taetle R and List AF:
Treatment with low-dose oral etoposide in patients with
myelodysplastic syndromes. Leuk Res. 22:7–12. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Hainsworth JD: Extended-schedule oral
etoposide in selected neoplasms and overview of administration and
scheduling issues. Drugs. 58(Suppl 3): 51–56. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Zucchetti M, Pagani O, Torri V, et al:
Clinical pharmacology of chronic oral etoposide in patients with
small cell and non-small cell lung cancer. Clin Cancer Res.
1:1517–1524. 1995.PubMed/NCBI
|
|
36.
|
Lowis SP, Newell DR and Pearson AD:
Exposure and schedule dependency of etoposide in neuroblastoma and
leukaemia cells in vitro. Eur J Cancer. 31A:622–626. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Dombernowsky P and Nissen NI: Schedule
dependency of the antileukemic activity of the
podophyllotoxin-derivative VP 16-213 (NSC-141540) in L1210
leukemia. Acta Pathol Microbiol Scand [A]. 81:715–724.
1973.PubMed/NCBI
|
|
38.
|
Schirner M, Hoffmann J, Menrad A and
Schneider MR: Antiangiogenic chemotherapeutic agents:
characterization in comparison to their tumor growth inhibition in
human renal cell carcinoma models. Clin Cancer Res. 4:1331–1336.
1998.
|
|
39.
|
Nilsson UW and Dabrosin C: Estradiol and
tamoxifen regulate endostatin generation via matrix
metalloproteinase activity in breast cancer in vivo. Cancer Res.
66:4789–4794. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Ma L, del Soldato P and Wallace JL:
Divergent effects of new cyclooxygenase inhibitors on gastric ulcer
healing: shifting the angiogenic balance. Proc Natl Acad Sci USA.
99:13243–13247. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Nagashima M, Asano G and Yoshino S:
Imbalance in production between vascular endothelial growth factor
and endostatin in patients with rheumatoid arthritis. J Rheumatol.
27:2339–2342. 2000.PubMed/NCBI
|
|
42.
|
Yu JL, Rak JW, Coomber BL, Hicklin DJ and
Kerbel RS: Effect of p53 status on tumor response to antiangiogenic
therapy. Science. 295:1526–1528. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Yap R, Veliceasa D, Emmenegger U, et al:
Metronomic low-dose chemotherapy boosts CD95-dependent
antiangiogenic effect of the thrombospondin peptide ABT-510: a
complementation anti-angiogenic strategy. Clin Cancer Res.
11:6678–6685. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Girnun GD, Naseri E, Vafai SB, et al:
Synergy between PPARgamma ligands and platinum-based drugs in
cancer. Cancer Cell. 11:395–406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Khafif A, Canfield VA, Syzek EJ and Medina
JE: Results of phase I–II trial of concomitant hyperfractionated
radiation and oral etoposide (VP-16) in patients with unresectable
squamous cell carcinoma of the head and neck. Am J Otolaryngol.
24:1–5. 2003.
|
|
46.
|
Vaishampayan U, Fontana J, Du W and
Hussain M: Phase II trial of estramustine and etoposide in
androgen-sensitive metastatic prostate carcinoma. Am J Clin Oncol.
27:550–554. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Bocci G, Tuccori M, Emmenegger U, et al:
Cyclophosphamidemethotrexate ‘metronomic’ chemotherapy for the
palliative treatment of metastatic breast cancer. A comparative
pharmacoeconomic evaluation. Ann Oncol. 16:1243–1252. 2005.
|
|
48.
|
Vanchieri C: When will the U.S. flinch at
cancer drug prices? J Natl Cancer Inst. 97:624–626. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Emmenegger U, Morton GC, Francia G, et al:
Low-dose metronomic daily cyclophosphamide and weekly tirapazamine:
a well-tolerated combination regimen with enhanced efficacy that
exploits tumor hypoxia. Cancer Res. 66:1664–1674. 2006. View Article : Google Scholar
|
|
50.
|
Verdrengh M and Tarkowski A: Impact of
topoisomerase II inhibition on cytokine and chemokine production.
Inflamm Res. 52:148–153. 2003. View Article : Google Scholar : PubMed/NCBI
|