Introduction
The prevalence of metabolic syndrome associated with
visceral obesity is increasing worldwide, resulting in a high risk
of developing complications, such as diabetes and cardiovascular
diseases. These pathologies are pathophysiologically associated
with atherosclerosis and non-alcoholic fatty liver disease (NAFLD),
which exhibit common features such as lipid accumulation and
inflammation (1,2). These features develop silently over
several years, and are the most common causes of cardiovascular and
chronic liver diseases.
Atherosclerosis and NAFLD progression are both
accompanied by inflammation development which involves several
factors, including various chemokines (monocyte chemoattractant
protein 1; MCP-1), cytokines (tumor necrosis factor-α; TNF-α),
interleukin-6 (IL-6) and C-reactive protein (CRP) (3–6).
NAFLD and atherosclerosis are now recognized as two aspects of a
shared disease, involving the local presence of activated
macrophages (7). Macrophages are
able to accumulate large amounts of lipids, transform into foam
cells and drive atherogenesis (8,9). The
same process of accumulation of lipid-loaded macrophages was
observed in NAFLD, where hyperlipidemic mice demonstrated bloated
foamy Kupffer cells (KCs) upon high-fat feeding (10). Consequently, drugs targeting lipid
homeostasis and inflammation may have great potential to improve
NAFLD and reduce atherosclerosis development.
Peroxisome proliferator-activated receptors (PPARs)
are nuclear receptors that control the expression of genes involved
in both carbohydrate and lipid metabolism, and could be valuable as
additional drug targets (11).
PPARα regulates the expression of genes encoding proteins that are
involved in lipid metabolism, fatty acid oxidation and glucose
homeostasis, thereby improving markers for atherosclerosis and
insulin resistance. In addition, PPARα exerts anti-inflammatory
effects both in the vascular wall and the liver (12). PPARγ agonists improve insulin
sensitivity and induce glycaemic control in diabetic animals as
well as in patients with type 2 diabetes (13). Thus, compounds that stimulate both
PPARα and PPARγ (dual PPARα/γ agonists) should additively improve
both lipid and glucose abnormalities in animal models and human
subjects with insulin resistance (IR) and/or type 2 diabetes
(T2DM). However, the effects of dual PPARα/γ agonists on NAFLD have
not been thoroughly studied.
The aim of the present study was to determine
whether dual PPARα/γ agonist tesaglitazar attenuates NAFLD and
atherosclerosis development in diabetic low-density lipoprotein
receptor-deficient (LDLr−/−) mice. Female
LDLr−/− mice were induced by a high-fat diet (HFD)
combined with a low-dose STZ injection to develop an animal model
of T2DM, and to induce NAFLD and atherosclerotic lesions.
Materials and methods
Generation of the diabetic model and
treatment
All animal procedures were performed in compliance
with ‘The Guide for the Care of Use of Laboratory Animals’
published by the National Institute of Health (NIH Publication No.
85-23, revised 1996) and approved by the Animal Care Committee of
Shanghai Tenth People’s Hospital, Tongji University School of
Medicine. The mice were housed five per cage and allowed access to
the appropriate diet and autoclaved water.
Three-week-old female LDLr−/− mice with a
C57BL/6 genetic background were created by homologous recombination
(Jackson Laboratory, Bar Harbor, ME, USA) and were fed an HFD (20%
fat, 20% sugar and 1.25% cholesterol). After 6 weeks, the
LDLr−/− mice underwent an intraperitoneal glucose
tolerance test (IPGTT). Mice exhibiting IR were injected once with
low-dose streptozotocin (STZ, 75–80 mg/kg i.p.) to induce partial
insulin deficiency. Two weeks after the STZ injection, most
HFD/STZ-treated mice displayed hyperglycemia, IR and glucose
intolerance, as previously reported (14). At age 11 weeks, animals with
similar degrees of hyperglycemia and body weight were randomly
divided into untreated (DM, n=15) and tesaglitazar-treated
(DM+tesaglitazar, n=15) groups. The mice fed an HFD were used as a
nondiabetic control group (n=15). For tesaglitazar treatment, the
mice received a daily oral dose of tesaglitazar (20 μg/kg/day).
These drugs were dissolved in water and administered by oral
gavage. The dose of drug was calculated based on previous studies
(15,16). After 6 weeks of treatment, the mice
were sacrificed under isoflurane anesthesia, and tissues were
removed and frozen immediately in liquid nitrogen or fixed with
paraffin. Blood samples were collected from the mice for
measurement of serum parameters.
Biochemical studies
Blood samples were collected from mice at the
beginning of the experiment, before pharmacological triggering and
at the end of the experiment. Fasting serum glucose, total
cholesterol (TC), triglyceride (TG) and high-density
lipoprotein-cholesterol (HDL-C) were measured using commercial kits
(Sigma, St. Louis, MO, USA). Serum MCP-1, TNF-α, IL-6 and CRP were
measured using commercial enzyme-linked immunosorbent assay (ELISA)
kits purchased from R&D Systems, Inc. (Minneapolis, MN,
USA).
Analysis of atherosclerotic lesions
After sacrifice by cervical dislocation, hearts were
fixed with 4% phosphate-buffered paraformaldehyde (PAF), and 10-μm
aortic sinus sections were cut followed by quantitative analysis of
lipid deposition by Oil Red O staining. Aortic sinuses were also
stained with rat monoclonal anti-mouse macrophage Mac3 (1:100, BD
Biosciences, Heidelberg, Germany), followed by detection with
biotinylated secondary antibody and streptavidin-horseradish
peroxidase. Images were captured using a JVC 3-charge-coupled
device video camera (Sony, Tokyo, Japan) and analyzed using the
computer-assisted Quips Image analysis system (Leica Microsystems
GmbH, Germany).
Histological analysis of the liver
At sacrifice, livers were perfused with phosphate
buffered saline (PBS) solution via the portal vein. After removal
of the liver, a section of approximately 4 mm2 was fixed
in PAF and embedded in paraffin. Paraffin-embedded sections (5 μm)
were stained with hematoxylin and eosin (H&E) to evaluate
steatosis and inflammation. Frozen-liver sections (7 μm) were
stained with rat monoclonal anti-mouse macrophage F4/80 (1:500,
Serotec, Oxford, UK), followed by detection with biotinylated
secondary antibody and streptavidin-horseradish peroxidase, to
evaluate macrophage infiltration. Analysis of lipid deposition was
performed by Oil Red O staining of 7-μm frozen-liver sections, with
H&E staining for visualization of the nuclei.
Hepatic lipid analysis
Frozen liver tissue (50 mg) was homogenized in SET
buffer (1 ml; sucrose 250 mM, EDTA 2 mM and Tris 10 mM), followed
by two freeze-thaw cycles, three cyles through a 27-gauge syringe
needle and a final freeze-thaw cycle. TG and TC were measured as
described above.
Real-time quantitative reverse
transcription-polymerase chain reaction
Total RNA was isolated from mouse livers using the
acid guanidinium thiocyanate/phenol/chloroform method. RNA was
reverse-transcribed using Moloney murine leukemia virus reverse
transcriptase and random hexamer primers (Invitrogen, Carlsbad, CA,
USA). mRNA levels were quantified by real-time quantitative PCR on
a MX-3000 apparatus (Agilent) using the Brilliant SYBR Green QPCR
master mix (Qiagen, Shanghai, China). The following primers were
constructed: TNF-α, 5′CGGAGTCCGGGCAGGT3′ (forward) and
5′GCTGGGTAGAGAATGGATGAACA3′ (reverse); MCP-1,
5′CAGCCAGATGCAGTTAACGC3′ (forward) and 5′GCCTACTCATTGGGATCATCTTG3′
(reverse); IL-6, 5′TGAGGAGACTTGCCTG3′ (forward) and
5′ACAACAACAATCTGAGGTGCC3′ (reverse). Individual gene expression was
normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
expression. All quantifications were performed in triplicate
samples for three separate experiments.
Statistical analysis
Values are presented as means ± SD unless indicated
otherwise. All data analysis was performed with the use of GraphPad
PRISM statistical software 5.0 (San Diego, CA, USA). The
statistical significance of differences between groups was
determined by one-way ANOVA followed by the Dunnett’s multiple
comparison test, or Student’s t-test when comparisons were made
between 2 groups. P<0.05 was considered to indicate a
statistically significant result.
Results
Effect of tesaglitazar on metabolic
parameter change
As shown in Table
I, at the beginning of the experiment, there was no significant
differences in serum TC, TG and glucose levels, and body weight
among the three groups of mice. Prior to pharmacological
triggering, all diabetic mice had higher serum TC and TG and
glucose levels than levels in the control group (P<0.001). The
mean body weight was significantly higher for diabetic mice than
HFD-fed mice at the age of 17 weeks (P<0.01). After 6 weeks of
oral tesaglitazar treatment, serum TC, TG and glucose levels
decreased in the diabetic mice (P<0.001). In addition,
tesaglitazar treatment led to a significant increase in serum HDL
levels in the diabetic mice (P<0.001). However, tesaglitazar
treatment caused an increase in body weight (P<0.01).
 | Table IAverage weights, cholesterol,
triglyceride and glucose levels (n=15, mean ± SD). |
Table I
Average weights, cholesterol,
triglyceride and glucose levels (n=15, mean ± SD).
| Group | Body weight (g) | Glu (mg/dl) | TC (mg/dl) | TG (mg/dl) | HDL (mg/dl) |
|---|
| Baseline (at age 3
weeks) | | | | | |
| Control | 22.3±1.4 | 118.7±17 | 487.4±82 | 175.6±34 | |
| Diabetic | 22.1±1.1 | 114.1±15 | 483.1±90 | 173.2±27 | |
| Before intervention
(at age 11 weeks) | | | | | |
| Control | 30.5±1.6 | 124.1±15 | 1025.6±74 | 395.6±10 | |
| Diabetic | 30.9±1.2 | 273.7±84b | 1351.3±126b | 816.0±88b | |
| Tesaglitazar | 30.7±1.4 | 272.5±79 | 1354.7±123 | 814.2±85 | |
| After intervention
(at age 17 weeks) | | | | | |
| Control | 39.6±1.2 | 130.5±17 | 1323.0±65 | 624.0±30 | 117.2±8 |
| Diabetic | 41.2±1.7a | 300.5±80b | 2114.3±118b | 1113.2±58b | 92.3±6b |
| Tesaglitazar | 43.5±2.1c | 152.6±23d | 1478.7±123d | 715.6±33d | 105.7±3d |
Effect of tesaglitazar on serum
inflammatory markers
All diabetic mice had higher serum MCP-1, TNF-α,
IL-6 and CRP levels than the control group (all P<0.001). As
shown in Table II, after 6 weeks
of oral tesaglitazar treatment, serum inflammatory marker levels
were significantly reduced in diabetic mice (P<0.001, P<0.01,
P<0.05).
| Table IISerum inflammatory markers as measured
by enzyme-linked immunosorbent assay in mice of the three groups
after 6-week intervention (n=15, mean ± SD). |
Table II
Serum inflammatory markers as measured
by enzyme-linked immunosorbent assay in mice of the three groups
after 6-week intervention (n=15, mean ± SD).
| Groups | MCP-1 (pg/ml) | TNF-α (ng/ml) | IL-6 (pg/ml) | CRP (μg/ml) |
|---|
| Control | 87.2±12.3 | 2.1±0.6 | 129.6±14.5 | 2.1±0.3 |
| Diabetic | 122.4±23.1b | 3.4±0.2b | 205.2±23.7b | 3.4±0.2b |
| Tesaglitazar | 102.7±19.6c | 2.7±0.4e | 174.3±26.1d | 2.0±0.4e |
Effect of tesaglitazar on atherosclerotic
lesions
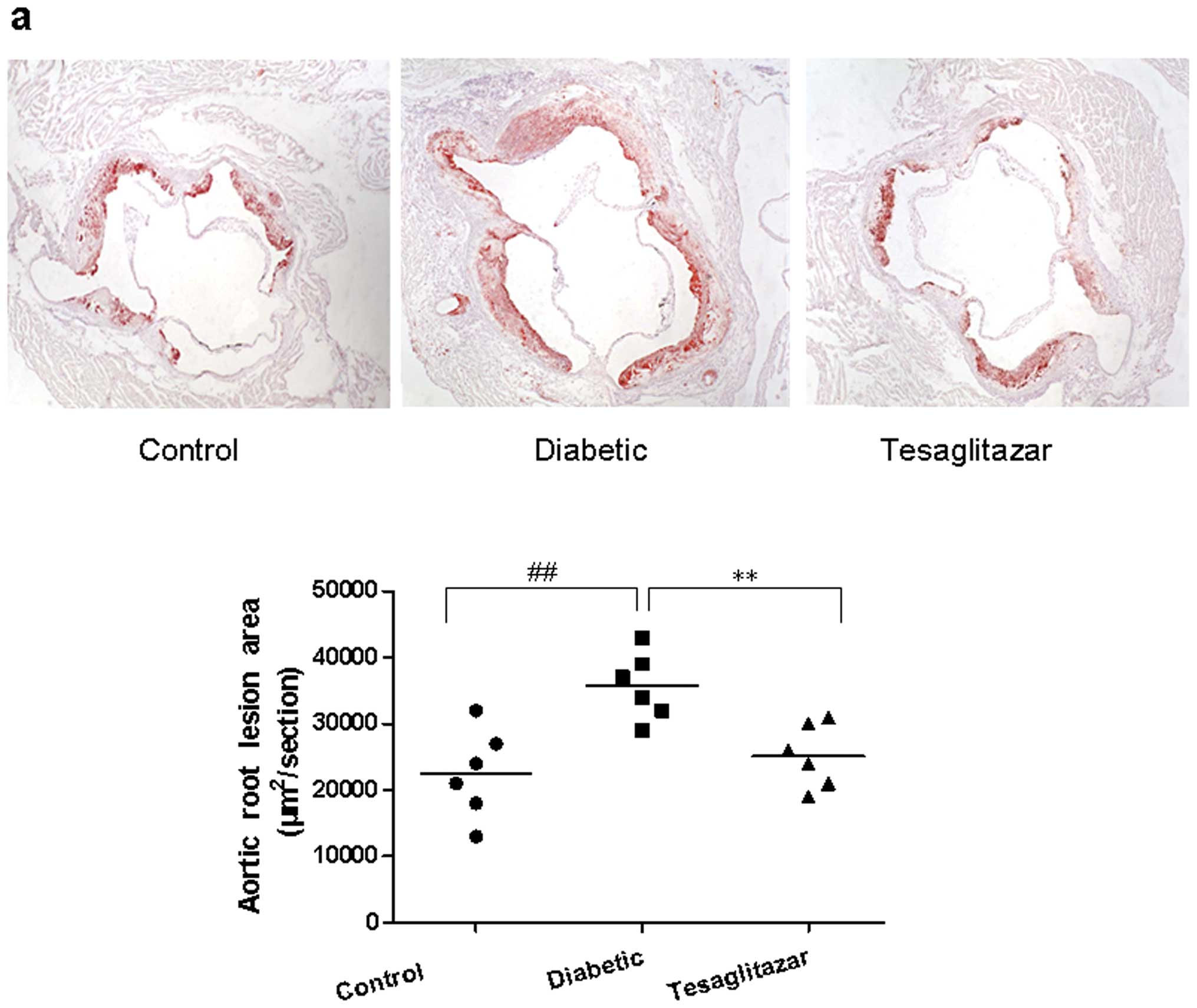
Oil-red O staining of atherosclerotic plaques was
red and widely observed in the diabetic mice. Compared with the
size of plaque in the control mice, plaques were increased in the
diabetic mice (P<0.01) but significantly diminished in the
tesaglitazar-treated diabetic mice (P<0.01; Fig. 1a). Macrophage expression in the
atherosclerotic plaques was identified by immunohistochemical
staining of Mac3. The Mac3-positive area was larger in the diabetic
mice group compared with the tesaglitazar-treated diabetic mice
(P<0.01) and control mice groups (P<0.001; Fig. 1b).
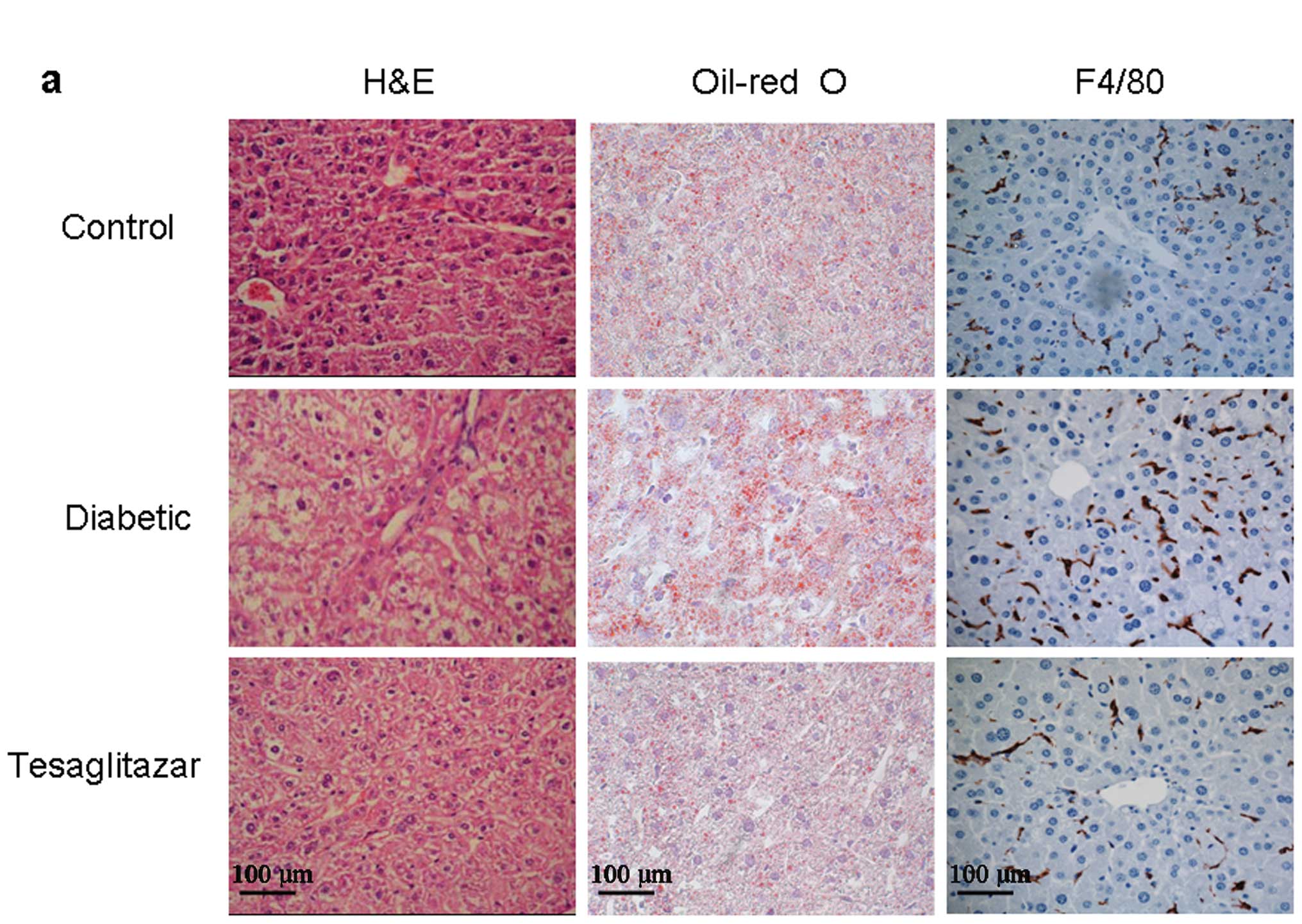
Effect of tesaglitazar on NAFLD
To investigate the inhibitory effect of tesaglitazar
on NAFLD, we analyzed the histology of liver tissue using H&E
staining, Oil-red O staining and F4/80 immunohistochemistry. As
shown in Fig. 2a and b, marked
microvesicular steatosis accompanied by a partial mild inflammation
was observed in diabetic mice. However, the degree of hepatic fat
accumulation was substantially alleviated by tesaglitazar treatment
and the number of infiltrating macrophages (F4/80-positive cells)
was significantly reduced in the tesaglitazar-treated diabetic mice
group (P<0.01). In addition, tesaglitazar treatment reduced
total hepatic cholesterol and triglyceride content (both P<0.01;
Fig. 2c).
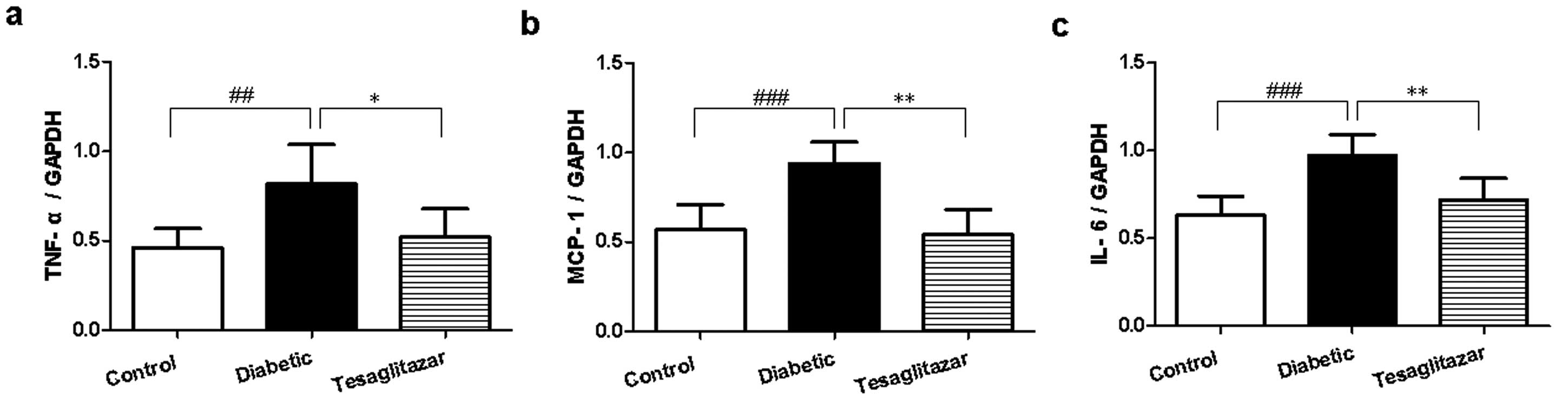
Effect of tesaglitazar on inflammatory
genes in the liver
In comparison with control mice, TNF-α, MCP-1 and
IL-6 mRNA expression was significantly increased in the liver of
diabetic mice (P<0.01, P<0.001, P<0.001), but this was
reduced following 6-week treatment with tesaglitazar (P<0.01,
P<0.05; Fig. 3).
Discussion
Ubiquitously expressed, dual PPARα/γ agonists have
been implicated in lipid metabolism and energy homeostasis.
Previous studies have established that tesaglitazar treatment
reduces atherosclerosis in a mouse model of hyperlipidemia by
reducing both lipid content and inflammation in the aorta (15,16),
yet the details regarding the underlying mechanism of tesaglitazar
in NAFLD remain less clear.
To address this issue, we examined the effect of
tesaglitazar on HFD-induced hepatic steatosis in diabetic
LDLr−/− mice. According to our data, lipid accumulation
was significantly reduced by treatment with tesaglitazar when
compared with that of the diabetic mice. These data indicate the
potential of tesaglitazar as a therapeutic agent for HFD-induced
fatty liver.
In the present study, the effects of tesaglitazar on
serum lipid concentrations were similar to those in previous
studies (15,16), namely TC and TG reduction, and
HDL-C increase. Tesaglitazar improved serum glucose disorders
observed in this animal model.
NAFLD and atherosclerosis are characterized by lipid
accumulation and chronic inflammation (17,18).
Numerous studies have reported NAFLD as an important cardiovascular
risk factor (19,20), Kleemann et al demonstrated
that after 10 weeks of a high cholesterol diet (1% cholesterol),
the lesion area in female APOE*3-Leiden (E3L) mice was correlated
with plasma levels of serum amyloid A protein (SAA), suggesting
that the hepatic inflammatory response is involved in the formation
of early atherosclerotic lesions (21). Due to the prominent role of the
liver in the uptake of lipids, the hepatic inflammatory response in
hyperlipidemic models precedes plaque formation. Increasing
evidence suggests a shared metabolic and inflammatory factors
associated with macrophage activation in atherosclerosis and NAFLD
(22,23) and thereby supports the proposal
that NAFLD and atherosclerosis are actually two aspects of the same
disease.
As inflammation plays a pivotal role in both
atherosclerosis and NAFLD, an important pharmacological objective
in both disorders is to target inflammatory activation directly.
The role of PPARs as anti-inflammatory mediators has been
well-established. In the present study, we demonstrated that dual
PPARα/γ agonist tesaglitazar inhibited macrophage infiltration both
in the aortic root and in the liver. Our data also demonstrated
that treatment with tesaglitazar decreased serum concentrations of
MCP-1, TNF-α, IL-6 and CRP. Tesaglitazar also reduced the mRNA
levels of TNF-α, MCP-1 and IL-6 in the liver of diabetic mice.
These inflammatory markers were previously shown to be involved in
the inflammatory cascade, for example, IL-6 is a multifunctional
cytokine that plays a critical role in the acute-phase inflammatory
response and its expression is known to be stimulated upon
TNF-α-mediated activation of NF-κB and promotes the expression of
intercellular adhesion molecule 1 (ICAM-1) and CRP synthesis. IL-6
has also been shown to stimulate macrophages to secrete MCP-1
(24).
Due to the complexity of the mechanisms that cause
tissue lipid accumulation, we were unable to identify a single
major factor or mechanism responsible for the observed inhibitory
effect of tesaglitazar on hepatic lipid accumulation in our
experiment. There are numerous possible mechanisms that may explain
the observed effect of tesaglitazar in the present study and such
mechanisms include food intake, energy expenditure, or fat
accumulation and expression of fatty acid oxidation enzymes in
other tissues. We could not test all the hypotheses and that was
the major limitation of the present study. However, our findings
suggest that dual PPARα/γ agonist tesaglitazar inhibits the
development of NAFLD and atherosclerosis in a diabetic mouse model
by regulating glucose and lipid metabolism, and the inflammatory
response. Additional studies are required to clarify the mechanisms
by which tesaglitazar induces these effects in vitro.
Acknowledgements
This research was supported by the
National Natural Science Foundation of China (Grant No.
81200198).
References
|
1
|
Menghini R, Casagrande V, Menini S, et al:
TIMP3 overexpression in macrophages protects from insulin
resistance, adipose inflammation, and nonalcoholic fatty liver
disease in mice. Diabetes. 61:454–462. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scorletti E, Calder PC, Byrne CD, et al:
Non-alcoholic fatty liver disease and cardiovascular risk:
metabolic aspects and novel treatments. Endocrine. 40:332–343.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baeck C, Wehr A, Karlmark KR, et al:
Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes
liver macrophage infiltration and steatohepatitis in chronic
hepatic injury. Gut. 61:416–426. 2012. View Article : Google Scholar
|
|
4
|
Serino M, Menghini R, Fiorentino L, et al:
Mice heterozygous for tumor necrosis factor-alpha converting enzyme
are protected from obesity-induced insulin resistance and diabetes.
Diabetes. 56:2541–2546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wunderlich FT, Ströhle P, Könner AC, et
al: Interleukin-6 signaling in liver-parenchymal cells suppresses
hepatic inflammation and improves systemic insulin action. Cell
Metab. 12:237–249. 2010. View Article : Google Scholar
|
|
6
|
Hanley AJ, Williams K, Festa A, et al:
Liver markers and development of the metabolic syndrome: the
insulin resistance atherosclerosis study. Diabetes. 54:3140–3147.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bieghs V, Rensen PC, Hofker MH and
Shiri-Sverdlov R: NASH and atherosclerosis are two aspects of a
shared disease: central role for macrophages. Atherosclerosis.
220:287–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gui T, Shimokado A, Sun Y, et al: Diverse
roles of macrophages in atherosclerosis: from inflammatory biology
to biomarker discovery. Mediators Inflamm.
2012:6930832012.PubMed/NCBI
|
|
9
|
Truman JP, Al Gadban MM, Smith KJ, et al:
Differential regulation of acid sphingomyelinase in macrophages
stimulated with oxidized low-density lipoprotein (LDL) and oxidized
LDL immune complexes: role in phagocytosis and cytokine release.
Immunology. 136:30–45. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wouters K, van Gorp PJ, Bieghs V, et al:
Dietary cholesterol, rather than liver steatosis, leads to hepatic
inflammation in hyperlipidemic mouse models of nonalcoholic
steatohepatitis. Hepatology. 48:474–486. 2008. View Article : Google Scholar
|
|
11
|
Evans JL, Lin JJ and Goldfine ID: Novel
approach to treat insulin resistance, type 2 diabetes, and the
metabolic syndrome: simultaneous activation of PPARalpha,
PPARgamma, and PPARdelta. Curr Diabetes Rev. 1:299–307. 2005.
View Article : Google Scholar
|
|
12
|
Zandbergen F and Plutzky J: PPARalpha in
atherosclerosis and inflammation. Biochim Biophys Acta.
1771:972–982. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ciudin A, Hernandez C and Simó R: Update
on cardiovascular safety of PPARgamma agonists and relevance to
medicinal chemistry and clinical pharmacology. Curr Top Med Chem.
12:585–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mu J, Woods J, Zhou YP, et al: Chronic
inhibition of dipeptidyl peptidase-4 with a sitagliptin analog
preserves pancreatic beta-cell mass and function in a rodent model
of type 2 diabetes. Diabetes. 55:1695–1704. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zadelaar AS, Boesten LS, Jukema JW, et al:
Dual PPARalpha/gamma agonist tesaglitazar reduces atherosclerosis
in insulin-resistant and hypercholesterolemic ApoE*3Leiden mice.
Arterioscler Thromb Vasc Biol. 26:2560–2566. 2006.PubMed/NCBI
|
|
16
|
Chira EC, McMillen TS, Wang S, et al:
Tesaglitazar, a dual peroxisome proliferator-activated receptor
alpha/gamma agonist, reduces atherosclerosis in female low density
lipoprotein receptor deficient mice. Atherosclerosis. 195:100–109.
2007. View Article : Google Scholar
|
|
17
|
Ma KL, Ruan XZ, Powis SH, et al:
Inflammatory stress exacerbates lipid accumulation in hepatic cells
and fatty livers of apolipoprotein E knockout mice. Hepatology.
48:770–781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Woollard KJ and Geissmann F: Monocytes in
atherosclerosis: subsets and functions. Nat Rev Cardiol. 7:77–86.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Targher G, Marra F and Marchesini G:
Increased risk of cardiovascular disease in non-alcoholic fatty
liver disease: causal effect or epiphenomenon? Diabetologia.
51:1947–1953. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bhatia LS, Curzen NP, Calder PC and Byrne
CD: Non-alcoholic fatty liver disease: a new and important
cardiovascular risk factor? Eur Heart J. 33:1190–1200. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kleemann R, Verschuren L, van Erk MJ, et
al: Atherosclerosis and liver inflammation induced by increased
dietary cholesterol intake: a combined transcriptomics and
metabolomics analysis. Genome Biol. 8:R2002007. View Article : Google Scholar
|
|
22
|
Maina V, Sutti S, Locatelli I, et al: Bias
in macrophage activation pattern influences non-alcoholic
steatohepatitis (NASH) in mice. Clin Sci (Lond). 122:545–553. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lingrel JB, Pilcher-Roberts R, Basford JE,
et al: Myeloid-specific Krüppel-like factor 2 inactivation
increases macrophage and neutrophil adhesion and promotes
atherosclerosis. Circ Res. 110:1294–1302. 2012.
|
|
24
|
Amar J, Fauvel J, Drouet L, et al:
Interleukin 6 is associated with subclinical atherosclerosis: a
link with soluble intercellular adhesion molecule 1. J Hypertens.
24:1083–1088. 2006. View Article : Google Scholar : PubMed/NCBI
|