Introduction
Craniocerebral injury is a common serious problem in
neurosurgery, with extremely high mortality and disability rates.
In craniocerebral injury, traumatic lesions show hemorrhagic
necrosis and surrounding nerve cells exhibit apoptosis, when
mediators of inflammation are released. Inflammatory cell
infiltration and brain edema are actively involved in the
pathophysiological mechanism, and the protection of the nerve cells
still in a reversible state around traumatic lesions has become a
focus of research in recent years.
Erythropoietin (EPO) has been shown to promote
neuro-protection in hypoxic-ischemic cerebral insults, through the
regulation of neurogenesis and preventing neuronal apoptosis
(1–3). EPO, a vital compound of erythroid
differentiation, is also involved in non-hematopoietic
tissue-protective pathways, demonstrating antiapoptotic,
anti-inflammatory, angiogenetic and neurotrophic properties
(4). Hypoxia has been proven to
increase EPO production in kidneys, the brain, the testis, the
liver and the spleen (5–7). In the brain, EPO is highly expressed
after a neuropathological insult (5). Astrocytes produce EPO after its
hypoxia-induced upregulation, while neurons express EPO receptors
(2,7). EPO has also been demonstrated to
promote neuroprotection after systemic administration, even in
severe cerebral ischemia (8), to
enhance neurological recovery in traumatic brain and spinal cord
injury (5,9–12),
and to prevent the loss of autoregulation of cerebral blood flow
(13). The neuroprotection and
restructuring of cerebral tissue after a neuropathological trauma
is likely to result in the use of EPO in clinical practice to limit
neuronal damage (14).
This study was aimed to investigate the impact of
EPO on nerve cell apoptosis in rats subsequent to experimental
brain injury, the release of inflammatory mediators, inflammatory
cell infiltration and brain edema.
Materials and methods
Experimental materials
The apoptosis detection kit, as well as the monocyte
chemotactic protein-1 (MCP-1) monoclonal and CD68 polyclonal
antibodies were purchased from Crystal US Biological (Downers
Grove, MO, USA), the erythropoietin was purchased from Kirin Co.,
Ltd (Tokyo, Japan), while the head blow device was constructed
in-house.
Animal grouping
Ninety healthy male SD rats (provided by the Animal
Center of the Soochow University School of Medicine), weighing
250–300 g, were randomized into a sham operation group, a control
group and an EPO group (n=10/group). Five rats of each group were
used to measure the number of apoptotic, MCP-1+ and
CD68+ cells, while the remaining 5 were measured for
brain water content.
Production of head blow device
The improved Feeney’s method was employed (15) while following the free-falling
principle to produce a head blow device, comprising a collision
bar, a drop hammer, a peripheral casing and fixed devices. The head
end of the collision bar had a diameter of 4.5 mm and a height of 3
mm, while the drop hammer weight was 20 g. The peripheral casing
had a height of 30 cm, and the hit height could be randomly
adjusted.
Animal models
The improved method of Feeney et al (15) was employed to produce a
free-falling model of brain contusion. The anesthetized rats were
injected in the stomach using 1% pentobarbital at the proportion of
30 mg/kg, their heads were secured in a prone position and a
sagittal scalp incision was made to expose the left parietal bone,
and a hole of 2 mm was placed by a drill in front of the lambdoid
suture and 2 mm to the left of the midline, the bone was expanded
by a window of 5×5 mm; the impact force of the falling body was
20×30 g/cm that caused brain injury to the left hemisphere. After
stopping the bleeding, bone wax was used to seal the bone window
and the scalp was sutured. Except for the brain contusion impact,
the same steps were repeated in the sham operation group. Having
successfully established the animal model, the EPO group was
injected with EPO 5,000 μ/kg, while the sham operation and the
control groups were injected with a normal amount of saline.
Subsequent to the operation the animals were caged individually.
The heads of the animals in each group were removed to excise the
brains 12, 48 and 120 h after the injury.
Preparation of the tissue sections
Rats in each group were anesthetized with an
overdose of 1% pentobarbital (40 mg/kg) at each time point. The
brains were excised immediately after and secured for 30 min with
4% paraformaldehyde buffer. Taking the contusion as the center,
four 5 μm-thick conventional paraffin sections were cut.
Subsequently, H&E, TUNEL and MCP-1 and CD68
immunohistochemistry stainings were conducted, respectively.
TUNEL staining
The slices were dewaxed by xylene, gradient alcohol
hydration was conducted and digestion was carried out for 20 min
with 20 μg/ml proteinase K, using 0.3% hydrogen peroxide and
methanol to block endogenous peroxidase for 30 min. Then the
sections were washed for 5 min (4°C) with 0.1% Triton X-100 and
sodium citrate buffer liquid. Subsequently, 50 μl TUNEL reactive
mixture was dripped onto the sections and they were incubated for 1
h at 37°C. After rinsing with phosphate-buffered saline (PBS), 50
μl converter POD was dripped onto the sections and was incubated
for 30 min at 37°C. The sections were then washed with PBS and
colored with 0.05% DAB for 15–20 min. The negative control
operations were the same as the above, with the exception of the
TUNEL reactive mixture. The apoptotic nuclei demonstrated brown
staining. Five high-magnification views (×400) from around the
contusion of each section were selected to count the number of
positive cells in each view, and the average value was
determined.
Immunohistochemistry of MCP-1 and
CD68
The paraffin-embedded brain tissue specimens were
cut into 5 μm-thick sections. The ABC method was applied and the
steps were as follows. i) Paraffin sections were prepared for brain
tissue and dewaxing; ii) they were washed in PBS twice, and placed
in 0.3% H2O2 methanol solution. The sections
were kept at room temperature for 20 min, then washed 3 times in
PBS solution to remove endogenous peroxidase activity. iii) The
slices were placed in an incubator at 37°C with 0.1% trypsin for 20
min, then washed 3 times in PBS. iv) Subsequent to adding normal
calf serum at a proportion of 1:10, the sections were kept at room
temperature for 20 min. v) Subsequent to adding the first antibody
at a working concentration, the sections were incubated at 37°C for
1 h, and washed 3 times in PBS solution. vi) After the addition of
the second biotinylated antibody at a working concentration, the
sections were incubated at 37°C for 1 h, and washed 3 times in PBS
solution. vii) Subsequent to adding ABC complex, the sections were
incubated at 37°C for 1 h, and washed 3 times in PBS solution.
viii) AB color was developed to brownish red, and viiii) after a
thorough washing in water, the sections were counterstained with
hematoxylin. x) The sections were dehydrated, made transparent and
mounted. PBS was used to replace the first antibody as a negative
control. Cells staining with a granular brown-red color in the
cytoplasm at a high magnification were considered positive cells.
The number of positive cells were then counted (5 fields of
view).
Determination of brain water content
Brain hemisphere tissue samples were taken from rats
in each group. Their wet weights was measured on electronic scales,
at room temperature (20–25°C) and in 70–90% humidity. Subsequently,
the samples were placed in an oven at 100±2°C for 24 h, and the dry
weight was measured. The water content in the brain was calculated
using the following formula: Wet weight - dry weight/wet weight x
100%.
Statistical analysis
The data are presented using the SPSS statistical
software, and the variable data were expressed as the means ± SD.
The data were analyzed by ANOVA and detection measures.
Results
H&E staining
The trauma control group exhibited unstructured
necrosis at different time points, showing edema of the hemorrhagic
focus and peripheral cells and the broadening of tissue space.
Numerous apoptotic neurons as well as the obvious infiltration of
inflammatory cells were visible in the center of the trauma and in
the peripheral edema. The center of the necrotic area and the
peripheral edema in the EPO group was smaller compared to the
control group, and was characterized by the alleviation of
peripheral edema, a decreasing number of apoptotic neurons and the
reduced infiltration of inflammatory cells. In the sham group only
a small number of apoptotic cells were detected, while inflammatory
cell infiltration was absent.
Evaluation of apoptosis by TUNEL
staining
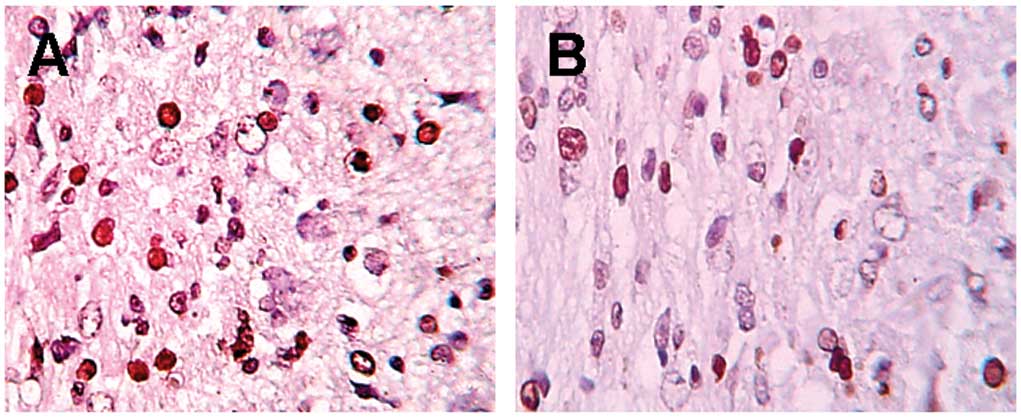
In TUNEL staining (Fig.
1), more apoptotic neurons were detected in the control group
at various time points. A clear decrease in apoptotic neurons
(P<0.01) was detected in the EPO group compared to the control
group, while only a few positive cells were detected in the sham
group (Table I).
 | Table IComparison of the apoptotic neurons in
the sham, control and EPO groups. |
Table I
Comparison of the apoptotic neurons in
the sham, control and EPO groups.
| Groups | No. of cases | 12 h | 48 h | 120 h |
|---|
| Sham | 5 | 1.56±0.68 | 1.40±0.80 | 1.68±0.82 |
| Control | 5 | 11.64±1.73 | 38.32±4.78 | 21.00±2.62 |
| EPO | 5 | 4.08±2.13a | 24.40±2.28a | 13.16±3.43a |
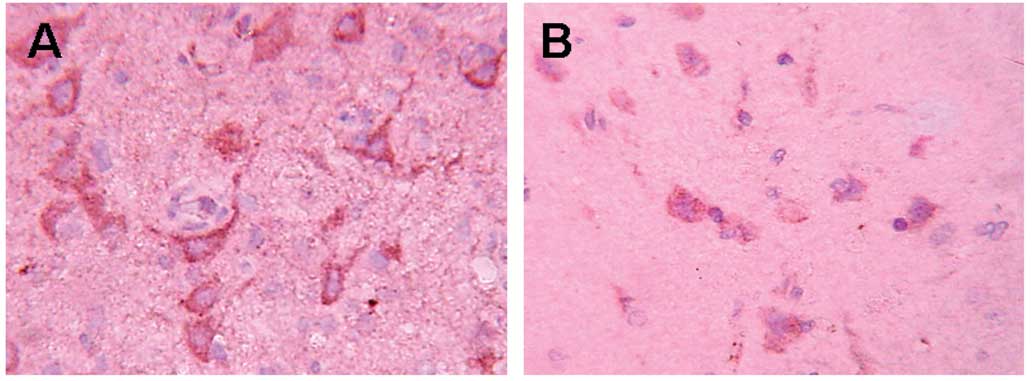
MCP-1 staining
In the control group (Fig. 2), a large number of
MCP-1+ cells were detected in the periphery of the
trauma at different time points. that was most obvious after 48 h.
In the EPO group the positive cells showed a clear decrease
(P<0.01) at different time points, compared to the control
group, while no positive cells were detected in the sham group
(Table II).
| Table IIComparison of the MCP-1+
cells in the sham, control and EPO groups. |
Table II
Comparison of the MCP-1+
cells in the sham, control and EPO groups.
| Groups | No. of cases | 12 h | 48 h | 120 h |
|---|
| Sham | 5 | - | - | - |
| Control | 5 | 14.00±2.12 | 33.20±2.71 | 20.80±2.24 |
| EPO | 5 | 4.32±1.67a | 16.04±3.62a | 10.04±0.90a |
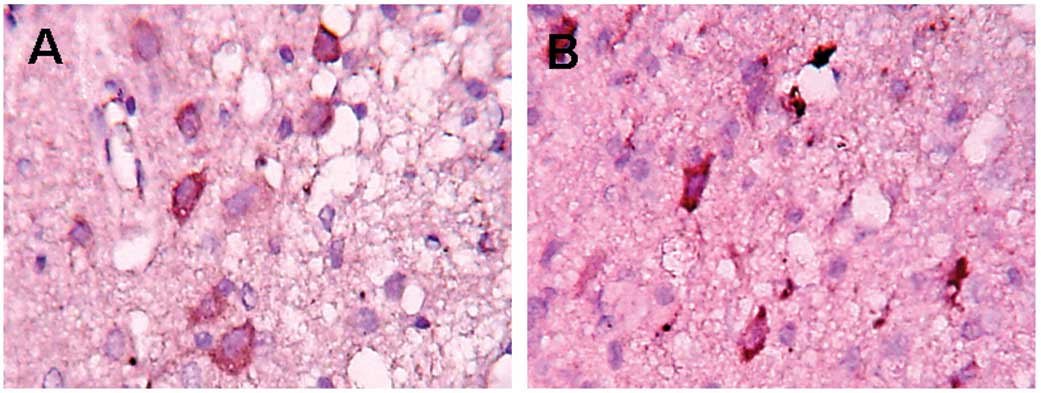
Infiltration of CD68+
cells
In the control group, the infiltration of
CD68+ cells was detected in the periphery of the trauma
12 h post-trauma. The largest number of positive cells were found
at 120 h. The positive cells in the EPO group showed a clear
decrease (P<0.05) compared to the control group (Fig. 3 and Table III).
| Table IIIComparison of the infiltration of
CD68+ cells in the sham, control and EPO groups. |
Table III
Comparison of the infiltration of
CD68+ cells in the sham, control and EPO groups.
| Groups | No. of cases | 12 h | 48 h | 120 h |
|---|
| Sham | 5 | 9.36±0.56 | 1.40±0.80 | 1.68±0.82 |
| Control | 5 | 18.68±1.96 | 26.08±3.17 | 39.12±3.70 |
| EPO | 5 | 12.60±2.75a | 16.24±3.14a | 27.12±4.78a |
Water content of brain tissue
Compared to the sham group, an increase of the water
content was observed in the brain tissue 12 h post-trauma, with the
highest content at 48 h. Cerebral edema was noted to subside after
120 h. Compared to the control group, the water content in the
brain tissue in the EPO group was the lowest (Table IV).
| Table IVComparison of the water content in
brain tissue in the sham, control and EPO groups. |
Table IV
Comparison of the water content in
brain tissue in the sham, control and EPO groups.
| Groups | No. of cases | 12 h | 48 h | 120 h |
|---|
| Sham | 5 | 77.02±0.74 | 77.08±1.23 | 77.49±0.30 |
| Control | 5 | 80.69±0.75 | 81.42±0.42 | 80.47±0.86 |
| EPO | 5 | 78.48±0.54a | 79.31±0.85a | 78.49±0.30a |
Discussion
EPO is a glycoprotein hormone, with a molecular
weight of 34 kDa. EPO existing in plasma consists of 165 amino
acids, with a high glycosylation degree containing mainly sialic
acid. It is located on human chromosome 7p22, including at least 5
introns. EPO exerts its effects by binding to its receptor EpoR,
and is produced mainly by the kidneys. In recent years, numerous
studies have shown that EPO and EpoR expression levels were
detected in human brain tissue and confirmed that the degree of
glycosylation in this type of EPO is different compared to the EPO
in serum. The former contains less sialic acid, while having a
stronger function. Astrocytes in the brain, neurons, microglia and
endothelial cells are able to produce EPO. RT-PCR has demonstrated
that mRNA of EPO and EpoR is widespread in the hippocampus and the
cerebral cortex, while the production of EPO is correlated with
blood, as well as the oxygen supply in brain tissue; when the brain
lacks blood or oxygen, the generating of EPO increases, protecting
neurons (16,17).
Research has shown that EPO has a strong effect on
neuronal apoptosis, as well as on the inflammatory response. Siren
et al (3) used an arterial
occlusive model in rat brain and found that apoptotic neurons in
the ischemic penumbral region in the experimental group (embolism
and intravenous injecting EPO) were markedly less or even
non-existent compared to the control group. Notwithstanding, the
infarct size was significantly reduced after a 24-h occlusion of
the brain artery. In pure or mixed medium neural cells, EPO
0.1–10.0 μ/ml blocked serum loss, as well as kainic acid
exposure-induced apoptosis. In their study conducted on
experimental spinal cord injury, Gorio et al (11) found that the sports function of the
5,000 U/kg EPO treatment group was significantly improved, while
the contusion focus was reduced by 25%. The motor neurons in the
central gray substance in the contusion area exhibited no
TUNEL+ cells, and the infiltrate inflammatory cells were
largely reduced, while the control group had a wide range of motor
neuron injuries, TUNEL+ expression and visible
inflammatory cell infiltrates. Agnello et al (18) found that in autoimmune myelitis,
EPO significantly reduced monocyte/macrophage infiltration in areas
of spinal cord inflammation, microglial cell activation, decreased
the production of IL-6, while delaying the appearance of the TNF-α
peak. In their studies, Chong et al (19) and Chen et al (20) found that endogenous EPO is not
sufficient to maintain cell survival during acute injury, while EPO
at a dose of 0.01-10 μ/ml for acute or chronic neuronal injury
provides broad neural protection and subsequent microglial
activation In this study, the EPO treatment group also showed an
obvious inhibitory effect, as well as an inflammatory reaction, in
the area around the injury. Cell apoptosis was significantly
reduced, while MCP-1+ and CD68+ cells also
decreased in number. In addition, this study also demonstrated that
EPO has the potential to reduce the water content of brain tissue,
indicating that EPO reduces traumatic brain edema. H&E staining
also confirmed that EPO affected brain injury compared to the
control group. The size of the injury area in the EPO group was
markedly reduced, the number of degenerated and necrotic nerve
cells and obvious infiltrating inflammatory cells was also
decreased, whereas the brain edema was palliated. The protection of
EPO on cell apoptosis after cerebral contusion involves the EpoR,
although its specific anti-apoptotic mechanism is not clear. The
research conducted on the mechanism of the production of
erythrocytopoiesis by EPO shows that EPO acts on EpoR-activated
tyrosine kinases JAK2 and JAK2 tyrosine phosphorylation of EpoR
residues, signals molecules in activated cells, such as signal
transducer and activator of transcription 5 (STAT5),
phosphatidylinositol 3-kinase (PI3K), extracellular
signal-regulated kinase 1,2 (ERKs), Ras protein/mitogen-activated
protein kinase (Ras/MAPK) and nuclear transcription factor-κB
(NF-κB). EPO also activates EpoR-JAK2-STAT5, EpoR-JAK2-PI3K,
EpoR-JAK2-ERKs, EpoR-JAK2-Ras/MAPK, EpoR-JAK2-NF-κB and other
pathways, while anti-apoptosis occurs through the upregulated
protective gene transcription and protective protein expression.
Digicaylioglu et al (21)
found that pre-treatment with EPO significantly reduces neural cell
apoptosis, while EpoR activation inhibits N-methyl-D-aspartate
(NMDA)- and NO-induced apoptosis. The mechanism of the activation
of JAK2 and JAK2 through EpoR, combined with the suppression of the
cytoplasmic transcription factor-κB (NF-IκB) to phosphorylate IκBα
tyrosine residues result in IκB degradation and NF-κB release.
NF-κB migrates into the center of the nucleus to activate target
genes, induce apoptosis and restrain the upregulation proteins XIAP
and c-IAP2. Moreover, XIAP and c-IAP2 block the final path of
activation of caspase in order to inhibit apoptosis. In addition,
NF-κB migration increases the activity of glutathione, manganese,
Cu and Zn-SOD to suppress neuronal apoptosis induced by the
accumulation of superoxide anions, such as
O2− and ONOO. Chong et al (22) demonstrated that EPO has an obvious
protective function on the neuronal damage-induced free radicals,
such as NO, and suggested EPO to be involved through the activation
of the extracellular signal-regulated kinase and protein kinase
Akt1 or protein kinase B, 1. Active Akt1 activates Bad through
phospho-serine 136 in a Bad protein, while the activated Bad
induces the fusion of anti-apoptotic BCL-2/BCL-XL in order to
inhibit neuronal apoptosis. Active Akt1 also restrains the
depolarization of free radical-induced mitochondrial transmembrane
potential, while the activation of caspases-8, -1 and -3 is known
to be the last channel of apoptosis.
By regulating microglial activation and controlling
cytokine release, EPO is highly involved in anti-inflammation. Due
to the depolarization of the mitochondrial membrane potential and
the release of cytochrome C, the injury to nerve cells induces the
activation of caspases-8, -1 and -3. In particular, the activation
of caspase-1 induces cell membrane phosphatidylserine exposure
through the digestion of cytoskeletal protein, while the exposed
phosphatidylserine participates in the activation and proliferation
of microglial cells (23). In
inflammatory responses of the central nervous system, the
microglial cell is the first and most important component of
inflammatory cells. Consequently, activated microglial cells are
likely to upregulate large numbers of cell surface receptors, while
releasing numerous pro-inflammatory factors, as well as toxic
substances, such as TNF-α, IL-1β, NO, superoxide and fatty acid
metabolism (24). Moreover, these
pro-inflammatory factors and toxic substances are also likely to
result in peripheral blood PMNs, monocytes/macrophages and
lymphocytes, as well as local activated microglial cells
infiltrating the area of the injury and around it. By releasing
lysosomal enzymes, oxygen metabolism, inflammatory mediators and
proinflammatory factors, activated microglial cells may worsen the
original inflammation while inducing the accumulation of more
inflammatory cells in the injury area, forming a vicious cycle that
results in the expansion of the scope of injury. By inhibiting
microglial activation and the release of its downstream
inflammatory factors, EPO thereby restrains the local inflammatory
response. In addition, EPO directly inhibits the pro-inflammatory
factors and the activation of IL-6, TNF-α and MCP-1 to inhibit the
inflammatory activity (3).
Immediately after brain injury, MCP-1 is expressed mainly by
astrocytes in the area around the injury, and afterwards mainly by
infiltrating monocytes/macrophages and activated microglia
(25). MCP-1 is the most important
cell factor leading to the activation of brain microglia,
peripheral blood monocytes/macrophages and lymphocyte infiltration
in the area around the injury, whereas the activated microglia and
other inflammatory cells produce large amounts of neurotoxic
factors, leading to the degeneration and necrosis of nerve cells.
By restraining the activity of MCP-1, EPO exhibits
anti-inflammatory properties.
The experimental results showed that the water
content in the cerebral hemisphere of the rats in the EPO treatment
group at each time point was markedly reduced (P<0.01) compared
to the control group, suggesting that EPO decreased traumatic brain
edema. EPO functions by directly inhibiting apoptosis in
microvascular endothelial cells to maintain the integrity of
vascular endothelia. Chong et al (26) confirmed that EPO prevents
hypoxia-induced vascular endothelial injury, while maintaining
mitochondrial membrane stability by directly activating protein
kinase B/Akt and inhibiting the cysteine protease caspases-8, -1
and -3 activity, in order to prevent the endothelial cell apoptosis
in turn. In addition, EPO clearly inhibited local inflammatory
reactions and reduced the local inflammatory vasoactive substances
and cytotoxic factors that are potentially involved in the
inhibition of brain edema. Martinez-Estrada et al (27) injected VEGF into the body of
animals to produce an animal model with a blood-brain barrier
opening and found that EPO markedly inhibited the damage caused by
blood-brain barrier permeability maintaining vascular tight
junctions between endothelial cells.
In conclusion, in this study, the protective effect
of EPO on injured brain tissue was achieved through one or a series
of ways, as described above, with a view to reduce cell apoptosis
around the edema contusion, the inflammation response, and cerebral
edema, while being involved in cerebral protection. In addition,
EPO demonstrated neurotrophic factors, promoting the
re-angiogenesis of blood vessels and accelerating the recovery of
the imbalance of the self-regulation of cerebral blood flow. EPO
has been widely used in clinical practice and has been proven to be
a safe medication with little negative effect. Ultimately, with
additional advances in research, EPO is likely to be used in
clinical application as a safe and effective neuroprotective
agent.
References
|
1
|
Bernaudin M, Marti HH, Roussel S, Divoux
D, Nouvelot A, MacKenzie ET and Petit E: A potential role for
erythropoietin in focal permanent cerebral ischemia in mice. J
Cereb Blood Flow Metab. 19:643–651. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sakanaka M, Wen TC, Matsuda S, Masuda S,
Morishita E, Nagao M and Sasaki R: In vivo evidence that
erythropoietin protects neurons from ischemic damage. Proc Natl
Acad Sci USA. 95:4635–4640. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siren AL, Fratelli M, Brines M, Goemans C,
Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C,
Capobianco A, et al: Erythropoietin prevents neuronal apoptosis
after cerebral ischemia and metabolic stress. Proc Natl Acad Sci
USA. 98:4044–4049. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brines M and Cerami A: Emerging biological
roles for erythropoietin in the nervous system. Nat Rev Neurosci.
6:484–494. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eid T and Brines M: Recombinant human
erythropoietin for neuroprotection: what is the evidence? Clin
Breast Cancer. 3(Suppl 3): S109–S115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tan CC, Eckardt KU, Firth JD and Ratcliffe
PJ: Feedback modulation of renal and hepatic erythropoietin mRNA in
response to graded anemia and hypoxia. Am J Physiol. 263:F474–F481.
1992.PubMed/NCBI
|
|
7
|
Marti HH, Wenger RH, Rivas LA, Straumann
U, Digicaylioglu M, Henn V, Yonekawa Y, Bauer C and Gassmann M:
Erythropoietin gene expression in human, monkey and murine brain.
Eur J Neurosci. 8:666–676. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kilic E, Kilic U, Soliz J, Bassetti CL,
Gassmann M and Hermann DM: Brain-derived erythropoietin protects
from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt
pathways. FASEB J. 19:2026–2028. 2005.PubMed/NCBI
|
|
9
|
Yatsiv I, Grigoriadis N, Simeonidou C,
Stahel PF, Schmidt OI, Alexandrovitch AG, Tsenter J and Shohami E:
Erythropoietin is neuroprotective, improves functional recovery,
and reduces neuronal apoptosis and inflammation in a rodent model
of experimental closed head injury. FASEB J. 19:1701–1703.
2005.
|
|
10
|
Boran BO, Colak A and Kutlay M:
Erythropoietin enhances neurological recovery after experimental
spinal cord injury. Restor Neuro Neurosci. 23:341–345.
2005.PubMed/NCBI
|
|
11
|
Gorio A, Gokmen N, Erbayraktar S, Yilmaz
O, Madaschi L, Cichetti C, Di Giulio AM, Vardar E, Cerami A and
Brines M: Recombinant human erythropoietin counteracts secondary
injury and markedly enhances neurological recovery from
experimental spinal cord trauma. Proc Natl Acad Sci USA.
99:9450–9455. 2002. View Article : Google Scholar
|
|
12
|
Lu D, Mahmood A, Qu C, Goussev A,
Schallert T and Chopp M: Erythropoietin enhances neurogenesis and
restores spatial memory in rats after traumatic brain injury. J
Neurotrauma. 22:1011–1017. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kalialis LV and Olsen NV: Erythropoietin –
a new therapy in cerebral ischemia? Ugeskr Laeger. 165:2477–2481.
2003.(In Danish).
|
|
14
|
Buemi M, Cavallaro E, Floccari F, Sturiale
A, Aloisi C, Trimarchi M, Grasso G, Corica F and Frisina N:
Erythropoietin and the brain: from neurodevelopment to
neuroprotection. Clin Sci (Lond). 103:275–282. 2002.PubMed/NCBI
|
|
15
|
Feeney DM, Boyeson MG, Linn RT, Murray HM
and Dail WG: Responses to cortical injury: I. Methodology and local
effects of contusions in the rat. Brain Res. 211:67–77. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mammis A, McIntosh TK and Maniker AH:
Erythropoietin as a neuroprotective agent in traumatic brain injury
Review. Surg Neurol. 71:527–531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hartley CE, Varma M, Fischer JP, Riccardi
R, Strauss JA, Shah S, Zhang S and Yang ZJ: Neuroprotective effects
of erythropoietin on acute metabolic and pathological changes in
experimentally induced neurotrauma. J Neurosurg. 109:708–714. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Agnello D, Bigini P, Villa P, Mennini T,
Cerami A, Brines ML and Ghezzi P: Erythropoietin exerts an
anti-inflammatory effect on the CNS in a model of experimental
autoimmune encephalomyelitis. Brain Res. 952:128–134. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chong ZZ, Kang JQ and Maiese K:
Erythropoietin fosters both intrinsic and extrinsic neuronal
protection through modulation of microglia, Akt1, Bad, and
caspase-mediated pathways. Br J Pharmacol. 138:1107–1118. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen G, Shi JX, Hang CH, Xie W, Liu J and
Liu X: Inhibitory effect on cerebral inflammatory agents that
accompany traumatic brain injury in a rat model: a potential
neuroprotective mechanism of recombinant human erythropoietin
(rhEPO). Neurosci Lett. 425:177–182. 2007. View Article : Google Scholar
|
|
21
|
Digicaylioglu M and Lipton SA:
Erythropoietin-mediated neuro-protection involves cross-talk
between Jak2 and NF-kappaB signalling cascades. Nature.
412:641–647. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chong ZZ, Kang JQ and Maiese K: Essential
cellular regulatory elements of oxidative stress in early and late
phases of apoptosis in the central nervous system. Antioxid Redox
Signal. 6:277–287. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiong Y, Mahmood A, Qu C, Kazmi H, Zhang
ZG, Noguchi CT, Schallert T and Chopp M: Erythropoietin improves
histological and functional outcomes after traumatic brain injury
in mice in the absence of the neural erythropoietin receptor. J
Neurotrauma. 27:205–215. 2010. View Article : Google Scholar
|
|
24
|
Gourmala NG, Buttini M, Limonta S, Sauter
A and Boddeke HW: Differential and time-dependent expression of
monocyte chemoattractant protein-1 mRNA by astrocytes and
macrophages in rat brain: effects of ischemia and peripheral
lipopolysaccharide administration. J Neuroimmunol. 74:35–44. 1997.
View Article : Google Scholar
|
|
25
|
Xiong Y, Lu D, Qu C, Goussev A, Schallert
T, Mahmood A and Chopp M: Effects of erythropoietin on reducing
brain damage and improving functional outcome after traumatic brain
injury in mice. J Neurosurg. 109:510–521. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chong ZZ, Kang JQ and Maiese K:
Erythropoietin is a novel vascular protectant through activation of
Akt1 and mitochondrial modulation of cysteine proteases.
Circulation. 106:2973–2979. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martínez-Estrada OM, Rodríguez-Millán E,
González-De Vicente E, Reina M, Vilaró S and Fabre M:
Erythropoietin protects the in vitro blood-brain barrier against
VEGF-induced permeability. Eur J Neurosci. 18:2538–2544.
2003.PubMed/NCBI
|