Introduction
Ischemic postconditioning (IPO) is defined as a
short series of repetitive cycles of brief reperfusion and
re-occlusion of the coronary artery applied immediately at the
onset of reperfusion. Its cardioprotective effects include the
reduction of infarct size and the improvement of coronary artery
endothelial dysfunction and neutrophil accumulation in the area at
risk (1,2). However, the effects of IPO are
determined by the time frame of the early reperfusion. A previous
study (3) has reported that the
infarct-sparing advantage of IPO and the reduction in
malondialdehyde (MDA) and dihydroethidium (DHE) fluorescence
intensity were lost when IPO was delayed for the 1-min period of
reperfusion. These data suggest that the early moments of
reperfusion during which immediate IPO is applied are crucial for
its protective effects to take place. Cohen et al (4) proposed that the time frame for IPO
was the first 2 min at the onset of reperfusion, as 1 min or
delayed IPO was ineffective. Hausenloy et al (5) suggested that IPO may be mediated
through the modulation of the mitochondrial permeability transition
pore (mPTP), whose opening in the first few minutes of myocardial
reperfusion mediates cell death. The significant burst of reactive
oxygen species (ROS) and Ca2+ overload during the first
minute of reperfusion are important manifestations of ischemic
reperfusion injury (6), whereas
IPO treatment reduces reperfusion injury by decreasing ROS
generation and attenuating mitochondrial Ca2+
concentrations. However, the exact mechanisms for IPO warrant
further investigation, as the ideal time frame for IPO has not yet
been fully elucidated.
Hydrogen sulfide (H2S), as an endogenous
material, has been characterized as the third gasotransmitter
besides nitric oxide (NO) and carbon monoxide (CO) (7). In the cardiovascular system,
H2S is predominantly generated by cystathionine-γ-lyase
(CSE) (7,8). CSE mRNA expression has been reported
to be higher in the rat myocardium than in the thoracic aorta, with
enzyme activity in the naïve myocardium being 19 nmol/min/g protein
(9). The level of H2S
detected in rat serum was 46 μM (10). Thus, the heart is constantly bathed
in a considerable amount of H2S generated by the cardiac
myocytes. Previously, it has been reported that the concentration
of endogenous H2S in plasma and myocardial tissue was
significantly decreased in isoproterenol-induced myocardial injury
(11,12). Additionally, H2S has
been shown to protect the heart from myocardial
ischemia-reperfusion (IR) injury in various studies (13–16).
Furthermore, endogenous H2S has been shown to mediate
the cardioprotection induced by IPO (17). Despite abundant support for
H2S in the cardioprotective effects of IPO, the
involvement of H2S in the early reperfusion phase of IPO
has not been studied. In the present study, we hypothesized that
increased endogenous H2S content in the coronary
effluent during early reperfusion is indispensible for the
beneficial effects of IPO.
Materials and methods
Materials
Our study conformed to the Guide for the Care and
Use of Laboratory Animals published by the US National Institutes
of Health (NIH Publications No. 85-23, revised 1996).
Male Sprague-Dawley rats (230–270 g; n=40) were
maintained in an air-filtered, temperature- (20–22°C) and
light-(12-h light/dark cycle) controlled room, with a relative
humidity of 50–52%. Rats were fed with standard commercial pellets
and water ad libitum. DL-propargylglycine (PAG) was obtained
from Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA).
Langendorff isolated heart model
Isolated heart experiments were performed as
previously described (18).
Sprague-Dawley rats were anesthetized with sodium pentobarbital (50
mg/kg). After a midline sternotomy, the hearts were rapidly excised
into ice-cold heparinized (5 U/ml) perfusate buffer. After removal
of the lung and surrounding tissue, the aorta was rapidly
cannulated with a 20-gauge, blunt-ended needle, and retrograde
coronary perfusion was initiated at a constant pressure of 80 mmHg
with modified Krebs-Henseleit buffer containing: 120 mM NaCl; 25 mM
NaHCO3; 11 mM D-Glucose; 4.7 mM KCl; 1.2 mM
MgSO4; 1.2 mM KH2PO4; and 2.5 mM
CaCl2, pH 7.4). The perfusate buffer was saturated with
a 95% O2 and 5% CO2 gas mixture at 37°C
before use. A latex balloon was inserted into the left ventricle
via the left atrium, inflated with distilled water and connected to
Maclab System (Maclab, AD Instruments, Ltd., Colarado Springs, CO,
USA). The left ventricular end diastolic pressure was set between 5
and 10 mm Hg. The balloon volume was unchanged throughout the
experiment. Cardiodynamic function for left ventricular developed
pressure (LVDP), rate pressure product (RPP), maximum gradient
during systoles (+dP/dtmax) and minimum gradient during
diastoles (−dP/dtmax) of the heart were continuously
monitored with a computer-based data acquisition system. Prior to
each experimental protocol, the isolated hearts were allowed to
stabilize for 20 min at 37°C. Hearts were excluded from further
study if after stabilization they failed to develop steady sinus
rhythm or their left ventricular developed pressures were <60 mm
Hg. Isolated rat hearts were perfused and stabilized for at least
20 min before recording data. Global ischemia was mimicked by
stopping the perfusion of Krebs-Henseleit buffer, i.e., perfusion
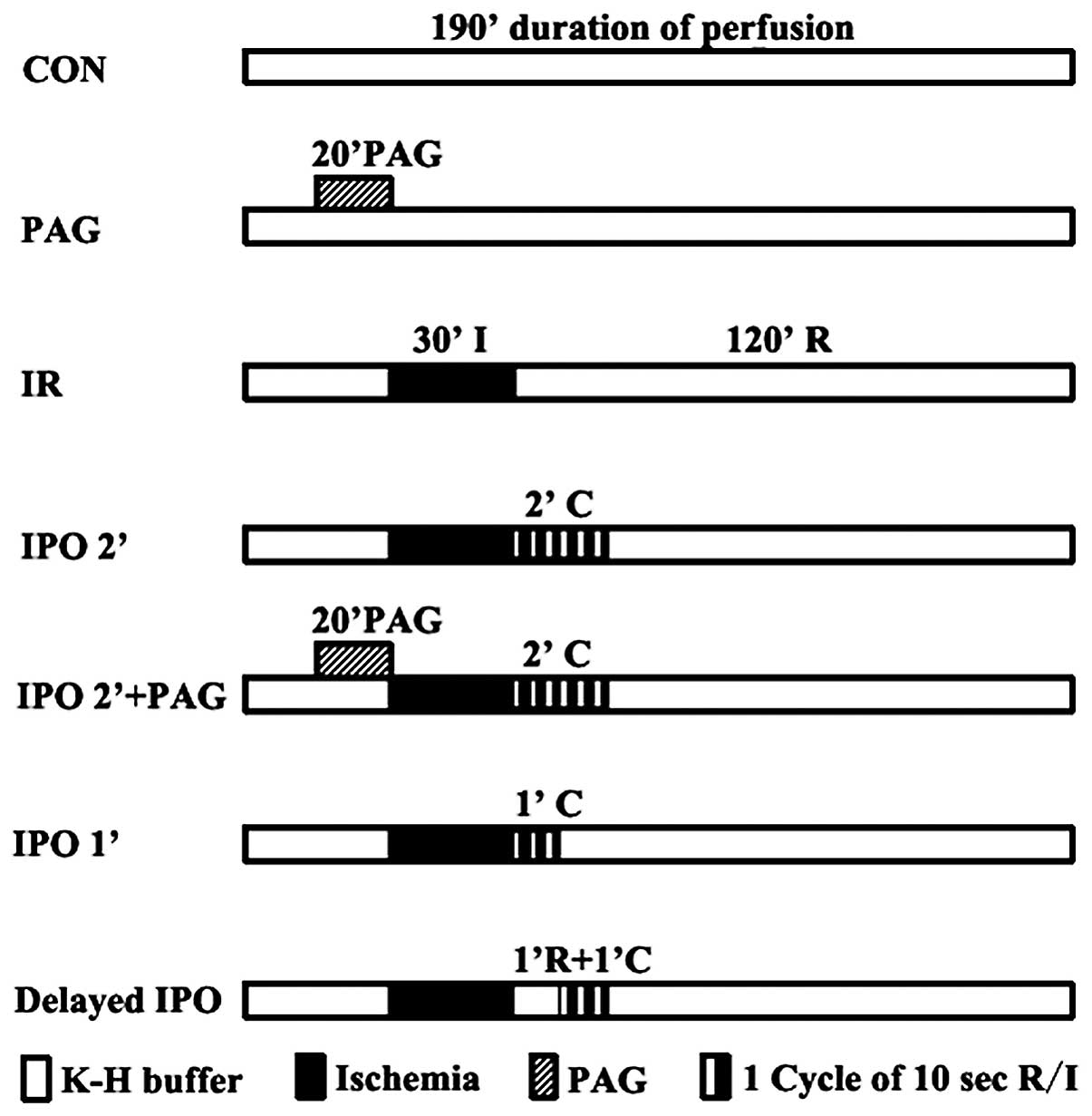
rate = 0 ml/min. The hearts were randomly divided into 7 groups
according to the perfusion protocol as shown in Fig. 1. The control (CON) and PAG groups
served as the negative controls in this study. Rat hearts were
randomly divided into 7 groups (n=5, each group): i) CON, 190 min
duration of perfusion; ii) PAG, treated with 2 mM PAG (the
inhibition of endogenous H2S synthesis) for 20 min after
20 min stabilization, followed by perfusion for 150 min; iii) IR,
after 40 min stabilization, hearts were administered 30 min global
ischemia, followed by reperfusion for 120 min; iv) IPO 2’, 6 cycles
of 10 sec reperfusion followed by 6 cycles of 10 sec ischemia
immediately upon reperfusion (2 min total intervention); v) IPO 2’
+ PAG: hearts were pre-treated with 2 mM PAG for 20 min prior to
global ischemia and IPO treatment for 2 min; vi) IPO 1’, the
algorithm of IPO was repeated for 3 cycles (1 min total
intervention); vii) delayed IPO, hearts were reperfused for 1 min,
after which the 6 cycles of IPO 2’ algorithm were applied.
H2S concentration
measurement
The coronary venous effluent at 1, 2, 3, 4, 5, 10,
20, 30, 60, 90 and 120 min of reperfusion was collected from each
group. Samples were diluted with deionised water (final volume, 500
μl) and added to a 1.5-ml tube containing zinc acetate (1%
w/v, 250 μl) to trap H2S (13,19).
Subsequently, N,N-dimethyl-p-phenylenediamine sulphate (20
μM; 133 μl) in 7.2 mol/l hydrogen chloride (HCl) was
added, followed by the addition of FeCl3 (30 μM;
133 μl) in 1.2 mol/l HCl. Thereafter, trichloroacetic acid
(10% w/v, 250 μl) was used to precipitate any proteins
present. Samples were cleared by centrifugation (10,000 × g) and
the A670 measured on aliquots from the resulting supernatant (300
μl) using an ultraviolet spectrometer 2450 (Shimadzu Corp.,
Kyoto, Japan). Various concentrations of sodium hydrogen sulfide
(NaHS) were used to plot standard curve and calculate the
concentration of H2S in samples
(μmol·l−1).
Infarct size
Infarct size was assessed by triphenyltetrazolium
chloride (TTC) staining (20). At
the end of the IR protocol, the hearts were cut into 2-mm
transverse slices. After incubation in 1% TTC in PBS (pH 7.4)
solution for 30 min (37°C), the sections were immersed in formalin
(4% w/v) for another 30 min. Images were scanned into a computer
and total ventricular area as well as infarct area were determined
by computerized planimetry (Adobe Photoshop, version CS3). The
infarct size was expressed as a percentage of the total ventricular
area (%).
Lactate dehydrogenase (LDH) content in
coronary efflux
The LDH content in the coronary effluent was used as
a biochemical marker of cardiomyocyte injury. For each heart, a
baseline sample of coronary effluent was collected from the
superfusate bath overflow during the final minute of equilibration
(prior to the onset of global ischemia). Coronary effluent was then
collected at the end of 120 min of reperfusion and a 1.5-ml aliquot
was stored on ice for assay within 24 h. LDH content was determined
with an automated spectrophotometric clinical assay using an ACE
Chemistry Analyzer (Alfa Wassermann Inc., West Caldwell, NJ,
USA).
Statistical analysis
All results are expressed as the means ± SD.
Statistical analysis was performed using SPSS 13.0 software for
Windows. Differences between groups were analyzed by one-way ANOVA
followed by the Student Newman-Keuls test. P<0.05 was considered
to indicate a statistically significant difference.
Results
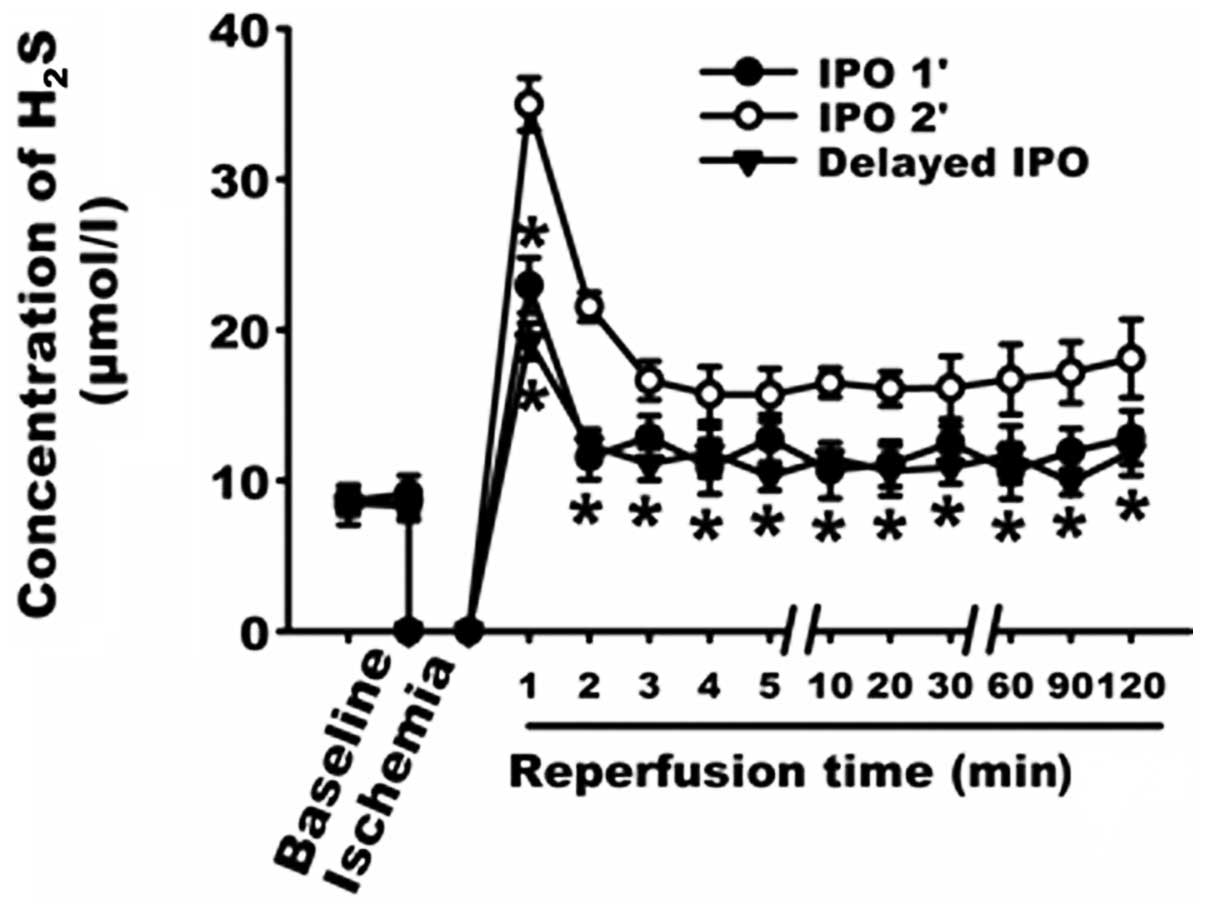
Changes in H2S concentration
in the coronary effluent
H2S is synthesized mainly by CSE in the
cardiovascular system. However, it is unclear whether
H2S can still be generated in ischemic heart. As shown
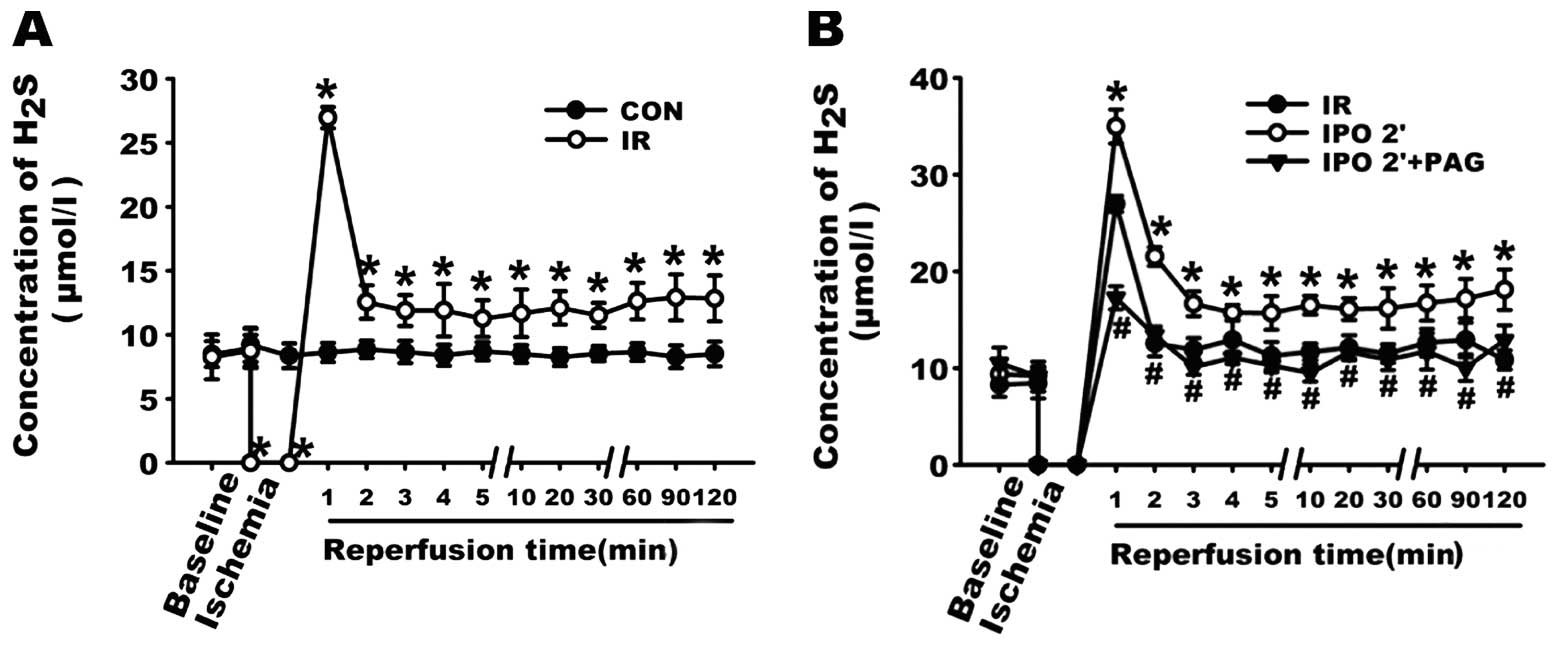
in Fig. 2A, the concentration of
H2S in the CON group was stable, ranging from 8.28±0.89
to 9.21±1.32 μmol/l. By contrast, after 30-min global
ischemia, the concentration of H2S in the coronary
effluent in the first minute of reperfusion reached a peak
(26.96±0.83 μmol/l) of almost 4-fold the level of the
baseline, which strongly suggests that during the 30 min of global
ischemia, H2S was still produced and accumulated in the
myocardium. On the basis of this result, we further examined the
changes in H2S concentration in the coronary effluents.
As shown in Fig. 2B, in the first
minute after the resumption of flow, the concentration of
H2S in the IR, IPO 2’, IPO 2’ + PAG group increased to
26.96±0.83, 34.97±1.74 and 17.26±1.20 μmol/l, respectively.
Compared with the IR group, the concentration of H2S in
the coronary effluent was significantly increased in the IPO 2’
group (P<0.05), indicating that the IPO 2’ treatment further
increased the level of H2S in the coronary effluent
during the first minute of reperfusion. However, in the IPO 2’ +
PAG group, pre-treatment with PAG for 20 min prior to global
ischemia abolished the effect of the IPO 2’ treatment on the
content of H2S during the first minute of reperfusion.
It should be noted that during the entire reperfusion process, the
concentration of H2S in the coronary effluent in the IPO
2’ group was significantly higher than that in the IR and IPO 2’ +
PAG group (all p<0.05).
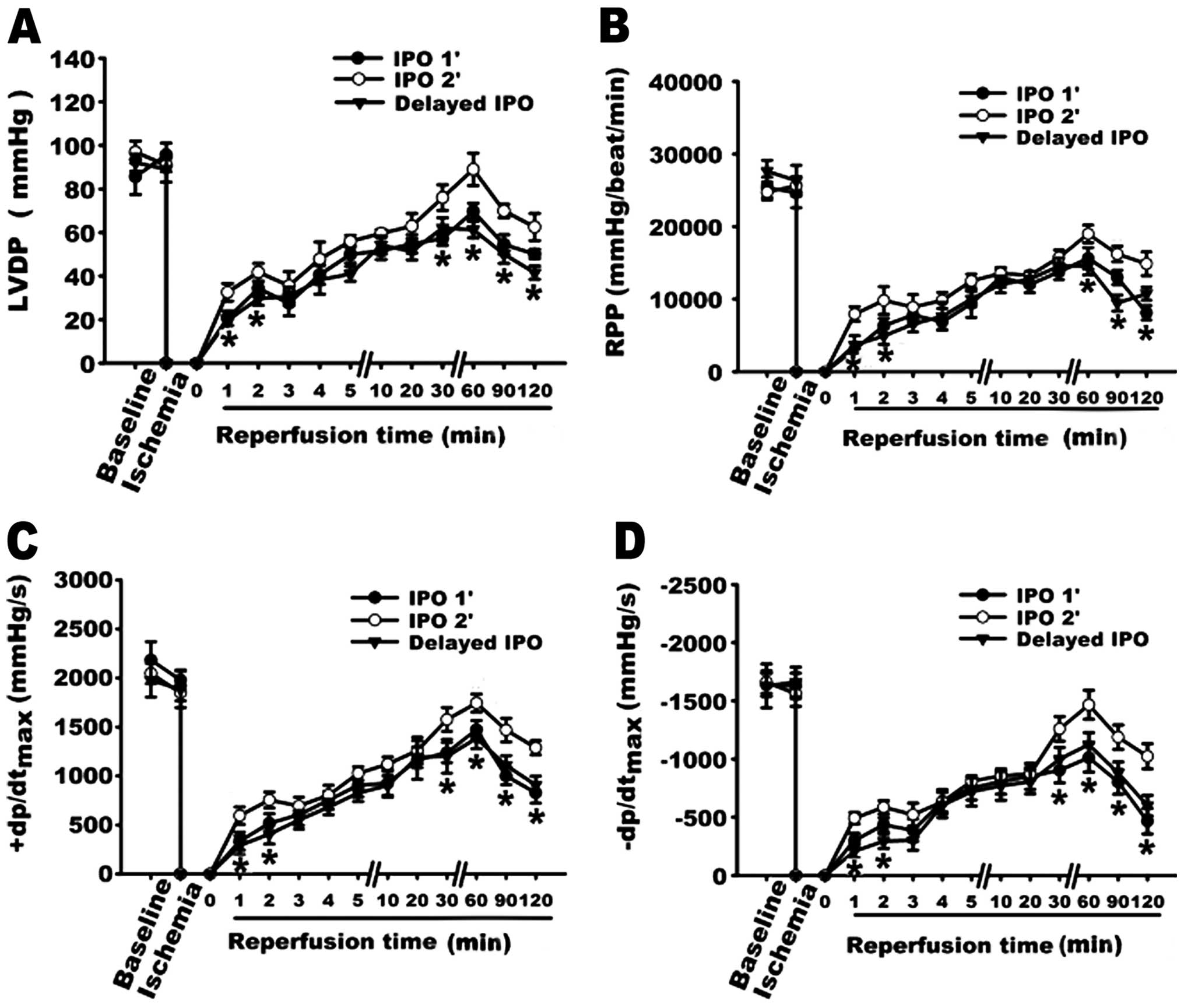
Changes in cardiodynamic function
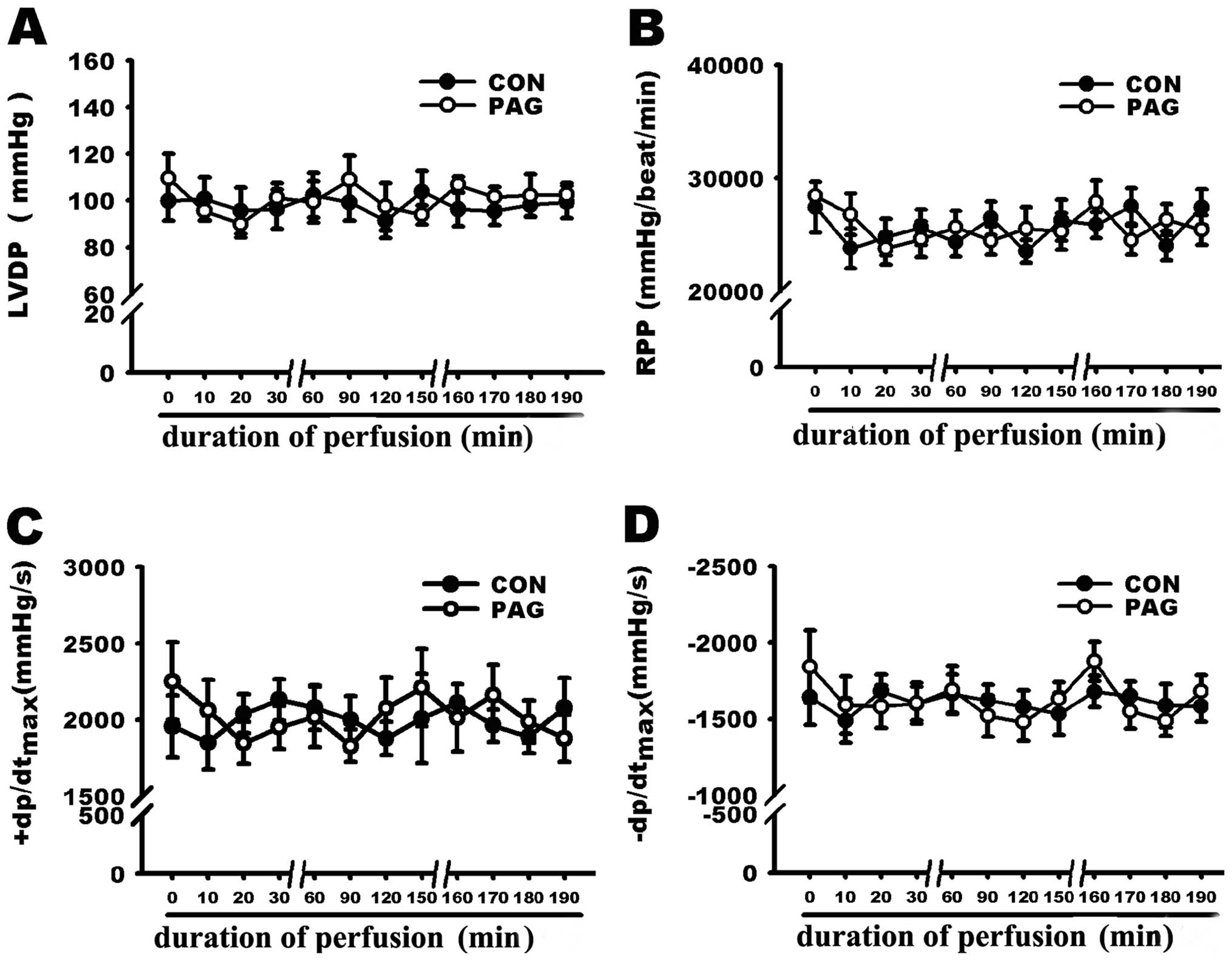
The effect of PAG on cardiodynamic function in the
isolated rat hearts was examined. As shown in Fig. 3, the administration of PAG (2 mM)
alone did not significantly influence cardiodynamic function
including LVDP, RPP, +dP/dtmax and −dP/dtmax.
PAG was therefore used in the following experiments to determine
the involvement of endogenous H2S in the
cardioprotection induced by IPO 2’ treatment.
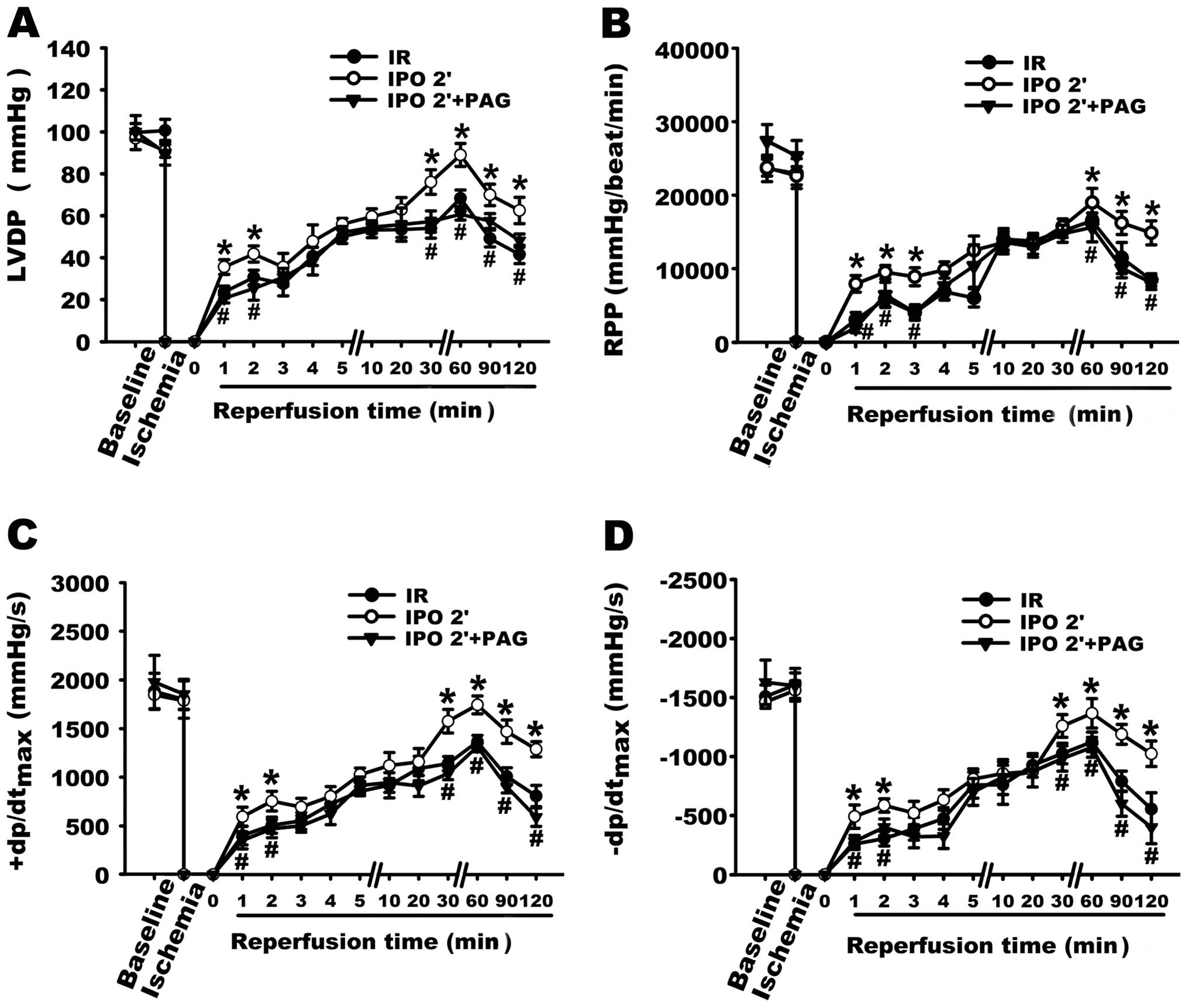
Cardiac mechanical function including LVDP, RPP,
±dp/dtmax was measured to determine the involvement of
endogenous H2S in the cardioprotection induced by the
IPO 2’ treatment. As shown in Fig.
4, the IPO 2’ treatment exerted a significant cardioprotective
effect with the recovery of cardiac function following ischemia
compared with that of IR. The recoveries at 120 min of LVDP, RPP,
+dP/dtmax and −dP/dtmax in IPO 2’ group were
1.50, 1.76, 1.60 and 1.84-fold of the IR group, respectively (all
P<0.05 compared with IR, n=5). However, pre-treatment with 2 mM
PAG 20 min prior to global ischemia, which inhibited the production
of endogenous H2S prior to and during ischemia,
significantly diminished the cardioprotective effect of the IPO 2’
treatment by reducing LVDP, RPP, +dP/dtmax and
−dP/dtmax to 0.77, 0.55, 0.46 and 0.39-fold of the IPO
2’ group, respectively (all P<0.05 compared with IPO 2’,
n=5).
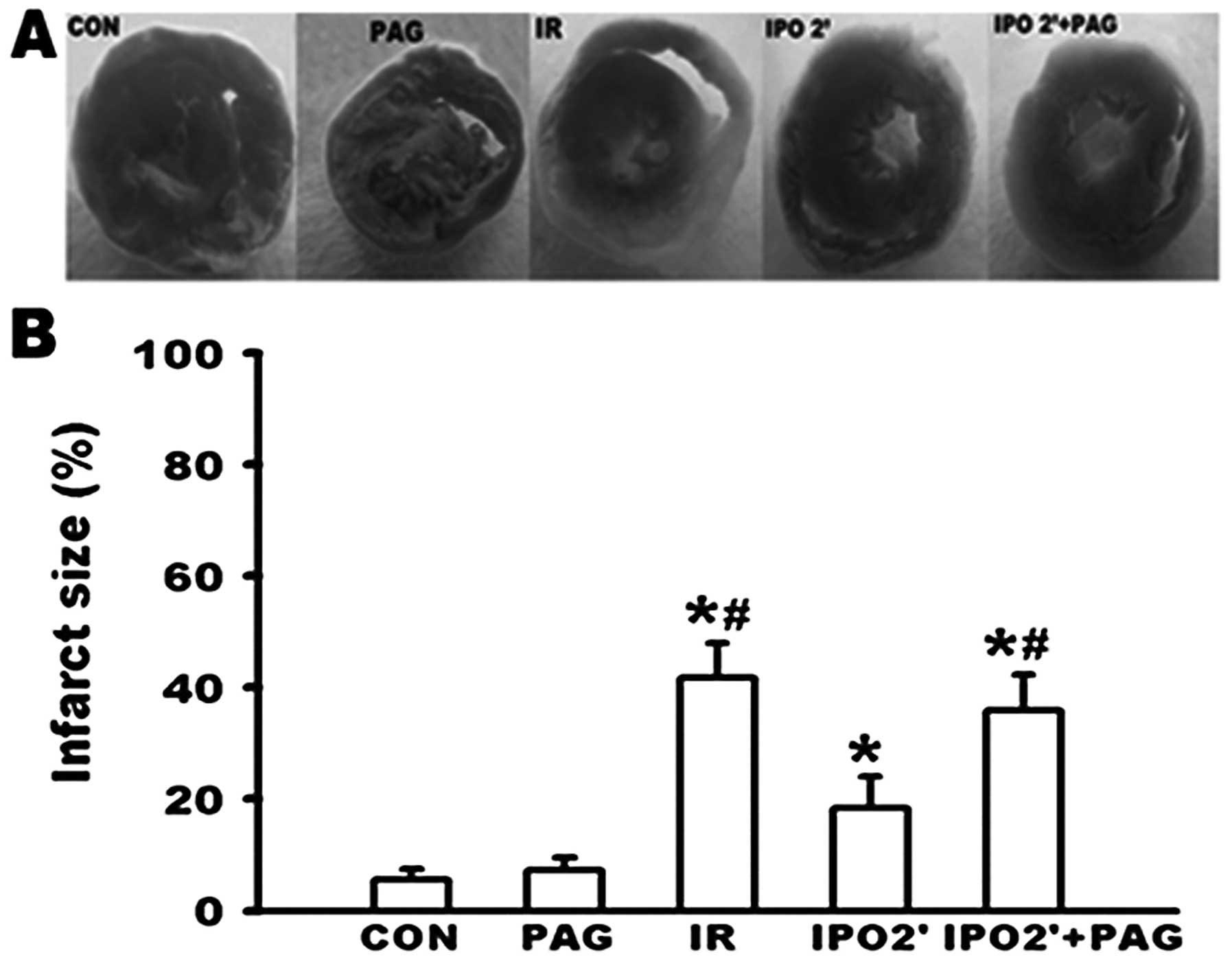
Changes in infarct size
As shown in Fig. 5,
no significant differences in infarct size were observed between
the CON and PAG group. While an infarct size of 41.8±6.2% was
caused in the IR group, the infarct size was significantly
decreased to 18.3±5.7% in the IPO 2’ group (P<0.05). However,
PAG pre-treatment in the IPO 2’ group resulted in an infarct size
of 35.9±6.4%. This was significantly increased when compared to the
IPO 2’ group (P<0.05), suggesting that PAG inhibited the
cardioprotection observed by the IPO 2’ treatment.
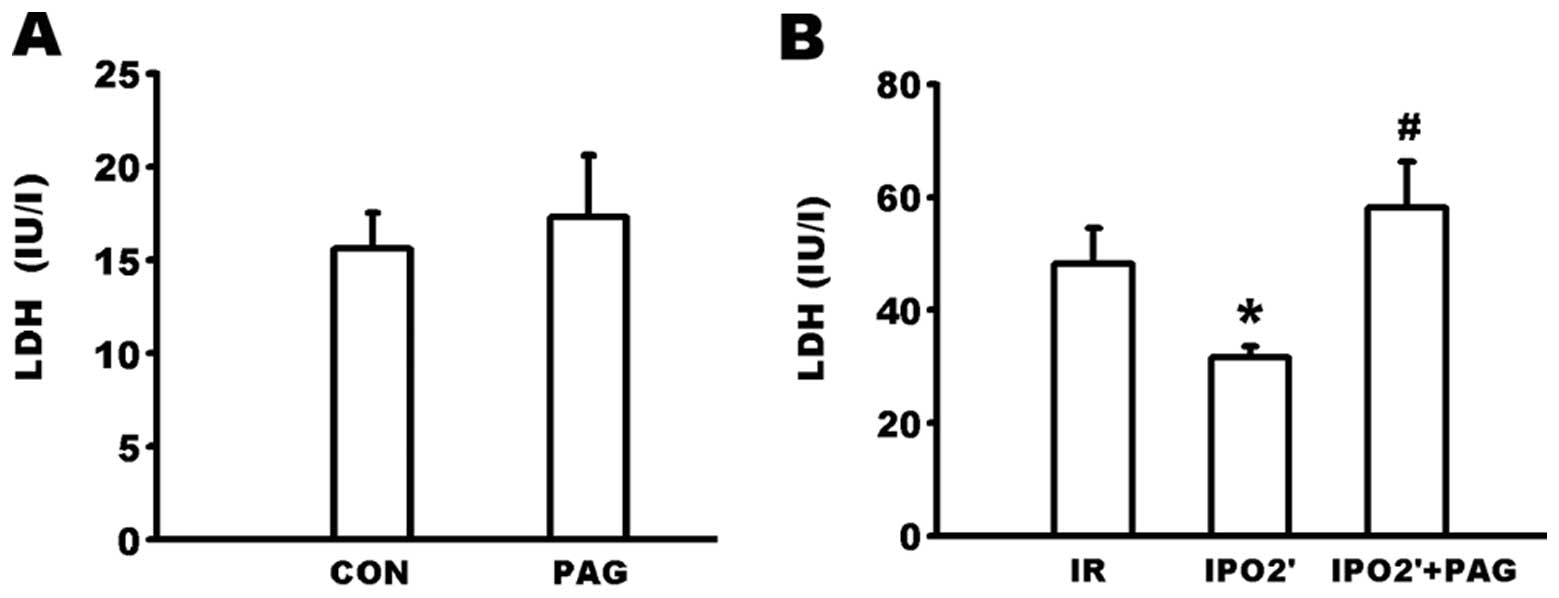
Changes in LDH levels in the coronary
effluent
As shown in Fig.
6A, there were no significant differences in LDH levels in the
coronary efflux between the CON and PAG groups. The LDH content in
the coronary effluent at 120 min of reperfusion (Fig. 6B) was significantly lower in the
IPO 2’ group compared to the IR group (31.60±1.95 vs. 48.20±6.32
IU/l, P<0.05). However, the LDH level was significantly
increased in the IPO 2’ + PAG group (58.20±8.06 IU/l) compared to
the IPO 2’ group (P<0.05).
Effect of IPO 1’ and Delayed IPO
As shown in Fig. 7,
the concentrations of H2S in the IPO 1’ and the delayed
IPO groups were decreased when compared to the IPO 2’ group. The
cardio-protective effects were reduced in the IPO 1’ and the
delayed IPO groups as represented by greater infarct size (Fig. 8) and lower recovery of
cardiodynamic function (Fig.
9).
Discussion
The main objective of this study was to explore the
role of endogenous H2S in the cardioprotection induced
by IPO during early reperfusion. We found that IPO 2’ (IPO lasting
for 2 min) significantly improved the heart contractile function
and reduced multiple manifestations of IR injury, including infarct
size and LDH levels. This is consistent with previous findings that
IPO protects the heart from lethal IR injury when assessed by
infarct size (3,21,22),
cardiodynamic performance (23–26)
and cellular injury biomarkers (6,27).
The myocardium generates a considerable amount of
H2S under physiological conditions (8,9);
however, H2S production is decreased upon ischemia
treatment (19). In the present
study, we demonstrated that the concentration of H2S was
stable throughout the entire experiment in the CON group, and its
concentration reached a peak during the first minute of reperfusion
in the IR group. This indicates that H2S accumulates in
the myocardium during the 30 min of global ischemia and is then
washed out immediately during early reperfusion. In addition,
during the first minute of reperfusion the concentration of
H2S in the IPO 2’ group was higher than that in the IR
group, suggesting that intermittent reperfusion delayed the washout
of H2S during early reperfusion. On the other hand,
H2S in aqueous solution is dissociated into
HS− and H+, which exist in equilibrium with
each other. The proportion of undissociated H2S under
standard temperature conditions at physiological pH (7.4) is 30–33%
(20,28), and the proportion increases as pH
decreases. As shown in a previous study, acidotic status induced by
ischemia is prolonged in IPO 2’ (4), thus the lower pH status induced by
IPO 2’ can further increase the concentration of H2S at
the onset of reperfusion.
It has been previously reported that the early phase
of reperfusion may be the key to cardioprotection induced by IPO
(3,4). Therefore, we designed other forms of
IPO, IPO starting 1 min after reperfusion (delayed IPO) or lasting
only 1 min (IPO 1’), to examine the important time frame of IPO 2’.
Our data demonstrated that IPO 1’ and delayed IPO failed to exert a
cardioprotective effect, and with both treatments the peak of the
H2S concentration decreased simultaneously during the
first minute of reperfusion. There may be 2 explanations for the
lower concentration of H2S during early reperfusion in
both groups. Firstly, there was not enough time for H2S
accumulation during early reperfusion since IPO 1’ lasted only 1
min. H2S was washed out immediately as the reperfusion
was initiated, which resulted in reduced retention of
H2S at the onset of the reperfusion in delayed IPO.
Secondly, during early reperfusion in both IPO 1’ and delayed IPO,
pH quickly returned to a neutral level. A lower pH is responsible
for the generation of increased amounts of H2S (4). Indeed, the time-relative changes of
H2S level are consistent with the critical time frame of
the IPO effect. Therefore, our data suggest that IPO lasting only 2
min and commencing at the onset of reperfusion could preserve
H2S in order to mediate the cardioprotection invoked by
IPO.
The accumulation of endogenous material during early
reperfusion is one of the important mechanisms of IPO-mediated
effects. The observations of Zhao et al (2) suggested that endogenous mechanisms
are put into action within the first few minutes of reperfusion
that attenuate reperfusion injury specifically. Kin et al
(25) reported that IPO delays the
washout of endogenous adenosine during the critical early moments
of reperfusion, thereby increasing intravascular adenosine
concentrations, which can activate adenosine receptors to elicit
protection against myocardial infarction. In addition, previous
studies (29,30) have shown that with IPO maneuvers
the heart releases autacoids that accumulate in an intermittent
manner during early reperfusion and trigger pathways leading to a
protected state. In the present study, we noted that the higher
concentration of H2S during early reperfusion in IPO 2’
was associated with improved recovery of contractile function,
decreased infarct size and LDH level. However, the inhibition of
endogenous H2S synthesis by PAG (the specific and
irreversible inhibitor of CSE) used 20 min prior to ischemic
treatment notably inhibited the generation of H2S and
abolished the cardioprotective effects exerted by IPO 2’. Our data
indicated that the increase in endogenous H2S during
early reperfusion contributes to the cardioprotective effects of
IPO 2’.
There is a significant burst of oxygen-derived free
radicals generated within the first minute of reperfusion peaking
4–7 min following the onset of reperfusion (6), which contribute to the IR injury.
H2S is a thiol that can interact with and ‘scavenge’
free radicals, including ONOO (31), H2O2 (12) and HOCl (32). Therefore, the potential cellular
mechanisms for H2S-mediated early cardioprotection
invoked by IPO 2’ may involve the reduction of the peak generation
of ROS occurring during the first minutes of reperfusion. In
addition, a previous study has shown that H2S, the
KATP channel opener, was involved in cardiac protection
(33). For this reason we presumed
that H2S may protect the ischemic myocardum by opening
KATP channels. However, the exact mechanisms remain
uncertain in the present study. It is not known whether deleterious
mechanisms are attenuated, or whether beneficial mechanisms are
triggered by H2S. This is a limitation of our present
study and warrants further investigation.
In conclusion, this study demonstrates that
endogenous H2S mediates the cardioprotection induced by
IPO during early reperfusion. H2S may be considered to
protect by ‘pharmacological IPO’ thus offering greater opportunity
for protection clinically, such as at the time of thrombolysis,
percutaneous transluminal coronary angioplasty (PTCA) and coronary
artery bypass grafting (CABG). This study provides an impetus for
further investigation into the synthesis of a drug that can release
H2S. If the certain protection of H2S were
proved clinically, then ischemia-reperfusion injury could be
attenuated by simply administering an oral medication that released
H2S.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (30570766, 30971169,
81170277; ZS Jiang) and Aid Program for Science and Techology
Innovative Research Team in Higher Educational Institutions
(2008-244; ZS Jiang) of Hunan Province.
References
|
1
|
Na HS, Kim YI, Yoon YW, Han HC, Nahm SH
and Hong SK: Ventricular premature beat-driven intermittent
restoration of coronary blood flow reduces the incidence of
reperfusion-induced ventricular fibrillation in a cat model of
regional ischemia. Am Heart J. 132:78–83. 1996. View Article : Google Scholar
|
|
2
|
Zhao ZQ, Corvera JS, Halkos ME, et al:
Inhibition of myocardial injury by ischemic postconditioning during
reperfusion: comparison with ischemic preconditioning. Am J Physiol
Heart Circ Physiol. 285:H579–588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kin H, Zhao ZQ, Sun HY, et al:
Postconditioning attenuates myocardial ischemia-reperfusion injury
by inhibiting events in the early minutes of reperfusion.
Cardiovasc Res. 62:74–85. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cohen MV, Yang XM and Downey JM: The pH
hypothesis of postconditioning: staccato reperfusion reintroduces
oxygen and perpetuates myocardial acidosis. Circulation.
115:1895–1903. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hausenloy DJ, Duchen MR and Yellon DM:
Inhibiting mitochondrial permeability transition pore opening at
reperfusion protects against ischaemia-reperfusion injury.
Cardiovasc Res. 60:617–625. 2003. View Article : Google Scholar
|
|
6
|
Sun HY, Wang NP, Kerendi F, et al: Hypoxic
postconditioning reduces cardiomyocyte loss by inhibiting ROS
generation and intracellular Ca2+ overload. Am J Physiol
Heart Circ Physiol. 288:H1900–1908. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang R: Two’s company, three’s a crowd:
can H2S be the third endogenous gaseous transmitter?
Faseb J. 16:1792–1798. 2002.
|
|
8
|
Szabo C: Hydrogen sulphide and its
therapeutic potential. Nat Rev Drug Discov. 6:917–935. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Geng B, Yang J, Qi Y, et al:
H2S generated by heart in rat and its effects on cardiac
function. Biochem Biophys Res Commun. 313:362–368. 2004.
|
|
10
|
Zhao W, Zhang J, Lu Y and Wang R: The
vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP)
channel opener. Embo J. 20:6008–6016. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yong QC, Pan TT, Hu LF and Bian JS:
Negative regulation of beta-adrenergic function by hydrogen
sulphide in the rat hearts. J Mol Cell Cardiol. 44:701–710. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Geng B, Chang L, Pan C, et al: Endogenous
hydrogen sulfide regulation of myocardial injury induced by
isoproterenol. Biochem Biophys Res Commun. 318:756–763. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Elrod JW, Calvert JW, Morrison J, et al:
Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury
by preservation of mitochondrial function. Proc Natl Acad Sci USA.
104:15560–15565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Johansen D, Ytrehus K and Baxter GF:
Exogenous hydrogen sulfide (H2S) protects against
regional myocardial ischemiareperfusion injury - evidence for a
role of K ATP channels. Basic Res Cardiol. 101:53–60. 2006.
|
|
15
|
Sivarajah A, McDonald MC and Thiemermann
C: The production of hydrogen sulfide limits myocardial ischemia
and reperfusion injury and contributes to the cardioprotective
effects of preconditioning with endotoxin, but not ischemia in the
rat. Shock. 26:154–161. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu YZ, Wang ZJ, Ho P, et al: Hydrogen
sulfide and its possible roles in myocardial ischemia in
experimental rats. J Appl Physiol. 102:261–268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yong QC, Lee SW, Foo CS, Neo KL, Chen X
and Bian JS: Endogenous hydrogen sulphide mediates the
cardioprotection induced by ischemic postconditioning. Am J Physiol
Heart Circ Physiol. 295:H1330–H1340. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morrison RR, Talukder MA, Ledent C and
Mustafa SJ: Cardiac effects of adenosine in A(2A) receptor knockout
hearts: uncovering A(2B) receptors. Am J Physiol Heart Circ
Physiol. 282:H437–H444. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bian JS, Yong QC, Pan TT, et al: Role of
hydrogen sulfide in the cardioprotection caused by ischemic
preconditioning in the rat heart and cardiac myocytes. J Pharmacol
Exp Ther. 316:670–678. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu YZ, Chong CL, Chuah SC, et al:
Cardioprotective effects of nitroparacetamol and paracetamol in
acute phase of myocardial infarction in experimental rats. Am J
Physiol Heart Circ Physiol. 290:H517–H524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Darling CE, Jiang R, Maynard M, Whittaker
P, Vinten-Johansen J and Przyklenk K: Postconditioning via
stuttering reperfusion limits myocardial infarct size in rabbit
hearts: role of ERK1/2. Am J Physiol Heart Circ Physiol.
289:H1618–1626. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang XM, Philipp S, Downey JM and Cohen
MV: Postconditioning’s protection is not dependent on circulating
blood factors or cells but involves adenosine receptors and
requires PI3-kinase and guanylyl cyclase activation. Basic Res
Cardiol. 100:57–63. 2005.
|
|
23
|
Crisostomo PR, Wang M, Wairiuko GM,
Terrell AM and Meldrum DR: Postconditioning in females depends on
injury severity. J Surg Res. 134:342–347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fantinelli JC and Mosca SM: Comparative
effects of ischemic pre and postconditioning on
ischemia-reperfusion injury in spontaneously hypertensive rats
(SHR). Mol Cell Biochem. 296:45–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kin H, Zatta AJ, Lofye MT, et al:
Postconditioning reduces infarct size via adenosine receptor
activation by endogenous adenosine. Cardiovasc Res. 67:124–133.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu M, Feng J, Lucchinetti E, et al:
Ischemic postconditioning protects remodeled myocardium via the
PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc
Res. 72:152–162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang HC, Zhang HF, Guo WY, et al: Hypoxic
postconditioning enhances the survival and inhibits apoptosis of
cardiomyocytes following reoxygenation: role of peroxynitrite
formation. Apoptosis. 11:1453–1460. 2006. View Article : Google Scholar
|
|
28
|
Dombkowski RA, Russell MJ and Olson KR:
Hydrogen sulfide as an endogenous regulator of vascular smooth
muscle tone in trout. Am J Physiol Regul Integr Comp Physiol.
286:R678–R685. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Penna C, Mancardi D, Rastaldo R, Losano G
and Pagliaro P: Intermittent activation of bradykinin B2 receptors
and mitochondrial KATP channels trigger cardiac postconditioning
through redox signaling. Cardiovasc Res. 75:168–177. 2007.
View Article : Google Scholar
|
|
30
|
Weihrauch D, Krolikowski JG, Bienengraeber
M, Kersten JR, Warltier DC and Pagel PS: Morphine enhances
isoflurane-induced postconditioning against myocardial infarction:
the role of phosphatidylinositol-3-kinase and opioid receptors in
rabbits. Anesth Analg. 101:942–949. 2005. View Article : Google Scholar
|
|
31
|
Whiteman M, Armstrong JS, Chu SH, et al:
The novel neuro-modulator hydrogen sulfide: an endogenous
peroxynitrite ‘scavenger’? J Neurochem. 90:765–768. 2004.PubMed/NCBI
|
|
32
|
Whiteman M, Cheung NS, Zhu YZ, et al:
Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated
oxidative damage in the brain? Biochem Biophys Res Commun.
326:794–798. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hassouna A, Matata BM and Galinanes M:
PKC-epsilon is upstream and PKC-alpha is downstream of mitoKATP
channels in the signal transduction pathway of ischemic
preconditioning of human myocardium. Am J Physiol Cell Physiol.
287:C1418–C1425. 2004. View Article : Google Scholar : PubMed/NCBI
|