Introduction
The term pulmonary hypertension (PH) describes a
group of lung disorders characterized by a progressive increase in
pulmonary arterial pressure. The common pathological feature of PH
is pulmonary vascular remodeling. Persistent high blood pressure in
the pulmonary circulation results in right ventricular hypertrophy
and dilatation, leading to the eventual occurrence of right heart
failure. Along with the advancement in studies on PH in recent
years, the importance of endothelial progenitor cells (EPCs) has
been recognized among the possible factors involved in the
mechanism of PH. EPCs have been considered to be pivotal in
maintaining the normal structure and function of the vascular
endothelium (1–3), and the dysfunction of this process
may, to a certain degree, contribute to the occurrence of pulmonary
vascular remodeling in PH. It has been shown that the excessive
apoptosis of pulmonary artery endothelial cells in PH results in
the destruction of endothelial integrity (4). Furthermore, the endothelial repair
effect of EPCs is not able to be implemented effectively, due to
the downregulation in EPC number and function (5,6),
leading to an imbalance between the lesion and the repair of the
vascular endothelium and the further damage of pulmonary
arteries.
EPC transplantation has been shown to be effective
at preventing the progression of PH in laboratory rats (4,7).
However, this procedure is likely to be limited in clinical
practice due to difficulties in obtaining sufficient EPCs from
donors during the effective treatment period; therefore, a safe and
convenient protocol to improve EPCs in patients with PH is
preferable. As a regulator of granulocytes, granulocyte-colony
stimulating factor (G-CSF) has been used for decades, and exhibits
reliability in the clinic. Furthermore, the administration of G-CSF
in cardiovascular disease leads to the repair of the injured vessel
and myocardium by the mobilization of bone marrow EPCs and their
precursors (8). It has also
exhibited efficacy at preventing the progression of PH (9). However, studies of EPC number and
function following the administration of G-CSF in PH are lacking,
and the mechanism underlying the protective effect of G-CSF on PH
has not been fully elucidated. It has been demonstrated that nitric
oxide (NO), as a signaling molecule, is required for the
mobilization of bone marrow EPCs (10), while reducing the apoptosis of EPCs
(11), and participating in
angiogenesis and vasculogenesis (12,13).
It has been shown that the cardioprotective effect of G-CSF is also
mediated by the NO system (14,15).
Therefore, we proposed that G-CSF attenuates PH by the NO-mediated
upregulation of EPCs.
In the present study, we utilized a rat model of PH,
created by the subcutaneous injection of monocrotaline (MCT), and
treated the rats with recombinant human G-CSF (rhG-CSF).
Nω-nitro-L-arginine methyl ester (L-NAME) was used concurrently as
a negative intervention. The therapeutic effect, number and
function of circulating EPCs and the concentration of plasma NO
were evaluated, in order to enhance the understanding of the
protective mechanisms of rhG-CSF in PH.
Materials and methods
Animals
Male Sprague-Dawley (SD) rats, aged 8 weeks, were
purchased from Capital Medical University (Beijing, China) and
housed in specific pathogen-free units of the Division of
Laboratory Animals at Capital Medical University. Thirty-two rats
were randomly divided into four groups: the model [MCT and
phosphate-buffered saline (PBS)], rhG-CSF treatment (MCT and
rhG-CSF), L-NAME intervention (MCT, rhG-CSF and L-NAME) and control
(PBS) groups. Each group contained eight rats. PH was induced by a
single subcutaneous injection of MCT (60 mg/kg; Sigma, St. Louis,
MO, USA) (16), while PBS was
administered to the controls. rhG-CSF (50 μg/kg/day; Xiamen Amoytop
Biotech Co., Ltd., Xiamen, China) or PBS was subcutaneously
injected from day five to day seven after the injection of MCT.
Furthermore, L-NAME (4 mg/kg/day; Sigma) was intragastrically
administered at the same time as the rhG-CSF injection and
continued to day 21. All animal studies and protocols were approved
by the Institutional Animal Care and Use Committee of Capital
Medical University.
Examination of hemodynamics
At day 21, the rats were anesthetized by an
intraperitoneal injection of pentobarbital (50 mg/kg). A
polyethylene catheter was inserted into the right ventricle (RV)
via the right external jugular vein, and another was targeted at
the ascending aorta via the right carotid artery. Right ventricular
systolic pressure (RVSP) and mean aortic pressure (MAoP) were
recorded using a polygraph (Nihon Kohden Corporation, Tokyo,
Japan).
Numbers of EPCs in peripheral blood
Peripheral blood was collected from the right
external jugular vein into EDTA-containing tubes and 100 μl was
incubated with 2 μl fluorescein isothiocyanate (FITC)-conjugated
goat monoclonal antibody against mouse immunoglobulin G, and 5 μl
mouse monoclonal antibody against rat vascular endothelial growth
factor receptor (VEGFR)-2 (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA). Following red cell lysis and washing with PBS, the
cells were incubated for a further 20 min with 5 μl allophycocyanin
(APC)-conjugated mouse monoclonal antibody against rat CD45 (BD
Biosciences, Franklin Lakes, NJ, USA) and 5 μl phycoerythrin
(PE)-conjugated mouse monoclonal antibody against rat CD34 (BD
Biosciences). The samples were subsequently centrifuged for 5 min
at 300 × g and then resuspended in 500 μl PBS and evaluated using
flow cytometry (BD FACSCalibur Flow Cytometer; BD Biosciences, San
Jose, CA, USA). Isotype controls were run in parallel and ~100,000
events were recorded. Circulating EPCs were defined as cells
positive for CD34 and VEGFR-2, but negative for CD45 (17).
Plasma NO measurement
Peripheral blood was centrifuged for 15 min at 1,200
× g, prior to 50 μl plasma being collected into 96-well plates for
the measurement of NO. The Nitric Oxide Assay kit (Beyotime
Biotechnology, Haimen, China) was based on Greiss reagent. The
concentration of NO was determined by spectrophotometry (490 nm),
following the addition of 50 μl Greiss reagents I (1% sulfanilamide
in 0.1 mol/l HCl) and II [0.1% N-(1-naphthyl-ethylenediamine
dihydrochloride)].
EPCs cultured in vitro
Mononuclear cells were isolated from peripheral
blood using Ficoll density gradient centrifugation (20 min at 400 ×
g without brake) and suspended with endothelial cell growth
medium-2 (EGM-2; Lonza Group AG, Basel, Switzerland) in 24-well
culture plates pre-coated with fibronectin (Sigma), and incubated
at 37°C in a humidified environment with 5% carbon dioxide
(CO2). Unattached cells were removed by extensive
washing on day three, and the culture medium was replaced every two
days thereafter. Following seven days of culture, cells were
incubated with 10 μg/ml DiI-labeled acetylated low-density
lipoprotein (acLDL; Molecular Probes®, Invitrogen Life
Technologies, Carlsbad, CA, USA). Four hours later, the cells were
incubated with 10 μg/ml FITC-conjugated lectin from Ulex europeus
agglutinin-1 (FITC-UEA-1; Sigma) for 1 h, following fixation with
4% paraformaldehyde. The cells were subsequently examined using
laser scanning confocal microscopy. Differentiating EPCs were
identified by double fluorescence staining, as previously described
(18).
Function of EPCs in vitro
EPCs were detached using 0.25% trypsin following
seven days of culture. EPC functions, such as proliferation,
adhesion and migration, were assessed as described in a previous
study (18). With regard to
proliferation, the cells were cultured for 24 h in 96-well plates
at a density of 105 cells/ml (200 μl per well), prior to
20 μl MTT (5 g/l) being added for 4 h. Following this, the media
was discarded and 100 μl dimethylsulfoxide (DMSO) was added for 10
min. The absorbance was measured at a wavelength of 490 nm. For
adhesion, EPCs were incubated with EGM-2 in 24-well plates
pre-coated with fibronectin (5×104/well) for 30 min.
Subsequent to washing three times with PBS, the attached cells were
counted in a high power field (HPF). To assess the migratory
ability of the cells, a modified Boyden chamber (8-μm pore size)
was used. EPCs were suspended in 100 μl EGM-2 without cytokines,
plus 0.5% fetal bovine serum (FBS), in the upper chamber
(5×105/ml) and placed in a 24-well culture plate
containing 600 μl EGM-2. Following 24 h of incubation, the lower
membrane of the chamber was fixed with 4% paraformaldehyde.
Migrated cells were counted in a HPF, subsequent to staining with
0.1% crystal violet.
Histological examination
Lung tissues were removed from the rats subsequent
to sacrifice by decapitaiton and fixed in 10% paraformaldehyde for
24 h. Following this, serial paraffin sections (5-μm) were stained
with hematoxylin and eosin for light microscopy (magnification,
×400). The medial wall thickness of the pulmonary arteriole is
expressed as: Wall thickness (WT, %) = [(medial thickness ×
2)/external diameter] × 100 (9).
Statistical analysis
Data are presented as the mean ± standard deviation,
and were statistically analyzed using SPSS statistical software
(version 13.0; SPSS, Inc., Chicago, IL, USA). Differences were
compared using one-way analysis of variance (ANOVA) tests.
Correlations were calculated according to Pearson’s correlation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Characteristics of the experimental
rats
Approximately 10 days following the
co-administration of MCT and PBS, rats exhibited shortness of
breath and a reduction in locomotor activity and food intake
compared with controls. In addition, the body weight of the model
rats was shown to have decreased (172.3±19.6 versus 208.4±18.5 g,
P<0.05). The administration of rhG-CSF resulted in a marked
improvement in the status of the rats, and there were no
differences in body weight between the rhG-CSF treatment group
(203.7±19.7 g) and the controls (P>0.05), while only the level
of locomotor activity remained slightly decreased. However, under
the negative intervention of L-NAME, the body weight of the rats
(170.5±18.2 g) was markedly decreased compared with that of the
rhG-CSF treatment group (P<0.05).
Changes in hemodynamics and
histology
Twenty-one days following the injection of MCT, the
RVSP of the rats in the model group was increased compared with
that of the controls (48.13±2.85 versus 27.88±3.04 mmHg,
P<0.01), while the MAoP was decreased from 120.33±18.25 mmHg in
the control group to 97.24±17.52 mmHg in the model group. This
indicated the occurrence of PH. The administration of rhG-CSF led
to the RVSP of the rats being decreased significantly compared with
that of the model group (30.38±2.83 versus 48.13±2.85 mmHg,
P<0.01). No differences were detected between the rhG-CSF
treatment group and the controls (P>0.05). The MAoP of the rats
in the rhG-CSF treatment group (113.82±21.73 mmHg) was increased
compared with that of the model group (P<0.05), and was restored
to the level of the controls (P>0.05; Fig. 1E and F).
Histological examination indicated that medial
hypertrophy of the pulmonary arteriole smooth muscle was evident in
the model group (Fig. 1B) and that
the WT of the model group was increased compared with that of the
controls (31.74±3.09 versus 13.99±1.14%, P<0.01). The
administration of rhG-CSF was shown to markedly attenuate the
medial hypertrophy of the pulmonary arteriole smooth muscle
(Fig. 1C), while the WT of the
rhG-CSF treatment group (17.31±1.92%) was notably decreased
compared with that of the model group (P<0.01; Fig. 1G).
The protective effects of rhG-CSF on the MCT-induced
rat model of PH were markedly attenuated by the negative
intervention of L-NAME. The RVSP of rats in the L-NAME intervention
group (51.19±2.93 mmHg) was increased significantly compared with
that of the rhG-CSF treatment group (P<0.01), while the MAoP was
decreased (96.25±17.92 mmHg, P<0.01; Fig. 1E and F). Histological examination
indicated that medial hypertrophy of the pulmonary arteriole smooth
muscle was evident, similar to that observed in the model group
(Fig. 1D), and the WT
(31.97±3.22%) was increased compared with that of the rhG-CSF
treatment group (P<0.01; Fig.
1G).
EPC level in peripheral blood
The number of EPCs in the peripheral blood, assessed
using flow cytometry, was significantly lower in the model group 21
days subsequent to the injection of MCT compared with the controls
(0.016±0.007 versus 0.031±0.011%, P<0.01). The administration of
rhG-CSF was shown to notably increase the number of EPCs
(0.042±0.013%) compared with the numbers in the model and control
groups (P<0.01). The intervention of L-NAME attenuated the
effect of rhG-CSF on the circulating EPCs (0.015±0.007%, P<0.01;
Fig. 2A).
Plasma concentration of NO
The plasma concentration of NO was lower in the
model group (19.66±2.78 μmol/l) than in the control group
(54.31±3.81 μmol/l, P<0.01). The administration of rhG-CSF was
shown to significantly increase the plasma concentration of NO
compared with that in the model group (50.85±2.64 versus 19.66±2.78
μmol/l, P<0.01), and no differences in NO level were observed
between the rhG-CSF treatment and control groups (P>0.05). The
administration of L-NAME, which is an inhibitor NO synthase,
suppressed the upregulation of plasma NO mediated by rhG-CSF
(Fig. 2B). Furthermore, the plasma
concentration of NO in each group was positively correlated with
the number of circulating EPCs (P<0.05; Fig. 2C–E).
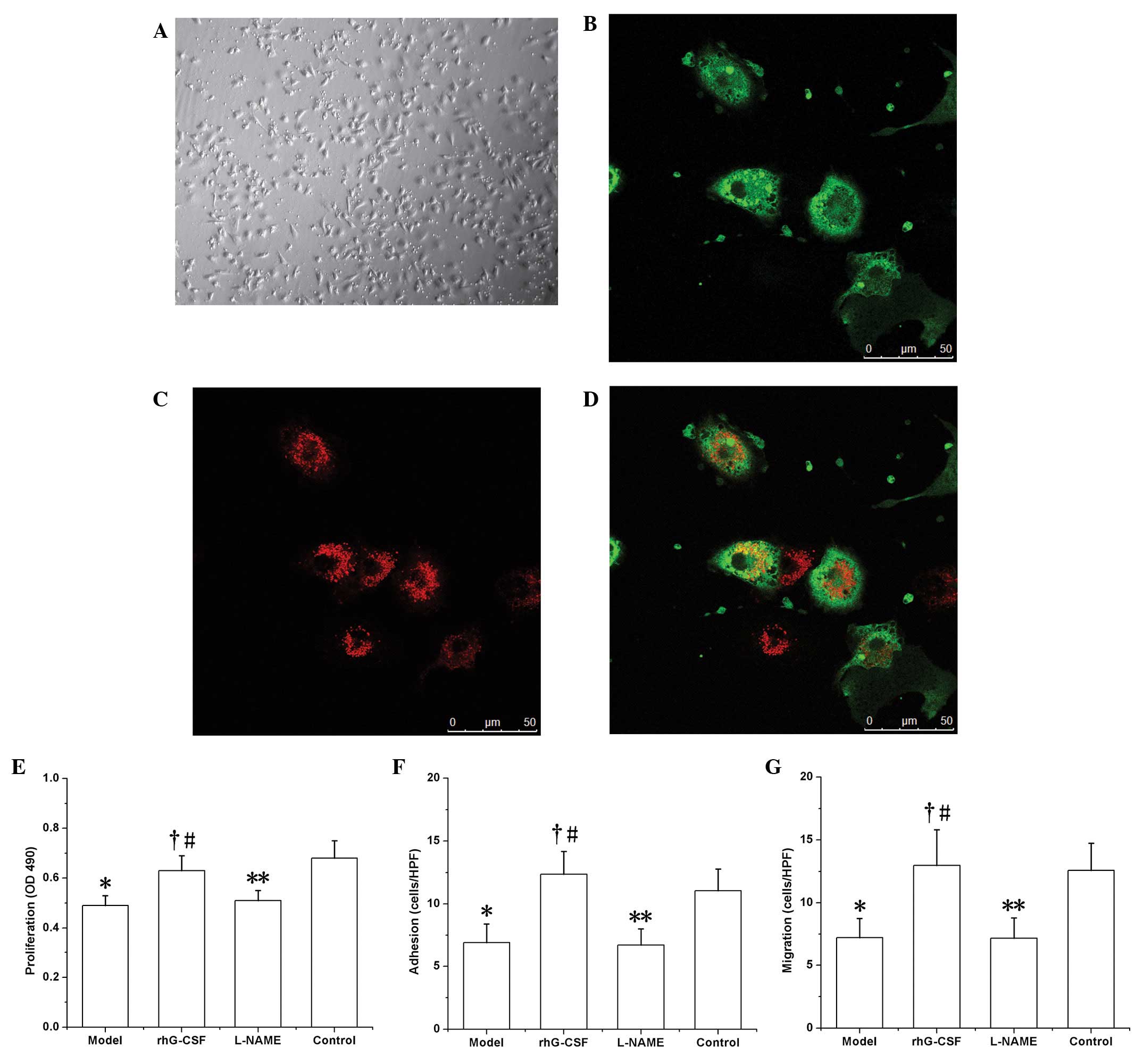
EPC growth in vitro
Following seven days of culture, abundant
spindle-like cells were observed to be adhered to the bottom of the
culture plate (Fig. 3A).
Subsequent to binding with FITC-UEA-1, these cells exhibited green
fluorescence (Fig. 3B), and red
fluorescence was exhibited following phagocytosis of DiI-acLDL
(Fig. 3C). The cells positive for
the two labels under laser scanning confocal microscopy (observed
as yellow fluorescence) were recognized as EPCs undergoing
differentiation (Fig. 3D).
EPC function in vitro
The functions of the EPCs were all downregulated in
the model group compared with those of the controls. In the test of
proliferation ability in vitro, the absorbance (at 490 nm;
OD) of the formazan supernatant (dissolved in DMSO) was 0.49±0.04
in the model group, which was decreased compared with that of the
control group (0.68±0.07, P<0.01). Similar results were observed
with regard to adhesion ability (6.93±1.47 cells/HPF in the model
group versus 11.05±1.73 cells/HPF in the control group, P<0.01)
and migratory ability (7.22±1.53 cells/HPF in the model group
versus 12.58±2.15 cells/HPF in the control group, P<0.01). In
the rhG-CSF treatment group, these functional indices were all
upregulated compared with those of the model group (proliferation,
OD 0.63±0.06; adhesion, 12.35±1.82 cells/HPF; migration, 12.97±2.84
cells/HPF; P<0.01; Fig. 3F–H).
Consistent with the effect of L-NAME on the number of EPCs in the
peripheral blood, the functions of the EPCs in the L-NAME
intervention group, i.e. proliferation (OD 0.51±0.04), adhesion
(6.73±1.28 cells/HPF) and migration (7.18±1.62 cells/HPF) were all
downregulated compared with those of the rhG-CSF treatment group
(P<0.01; Fig. 3E–G).
Discussion
In the present study, it was demonstrated that in a
rat model of PH, the number and function of circulating EPCs were
markedly decreased. Moreover, there was a downregulation in the
plasma concentration of NO, which was positively correlated with
the number of circulating EPCs. Administration of rhG-CSF elevated
the plasma level of NO, upregulated the number and function of
circulating EPCs and effectively improved pulmonary hemodynamics
and vascular reconstruction. Furthermore, the positive correlation
between the concentration of plasma NO and circulating EPCs was
also observed in the rhG-CSF treatment group. However, the
protective effects of rhG-CSF on PH were impaired by the
L-NAME-mediated downregulation of NO and the EPCs. These results
indicate that rhG-CSF prevents or attenuates the progression of
MCT-induced PH by improving vascular injury repair mechanisms via
the NO-mediated upregulation of EPCs.
PH is characterized by the persistent contraction
and medial hypertrophy of extensive pulmonary arterioles, which may
thus result in the abnormal elevation of pulmonary artery pressure.
Despite the fact that drug interference may improve the symptoms
and decrease the occurrence of heart attack, the effects are
partial and limited and right ventricular failure or mortality is
likely be the fate of numerous patients in the clinic. It is
therefore crucial to prevent the progression of the pathological
changes in the initial stages of this disease. Studies have shown
that the early pathological change in PH is the injury of the
arteriolar endothelium, resulting from excessive apoptosis of the
endothelial cells. This then induces vasodilatation dysfunction,
the over-proliferation of vascular smooth muscle cells and
fibroblasts, ultimately resulting in the occurrence of PH (19–21).
Thus, reconditioning the injured endothelium as early as possible
may prevent this course of the disease.
EPCs, which are co-precursors with hematopoietic
stem cells, have been suggested to be pivotal to the homeostasis
and repair of the vascular endothelium. Studies have shown that the
number of circulating EPCs was decreased in patients with PH
(5,6) and that PH was alleviated by the
transplantation of exogenous EPCs in an experimental animal model
(22,23), which suggested that the impaired
condition of the EPCs contributed to the occurrence of PH. An
appropriate strategy to upregulate EPCs may be used to prevent this
course effectively. In the present study, the changes in pulmonary
hemodynamics and histology that were evident following the
injection of MCT in SD rats appeared consistent with those in
patients with PH and were suited to performing further observations
on circulating EPCs. It was observed that the number of EPCs in the
peripheral blood decreased significantly in the PH models compared
with the number in the controls. In addition, when cultured in
vitro, the functions of EPCs, such as proliferation, adhesion
and migration, were downregulated. It is possible that the repair
processes secondary to the ongoing lesion of the vascular
endothelium may lead to the large consumption of circulating EPCs
and potentially exhaustion. Bone marrow EPCs and precursors, as the
reservoir of circulating EPCs, may also be impaired, in number and
function (24,25). A reduction in the protective
mechanism of EPCs therefore exacerbates vascular damage and
dysfunction.
Data have shown that G-CSF is able to increase the
number of circulating EPCs, upregulate the maturation and
proliferation capacity of EPCs (26) and then accelerate the repair of the
injured vessel and myocardium. Furthermore, G-CSF has exhibited a
protective effect in cardiovascular diseases (27). The mobilization of bone marrow stem
cells using G-CSF has also been shown to effectively to prevent the
progression of PH (9). In the
present study, it was demonstrated that, following the
administration of rhG-CSF, the number of EPCs in the peripheral
blood was increased significantly and the function of the EPCs was
upregulated. As a result, there was an improvement in the
pathological changes in the pulmonary artery, in addition to an
alleviation of pulmonary artery pressure.
To further investigate the possible mechanism
underlying the protective effect of G-CSF in PH, we measured the
plasma concentration of NO in each group. It was shown that the
level of plasma NO decreased significantly in the rat model of PH
compared with the controls, and that the administration of rhG-CSF
effectively induced the upregulation of NO and accelerated the
repair of the injured pulmonary artery endothelium by mobilizing
the bone marrow EPCs (10), while
reducing the apoptosis of EPCs (11). Furthermore, the level of NO was
demonstrated to be positively correlated with the number of
circulating EPCs in the PH model and rhG-CSF treatment groups.
However, the exact signaling pathway involved in the G-CSF-induced
upregulation of NO and EPCs was not elucidated in the current
study. The study by Ueda et al(14) demonstrated that G-CSF
phosphorylated and activated endothelial NO synthase (NOS) in the
acute stage of myocardial infarction and increased NO production,
thus inducing protective effects on the myocardium. Our observation
regarding the change in plasma NO levels in PH was consistent with
that of the study by Ueda et al, which suggests that the
same signaling pathway may be involved. Furthermore, the
administration of L-NAME, which acted as an NOS inhibitor in the
present study, was accompanied by reductions in the plasma
concentration of NO and in the number and function of circulating
EPCs, thus attenuating the protective effect of rhG-CSF in PH. This
further demonstrated the protective effects of the NO-mediated
upregulation of EPCs in PH.
In conclusion, the current study indicated that the
administration of rhG-CSF may represent a novel strategy for the
treatment of PH. The treatment may effectively prevent the disorder
by upregulating the number and function of circulating EPCs via the
NO system, and then accelerate the reparation of the pulmonary
artery endothelial lesion. However, the prospective efficacy and
side-effects of rhG-CSF remain important issues to be investigated
in advanced experimental and in vivo studies.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (30560159).
References
|
1
|
Asahara T, Murohara T, Sullivan A, Silver
M, van Der Zee R, Li T, Witzenbichler B, Schatteman G and Isner JM:
Isolation of putative progenitor endothelial cells for
angiogenesis. Science. 275:964–967. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fan Y, Shen F, Frenzel T, Zhu W, Ye J, Liu
J, Chen Y, Su H, Young WL and Yang GY: Endothelial progenitor cell
transplantation improves long-term stroke outcome in mice. Ann
Neurol. 67:488–497. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhattacharya V, McSweeney PA, Shi Q, Bruno
B, Ishida A, Nash R, Storb RF, Sauvage LR, Hammond WP and Wu MH:
Enhanced endothelialization and microvessel formation in polyester
grafts seeded with CD34+ bone marrow cells. Blood.
95:581–585. 2000.PubMed/NCBI
|
|
4
|
Zhao YD, Courtman DW, Deng Y, Kuqathasan
L, Zhang Q and Stewart DJ: Rescue of monocrotaline-induced
pulmonary arterial hypertension using bone marrow-derived
endothelial-like progenitor cells: efficacy of combined cell and
eNOS gene therapy in established disease. Circ Res. 96:442–450.
2005. View Article : Google Scholar
|
|
5
|
Diller GP, van Eijl S, Okonko DO, Howard
LS, Ali O, Thum T, Wort SJ, Bédard E, Gibbs JS, Bauersachs J, et
al: Circulating endothelial progenitor cells in patients with
Eisenmenger syndrome and idiopathic pulmonary arterial
hypertension. Circulation. 117:3020–3030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Junhui Z, Xingxiang W, Guosheng F, Yunpeng
S, Furong Z and Junzhu C: Reduced number and activity of
circulating endothelial progenitor cells in patients with
idiopathic pulmonary arterial hypertension. Respir Med.
102:1073–1079. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nagaya N, Kangawa K, Kanda M, Uematsu M,
Horio T, Fukuyama N, Hino J, Harada-Shiba M, Okumura H, Tabata Y,
et al: Hybrid cell-gene therapy for pulmonary hypertension based on
phagocytosing action of endothelial progenitor cells. Circulation.
108:889–895. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ohki Y, Heissig B, Sato Y, Akiyama H, Zhu
Z, Hicklin DJ, Shimada K, Ogawa H, Daida H, Hattori K and Ohsaka A:
Granulocyte colony-stimulating factor promotes neovascularization
by releasing vascular endothelial growth factor from neutrophils.
FASEB J. 19:2005–2007. 2005.
|
|
9
|
Maruyama H, Watanabe S, Kimura T, Liang J,
Nagasawa T, Onodera M, Aonuma K and Yamaguchi I: Granulocyte
colony-stimulating factor prevents progression of
monocrotaline-induced pulmonary arterial hypertension in rats. Circ
J. 71:138–143. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aicher A, Heeschen C, Mildner-Rihm C,
Urbich C, Ihling C, Technau-Ihling K, Zeiher AM and Dimmeler S:
Essential role of endothelial nitric oxide synthase for
mobilization of stem and progenitor cells. Nat Med. 9:1370–1376.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Laufs U, Werner N, Link A, Endres M,
Wassmann S, Jürgens K, Miche E, Böhm M and Nickenig G: Physical
training increases endothelial progenitor cells, inhibits neointima
formation, and enhances angiogenesis. Circulation. 109:220–226.
2004. View Article : Google Scholar
|
|
12
|
Fukumura D, Gohongi T, Kadambi A, Izumi Y,
Ang J, Yun CO, Buerk DG, Huang PL and Jain RK: Predominant role of
endothelial nitric oxide synthase in vascular endothelial growth
factor-induced angiogenesis and vascular permeability. Proc Natl
Acad Sci USA. 98:2604–2609. 2001. View Article : Google Scholar
|
|
13
|
Guthrie SM, Curtis LM, Mames RN, Simon GG,
Grant MB and Scott EW: The nitric oxide pathway modulates
hemangioblast activity of adult hematopoietic stem cells. Blood.
105:1916–1922. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ueda K, Takano H, Hasegawa H, Niitsuma Y,
Qin Y, Ohtsuka M and Komuro I: Granulocyte colony stimulating
factor directly inhibits myocardial ischemia-reperfusion injury
through Akt-endothelial NO synthase pathway. Arterioscler Thromb
Vasc Biol. 26:e108–113. 2006. View Article : Google Scholar
|
|
15
|
Shimada K, Okabe TA, Mikami Y, Hattori M,
Fujita M and Kishimoto C: Therapy with granulocyte
colony-stimulating factor in the chronic stage, but not in the
acute stage, improves experimental autoimmune myocarditis in rats
via nitric oxide. J Mol Cell Cardiol. 49:469–481. 2010. View Article : Google Scholar
|
|
16
|
Matsuda Y, Hoshikawa Y, Ameshima S, Suzuki
S, Okada Y, Tabata T, Sugawara T, Matsumura Y and Kondo T: Effects
of peroxisome proliferator-activated receptor gamma ligands on
monocrotaline-induced pulmonary hypertension in rats. Nihon Kokyuki
Gakkai Zasshi. 43:283–288. 2005.(In Japanese).
|
|
17
|
Chakroborty D, Chowdhury UR, Sarkar C,
Baral R, Dasgupta PS and Basu S: Dopamine regulates endothelial
progenitor cell mobilization from mouse bone marrow in tumor
vascularization. J Clin Invest. 118:1380–1389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu MG, Men LN, Zhao CY, Zhao X, Wang YX,
Meng XC, Shen DR, Meng BY, Zhang Q and Wang T: The number and
function of circulating endothelial progenitor cells in patients
with Kawasaki disease. Eur J Pediatr. 169:289–296. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gurbanov E and Shiliang X: The key role of
apoptosis in the pathogenesis and treatment of pulmonary
hypertension. Eur J Cardiothorac Surg. 30:499–507. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tuder RM, Marecki JC, Richter A,
Fijalkowska I and Flores S: Pathology of pulmonary hypertension.
Clin Chest Med. 28:23–42. 2007. View Article : Google Scholar
|
|
21
|
Budhiraja R, Tuder RM and Hassoun PM:
Endothelial dysfunction in pulmonary hypertension. Circulation.
109:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ormiston ML, Deng Y, Stewart DJ and
Courtman DW: Innate immunity in the therapeutic actions of
endothelial progenitor cells in pulmonary hypertension. Am J Respir
Cell Mol Biol. 43:546–554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takahashi M, Nakamura T, Toba T, Kajiwara
N, Kato H and Shimizu Y: Transplantation of endothelial progenitor
cells into the lung to alleviate pulmonary hypertension in dogs.
Tissue Eng. 10:771–779. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Valgimigli M, Rigolin GM, Fucili A, Porta
MD, Soukhomovskaia O, Malagutti P, Bugli AM, Bragotti LZ,
Francolini G, Mauro E, et al: CD34+ and endothelial
progenitor cells in patients with various degrees of congestive
heart failure. Circulation. 110:1209–1212. 2004.
|
|
25
|
Kissel CK, Lehmann R, Assmus B, Aicher A,
Honold J, Fischer-Rasokat U, Heeschen C, Spyridopoulos I, Dimmeler
S and Zeiher AM: Selective functional exhaustion of hematopoietic
progenitor cells in the bone marrow of patients with postinfarction
heart failure. J Am Coll Cardiol. 49:2341–2349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Powell TM, Paul JD, Hill JM, Thompson M,
Benjamin M, Rodrigo M, McCoy JP, Read EJ, Khuu HM, Leitman SF, et
al: Granulocyte colony-stimulating factor mobilizes functional
endothelial progenitor cells in patients with coronary artery
disease. Arterioscler Thromb Vasc Biol. 25:296–301. 2005.
View Article : Google Scholar
|
|
27
|
Kovacic JC, Muller DW and Graham RM:
Actions and therapeutic potential of G-CSF and GM-CSF in
cardiovascular disease. J Mol Cell Cardiol. 42:19–33. 2007.
View Article : Google Scholar : PubMed/NCBI
|