Introduction
With >500 microRNA (miRNA) genes identified
experimentally in the human genome and a plethora of
computationally-predicted mRNA targets, it is believed that these
small RNAs have a central role in diverse cellular and
developmental processes. Therefore, the aberrant expression of
miRNA genes may lead to human disease, including cancer. Several
studies have confirmed that miRNAs regulate cell proliferation and
apoptosis (1–3). Arsenic trioxide (ATO), an ancient
traditional Chinese medicine, has been successfully used as a
therapeutic agent for leukemia (4). Drug resistance and toxicity are major
concerns with this treatment. miRNAs are endogenous small
non-coding RNA molecules that may modulate cellular sensitivity to
anticancer drugs (5). miR-203 is
overexpressed in pancreatic adenocarcinoma cells and demonstrates a
correlation with poor prognosis in patients that have undergone
pancreatectomy. It has also been proposed as a tumor-suppressive
miRNA in hepatocellular carcinoma (6,7).
However, miR-203 has been rarely characterized in chronic
myelogenous leukemia (CML). In our previous study, a eukaryotic
expression vector carrying the hsa-miR-203 gene was successfully
constructed and PmiR-203 was able to effectively inhibit the
proliferation and promote the apoptosis of K562 cells (8). Therefore, we questioned whether
miR-203 modulates the chemosensitivity of leukemia cells. In the
present study, we evaluated the role of PmiR-203 and its effect on
ATO treatment. We aimed to provide mechanistic evidence for the
synergistic effects of PmiR-203 and ATO in K562 leukemia cells.
Consequently, the potential of miR-203 should be explored as a gene
therapy for the treatment of leukemia.
Materials and methods
Materials
Fetal calf serum (FCS) and Dulbecco’s modified
Eagle’s medium (DMEM)/F12 were obtained from Invitrogen Life
Technologies (Carlsbad, CA, USA). NotI, XhoI (Takara
Bio Inc., Shiga, Japan), T4 DNA Ligase (Takara Bio Inc.), RNasin
inhibitor (Promega Corporation, Madison, WI, USA), Hoechst 33258,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide
(PI; Sigma, St. Louis, MO, USA) were also used in this study.
Design and construction of the eukaryotic
expression vector expressing hsa-miR-203
The mature miRNA-203 sequences are available from
the miRNA Registry (5′-GTGAAATGTTTAGGACCACTAG-3′). In order to
prevent the formation of a termination signal, TTGGCCACTGACT was
selected as the region in a miRNA expression vector template. TGCT
was added to the 5′ positive-sense strand template of the miRNA
expression vector and GTCC was added to the 5′ antisense strand
template. Further, a non-specific sequence was designed and sent to
GenePharma Co., Ltd. (Shanghai, China) for synthesis and finally, a
eukaryotic hsa-miR-203 expression vector was successfully
constructed. The assay was performed as previously described
(8).
Cell line and DNA transfection
The K562 (CML) cell line (Shanghai Institute of Cell
Biology, China) was grown in DMEM/F12 containing 10% FCS at 37°C in
a humidified atmosphere with 5% CO2 in a Thermo FORMA
3110 incubator (Thermo Scientific, Marietta, OH, USA). K562 cells
in the exponential phase of growth were seeded in 96- or 24-well
plates (Costar, Cambridge, MA, USA) and transfected with the
plasmid using Lipofectamine 2000 reagent (Invitrogen; 1:1.2
volume/mass ratio of Lipofectamine 2000 to the plasmid) in
serum-free DMEM/F12 for 6 h. At the end of transfection, the cells
were incubated in medium containing 10% FCS. Transfection of
PmiR-203 into the K562 cells with a final concentration of 500
ng/μl was performed according to the manufacturer’s
instructions.
Plasmid construction and site-directed
mutagenesis
The 3′ untranslated regions (3′-UTRs) of the bcr/abl
genes were amplified by polymerase chain reaction (PCR) using
genomic DNA of the K562 cell line and cloned downstream of the
Renilla luciferase open reading frame in the psiCHECK-2 vector
(Promega) using XhoI and NotI restriction sites. The
bcr/abl primers (CCGCTCGAGCAGCAGTCAGGGGTCAGG and
ATAAGAATGCGGCCGCTTCTAATGTAAACACTG ATTTATTTA) were designed to bind
to position 1992 of the bcr/abl genomic sequence. Tandem mutations
were introduced to the seed region of the miR-203 binding site in
the primers; UGUAAAGU was substituted by AUCGAUC and the construct
was named bcr/abl-mut-UTR.
Luciferase reporter assay
For luciferase reporter assays, cells were seeded in
96-well plates and cotransfected with 25 nM miRNA mimics or 100 nM
hairpin inhibitors together with 15 ng/well psiCHECK-2 reporter
vectors. Forty-eight hours after transfection, luciferase activity
was measured using the dual-luciferase reporter assay system kit
(Promega) according to the manufacturer’s instructions and a Tecan
M200 luminescence reader (Tecan Group Ltd., Männedorf,
Switzerland). Values were double normalized to firefly luciferase
activity and to cells transfected with empty psiCHECK-2 control
vectors. The sequenc inclued Bcr/abl 3′UTR,
5′-UCUGAGUUCUUGAAGCAUUUCAA-3′, hsa-miR-203,
3′-GAUCACCAGGAUUUGUAAAGUG-5′ and Bcr/abl-mut 3′UTR,
5′-UCUGAGUUCUUGAAGAUCGAUCA-3′.
ATO sensitivity assay
The viability of K562 cells was determined by the
MTT assay. Briefly, cells at a density of 5×104 cells/
ml were transfected with PmiR-203 or a negative control (NC;
plasmid containing a scrambled sequence, 0.8 μg/ml) in the
presence of Lipofectamine 2000 and serum-free DMEM/F12 media for 6
h. The cells were plated in 96-well plates in medium containing 10%
FCS for another 48 h, with the presence of varying concentrations
of ATO (1.25, 2.5, 5.0, 10.0 and 20.0 μg/ml). Then, 20
μl MTT stock solution (5 mg/ml) was added to each well to a
final MTT concentration of 0.45 mg/ml and the plate was incubated
for 4 h at 37°C. The medium was then removed and dimethyl sulfoxide
(DMSO; 150 μl) was added to dissolve the blue formazan
crystals at room temperature for 30 min. The viability of the cells
was assessed by absorbance at 570 nm on a Bio-Rad microtiter plate
reader (Hercules, CA, USA). The IC50 values were
determined using IC50 software (Northwest A & F
University, Beijing, China).
Hoechst 33258 staining
K562 cells transfected with PmiR-203 (0.8
μg/ml) were incubated for 48 h. Additionally, K562 cells
treated with ATO (10 μg/ml) were also grown for 48 h. Then,
the cells were collected by centrifuging at 4°C for 6 min at 111.8
× g and washed in phosphate-buffered saline (PBS). The cells were
sprayed onto glass slides, air-dried and fixed with methanol for 10
min. Then, the cells were washed with buffer twice, stained with
Hoechst 33258 for 10 min, washed with distilled water and finally
air-dried again. The morphology of the K562 cells was observed
under a fluorescent microscope.
Detection of mitochondrial membrane
potential
K562 human CML cells were incubated in 12-well
flat-bottomed plates at a concentration of 1.0×106
cells/ml. Experimental groups: 1, control group with blank cells
(control); 2, negative control (NC, containing plasmid, Scramble
sequence 0.8 μg/ml); 3, PmiR-203; 4, ATO. K562 cells
transfected with PmiR-203 (0.8 μg/ml) were incubated for 48
h. Meanwhile, K562 cells treated with ATO (10 μg/ml) were
also grown for 48 h. The cells were collected by centrifugation at
4°C for 6 min at 111.8 × g and washed in PBS. The cells were
sprayed on glass slides, air-dried and fixed with methanol for 10
min, and then washed with buffer twice, stained with Hoechst 33258
stain for 10 min, washed with distilled water, and finally air
dried. The morphology of the K562 cells was observed under a
fluorescent microscope. After cultivation for 48 h, the cells were
collected and washed with PBS. They were then suspended in 500
μl JC-1 culture fluid, incubated in a 5% CO2
atmosphere at 37°C for 20 min and washed twice with incubation
buffer. The cells were then suspended in 500 μl incubation
buffer. A drop of the cell suspension was placed on a microscope
slide, covered with a cover glass and then observed under a
fluorescence microscope.
Flow cytometric analysis of the cell
cycle and apoptosis
K562 cells were seeded at a density of
1.0×105 cells/ml (500 μl/well) in 24-well plates
(Costar) and transfected with miR-203 (0.8 μg/ml) using
Lipofectamine 2000 reagent in serum-free DMEM/F12 for 6 h.
Following transfection, 500 μl appropriate growth medium
containing 20% FCS was added to each well with a total volume of
1,000 μl. Cells were continuously incubated for another 48 h
in the presence of 10.0 mg/ml ATO, then harvested, washed twice
with PBS and fixed with 70% ethanol. They were also treated with
RNase A (1 mg/ml) following the elimination of ethanol. Finally,
the cells were stained with PI solution (50 μg/ml). The cell
cycles were analyzed by flow cytometry according to the content of
DNA (Beckman Coulter Elite, Fullerton, CA, USA). Pretreatment of
the K562 cells was performed as described above. Viable cells were
collected and double-stained with FITC-conjugated Annexin V and PI.
For each sample, data from ∼10,000 cells were recorded in list mode
on logarithmic scales. Apoptosis and necrosis were analyzed by
quadrant statistics on PI-negative, Annexin V positive and PI and
Annexin V-positive cells.
Detection of caspase-3 and caspase-9
activity
Pretreatment of K562 cells was performed as follows:
1, control group with blank cells (control); 2, negative control
(NC,containing plasmid, Scramble sequence 0.8 μg/ml); 3,
PmiR-203 (0.8 μg/ml); 4, ATO (10 μg/ml); 5,
PmiR-203+ATO. The total RNA from treated cells was extracted in
Trizol (Invitrogen) and was quantified by an ultraviolet
spectrophotometer (UVP, Upland, CA, USA) at a wavelength of 260 nm.
The Bcr/abl expression level was determined by real-time PCR.
Reverse transcribed with an M-MLV reverse transcriptase. A 20
μl reverse transcription (RT) reaction was incubated at 37°C
for 30 min. Real-time PCR was performed using a PCR amplifier
(Bio-Rad). The PCR cycles were as follows. There was an initial
denaturation at 94°C for 3 min. The reaction was repeated for 40
cycles; each cycle consisted of denaturing at 94°C for 20 sec,
annealing at 50°C for 25 sec and synthesis at 72°C for 20 sec,
according to the manufacturer’s instructions. The GAPDH group was
used as the internal control. The expression level was calculated
using CT and 2−ΔΔCt. Then, the cells were lysed with
RIPA buffer in the presence of a proteinase inhibitor (Shenergy
Biocolor BioScience and Technology Co., Ltd., Shanghai, China). The
protein concentration was determined using bicinchoninic acid (BCA;
Bioss, Beijing, China) and adjusted to 2.0 μg/μl.
Then, 50 μl cell suspension containing 100 μg
proteins was extracted from each well. Lysis buffer (50 μl)
was used as the control. Next, 0.5 μl dithiothreitol (DTT)
was added for each 50 μl 2X reaction buffer and 50 μl
prepared 2X reaction buffer was added to each well. Additionally, 5
μl caspase-3 substrate or caspase-9 substrate was added to
the wells. The plates were incubated in the dark at 37°C for 4 h
and the A405 value was detected using an enzyme-labeled instrument
(Bio-Tek, Winooski, VT, USA).
Analysis of bcr/abl mRNA levels
Pretreatment of K562 cells was performed as
described above. The total RNA from the treated cells was extracted
using TRIzol (Invitrogen) and was quantified using an ultraviolet
spectrophotometer (UVP, Upland, CA, USA) at a wavelength of 260 nm.
The bcr/abl expression level was determined by real-time PCR using
M-MLV reverse transcriptase. A 20 μl reverse transcription
reaction was incubated at 37°C for 30 min. Real-time PCR was
performed using a PCR amplifier (Bio-Rad). The PCR cycles were as
follows: initial denaturation at 94°C for 3 min, then 40 cycles of
denaturing at 94°C for 20 sec, annealing at 50°C for 25 sec and
synthesis at 72°C for 20 sec, as per the manufacturer’s
instructions. The GAPDH gene was used as the internal control. The
expression level was calculated using cycle threshold (Ct) values
and the 2−ΔΔCt method.
Western blot analysis of cytochrome c
protein levels
The cells were analyzed in RIPA buffer in the
presence of a proteinase inhibitor. The protein concentration was
determined using BCA. Proteins (30 μg) were separated using
10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred to a nitrocellulose membrane. Membranes
were probed with primary antibodies against cytochrome c
(rabbit polyclonal; Cell Biotech, Tianjin, China) at room
temperature for 2 h, washed extensively with 0.1% Tween-20 in PBS
and incubated with secondary antibodies conjugated with horseradish
peroxidase at a dilution of 1:10,000 (Pharmingen, Becton Dickinson,
San Diego, California, USA), at room temperature for 3 h.
Development was performed using an enhanced chemiluminescence (ECL)
system (Amersham Pharmacia Biotech, Amersham, UK).
Statistical analysis
All results and data were confirmed in at least
three separate experiments. Data are expressed as mean ± standard
deviation (SD). Statistical comparisons were made by one-way
analysis of variance (ANOVA). P<0.05 was considered to indicate
a statistically significant difference.
Results
bcr/abl is directly regulated by
miR-203
To confirm bcr/abl as a direct target of miR-203 in
K562 leukemia cells, we constructed the dual-luciferase reporter
(psi-CHECK) containing either the miR-203 recognition sequence or a
mutated sequence from the 3′UTR of bcr/abl mRNA immediately
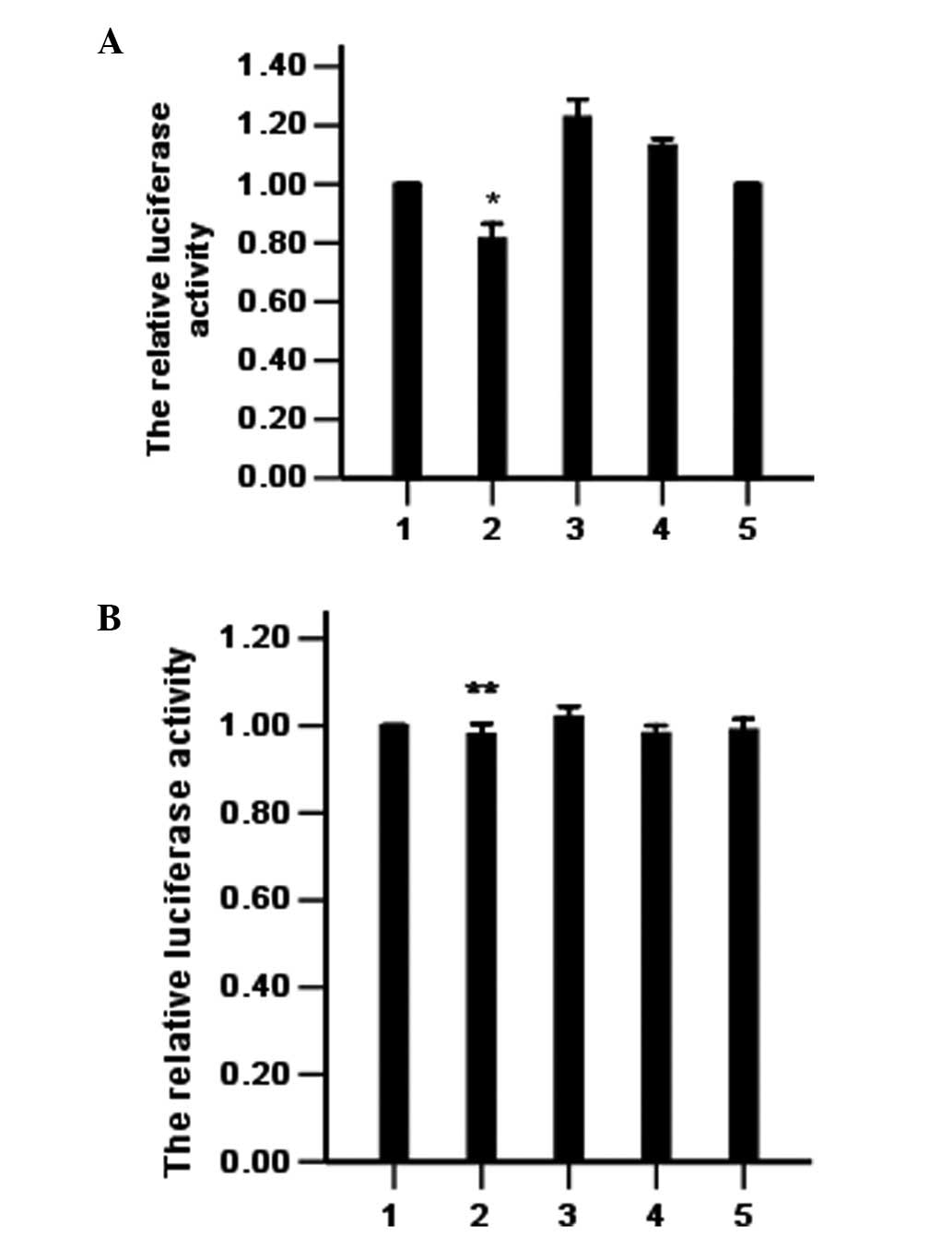
downstream of the luciferase gene. As shown in Fig. 1, transfection with miR-203 reduced
the activity of the bcr/abl-UTR reporter in the K562 cells but did
not affect the activity of the bcr/abl-mut-UTR reporter. These
results suggest that bcr/abl is a target of miR-203 in K562
cells.
PmiR-203 promotes ATO sensitivity in
leukemia cells
In this study, we determined the effect of PmiR-203
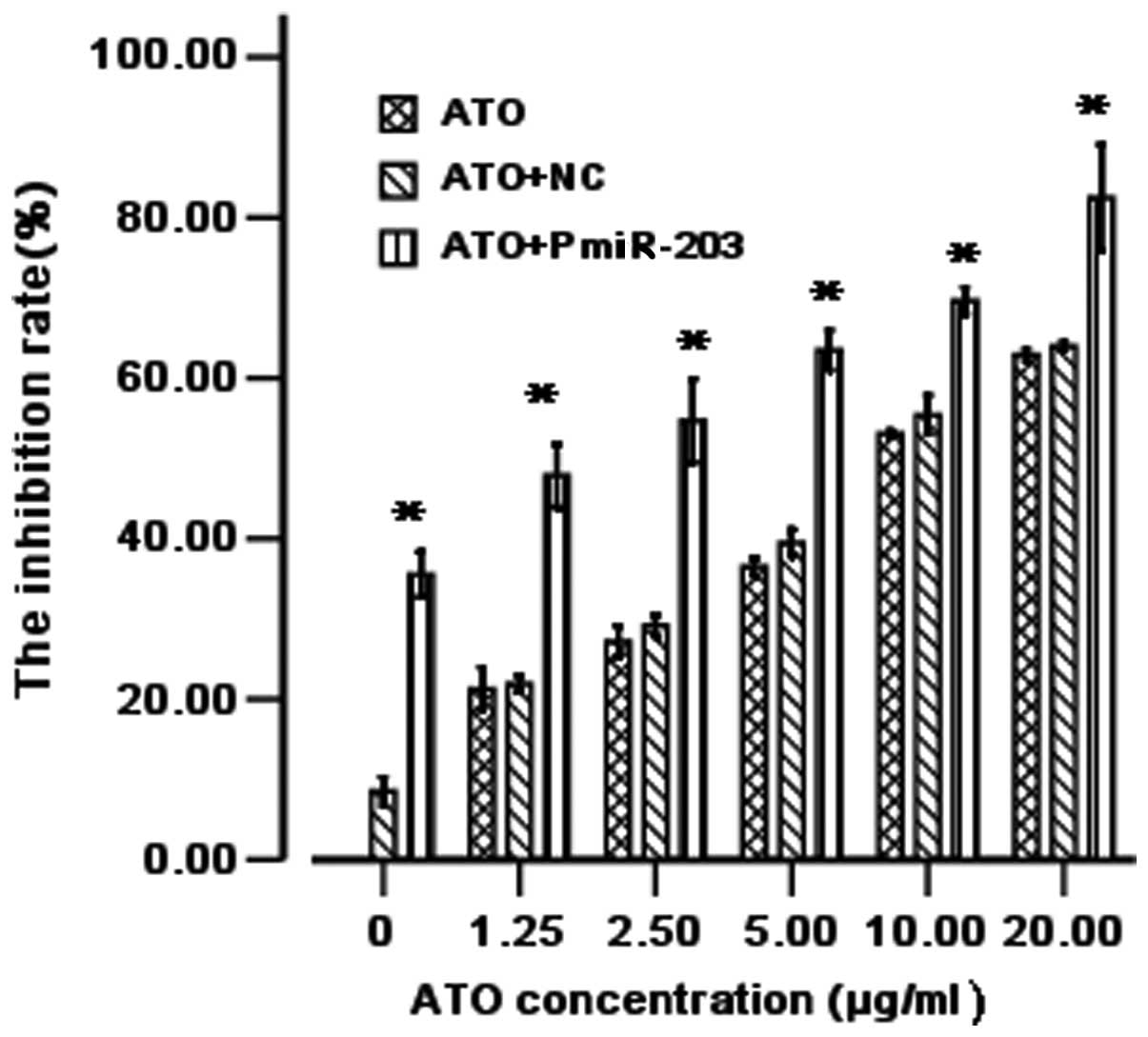
on cell viability (alone or in combination with ATO). As shown in
Fig. 2, PmiR-203 alone effectively
inhibited cell viability. The data indicate that PmiR-203 is also
able to increase the ATO-induced inhibitory effects on K562 cells.
Thus, PmiR-203 significantly decreases the IC50 values
of ATO. When used alone, the IC50 of ATO was 6.49
μg/ml and when used in combination with the NC, the
IC50 of ATO was 5.8 μg/ml. However, when used in
combination with PmiR-203, the IC50 of ATO was 2.45
μg/ml. The susceptibility of the cells increased 2.64-fold
(Fig. 2).
Cell morphology validates cell
apoptosis
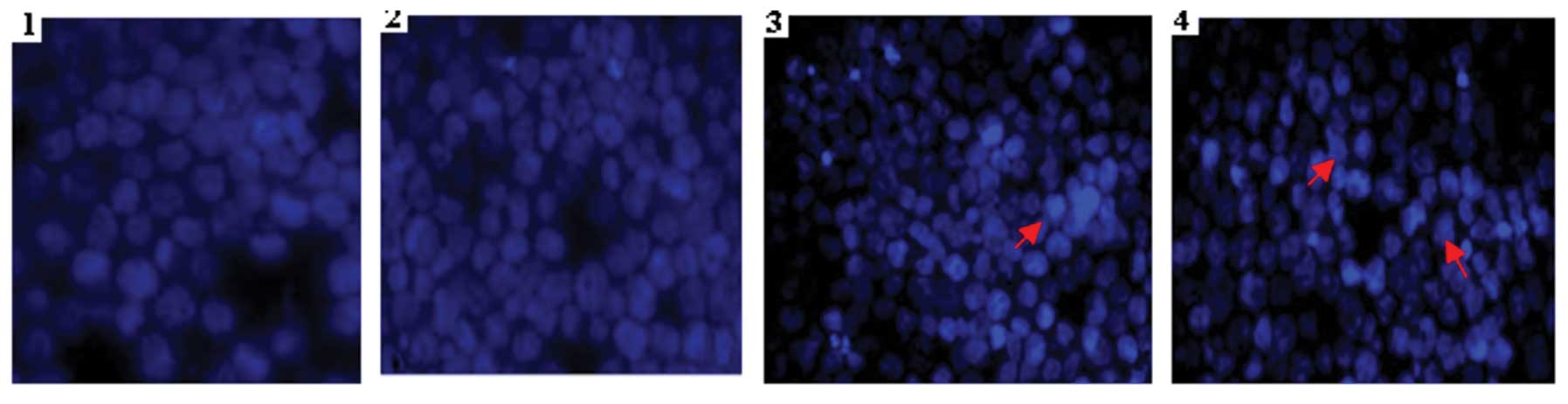
Hoechst 33258 staining (Fig. 3) also revealed that K562 cells in
the PmiR-203 and ATO groups acquired typical features of apoptosis,
including cell shrinkage, nuclear pyknosis and apoptotic bodies at
48 h post-transfection. The results confirmed that PmiR-203 and ATO
are able to induce cell apoptosis.
Detection of the mitochondrial membrane
potential
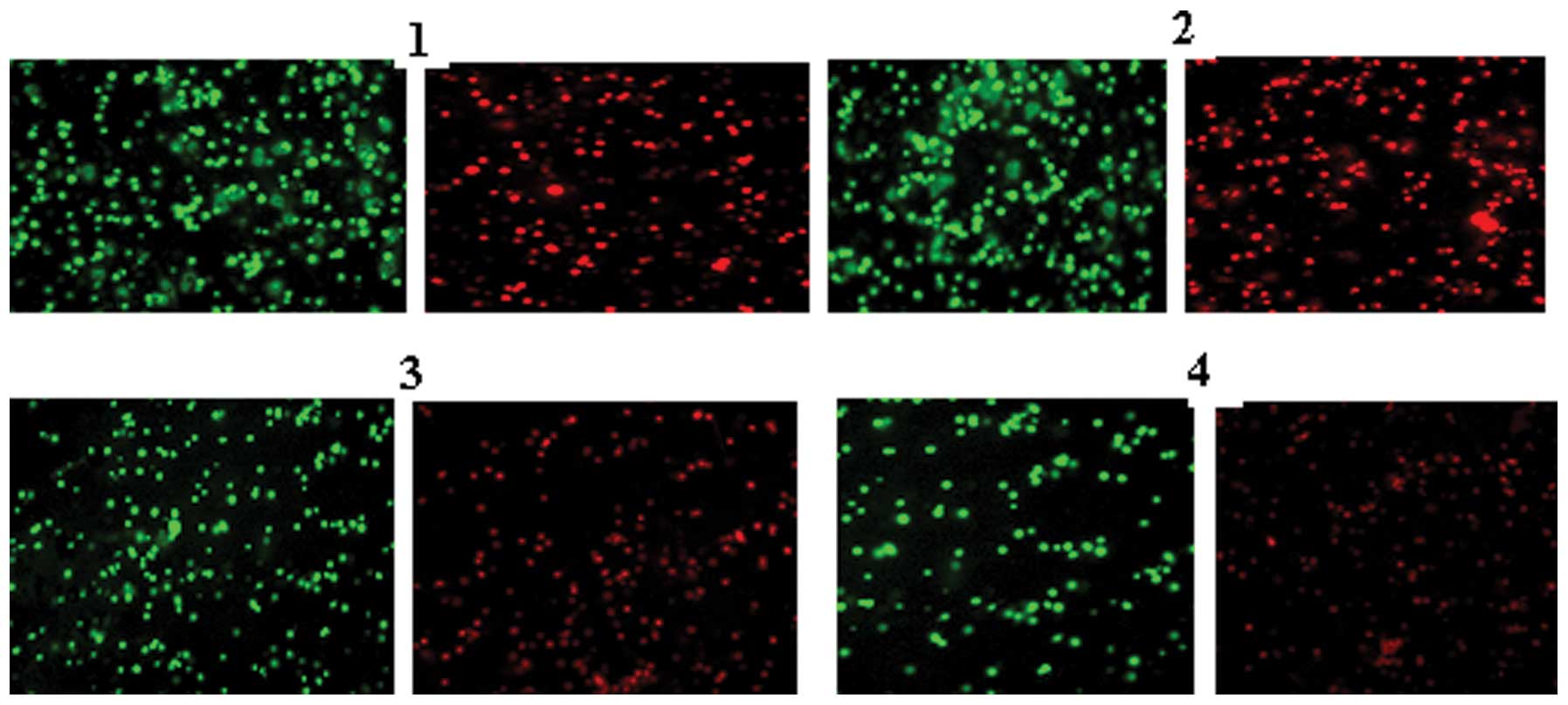
Under a bicolor filter, while normal cells show
high-intensity green and red fluorescence, apoptotic cells show
high-intensity green fluorescence and low-intensity red
fluorescence. The results of the negative control group were
similar to those of the cell control group. All the cells in the
former group demonstrated high-intensity green and red
fluorescence, while the cells in the PmiR-203+ATO group
demonstrated high-intensity green fluorescence and low-intensity
red fluorescence (Fig. 4).
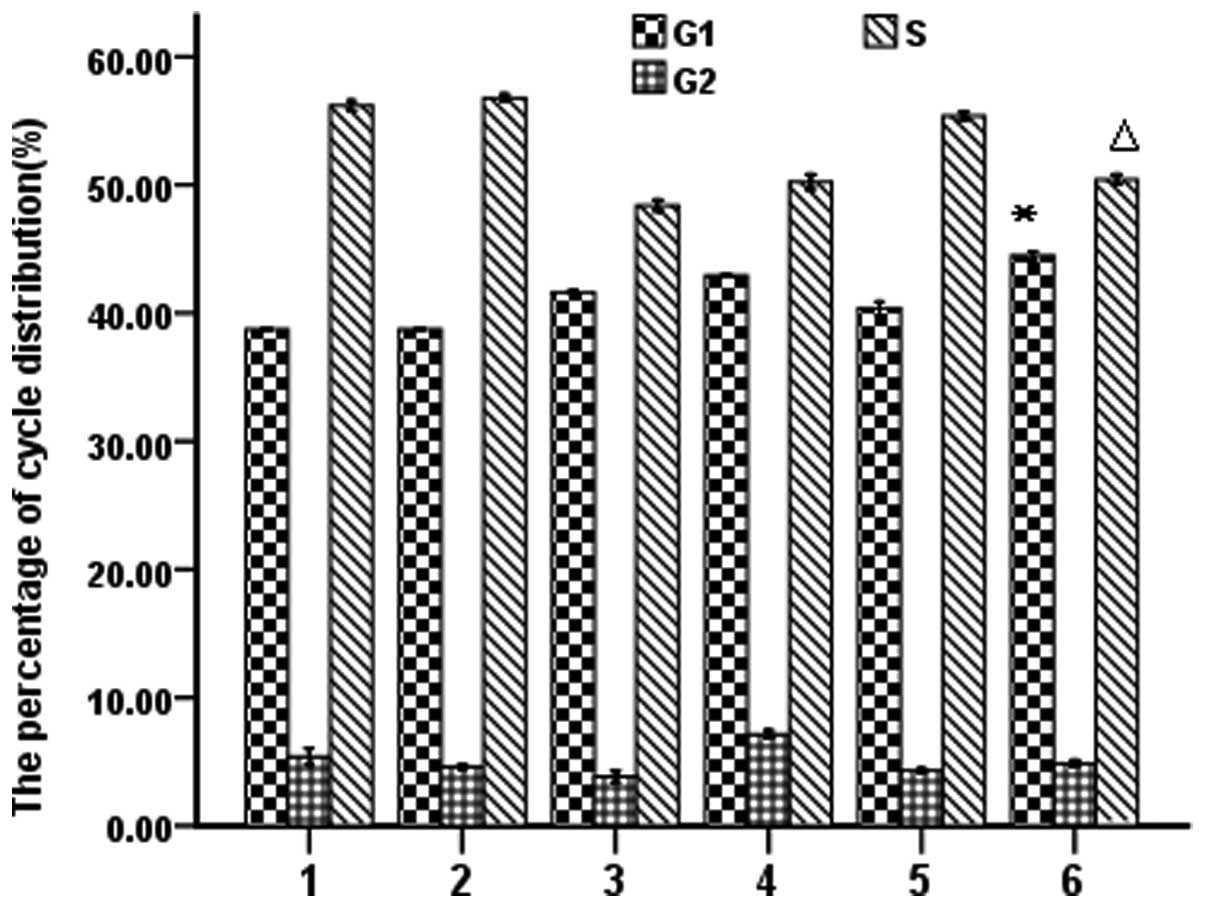
PmiR-203 regulates the cell cycle as
determined by flow cytometry
To explore the effects of PmiR-203 on the cell
cycle, PmiR-203 treatment alone or in combination with 10.0
μg/ml ATO was investigated in K562 cells. Cells were stained
with PI solution. Cell cycles were analyzed by flow cytometry
according to the DNA content. As shown in Fig. 5, PmiR-203 and ATO were able to
independently induce G1 phase arrest. The increased sub-G1-phase
cell population indicates that PmiR-203 and ATO induce apoptosis in
K562 cells (Fig. 5).
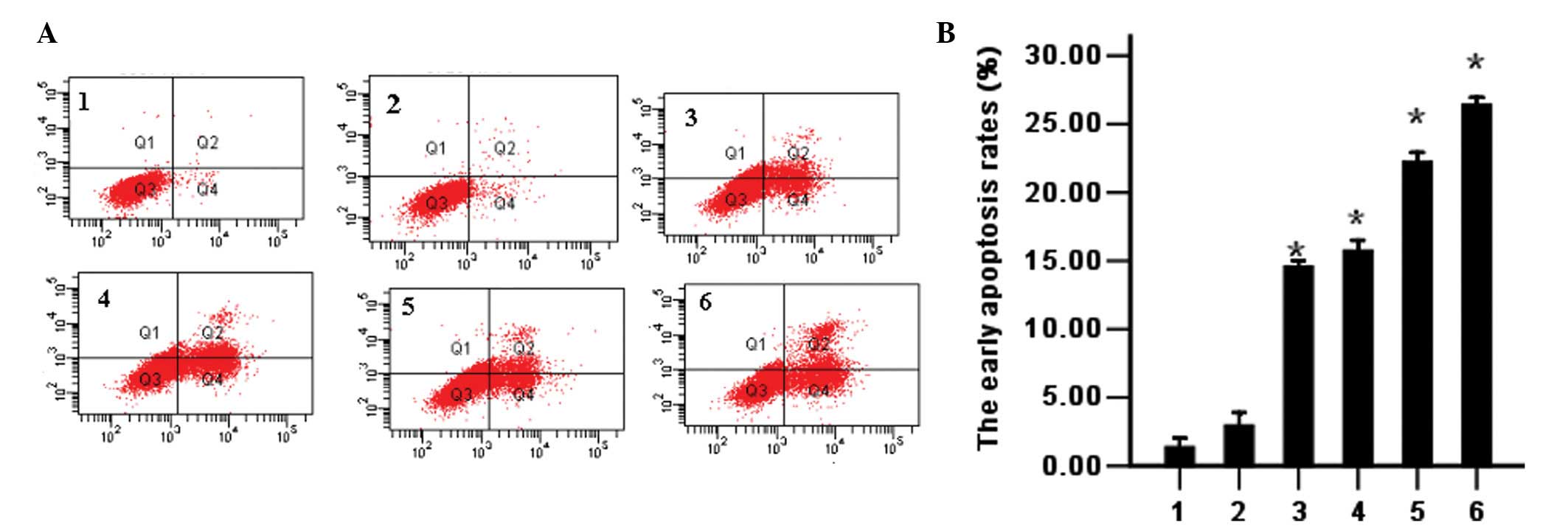
PmiR-203 induces cell apoptosis as
determined by flow cytometry.
PmiR-203 was used alone or in combination with ATO
in K562 cells. Apoptosis of the K562 cells was detected by flow
cytometry using double-staining with Annexin V and PI. The results
demonstrated that PmiR-203 alone induces cell apoptosis and
promotes the ATO-induced apoptosis of cells as compared with
controls (Fig. 6).
Detection of caspase-3 and caspase-9
activities using a colorimetric method
The caspase family plays an important role in the
mediation of apoptotic progress and caspase-3 and caspase-9 are
considered key performing elements in the process of apoptosis. The
activities of these proteins are associated with DNA fragmentation,
chromatin condensation and apoptotic body formation. Under normal
conditions, caspase-3 and caspase-9 exist in the cytoplasm in the
inactive pro-enzyme form; however, during apoptosis, they are
activated and split the corresponding substrate in the cytoplasm
and nucleus, finally leading to apoptosis. The activities of
caspase-3 and caspase-9 were significantly increased in the
PmiR-203, ATO and PmiR-203+ATO groups, indicating that PmiR-203 and
ATO activate caspase-3 and caspase-9 (Table I).
 | Table IActivities of caspase-3 and caspase-9
(n=3). |
Table I
Activities of caspase-3 and caspase-9
(n=3).
| Group | Caspase-3 | Caspase-9 |
|---|
| Control | 0.103±0.02 | 0.105±0.01 |
| NC | 0.216±0.01 | 0.260±0.03 |
| PmiR-203 | 0.452±0.01a | 0.490±0.09a |
| ATO | 0.434±0.03a | 0.484±0.04a |
| ATO+PmiR-203 | 0.623±0.05a | 0.636±0.06a |
| ATO+NC | 0.412±0.02a | 0.425±0.07a |
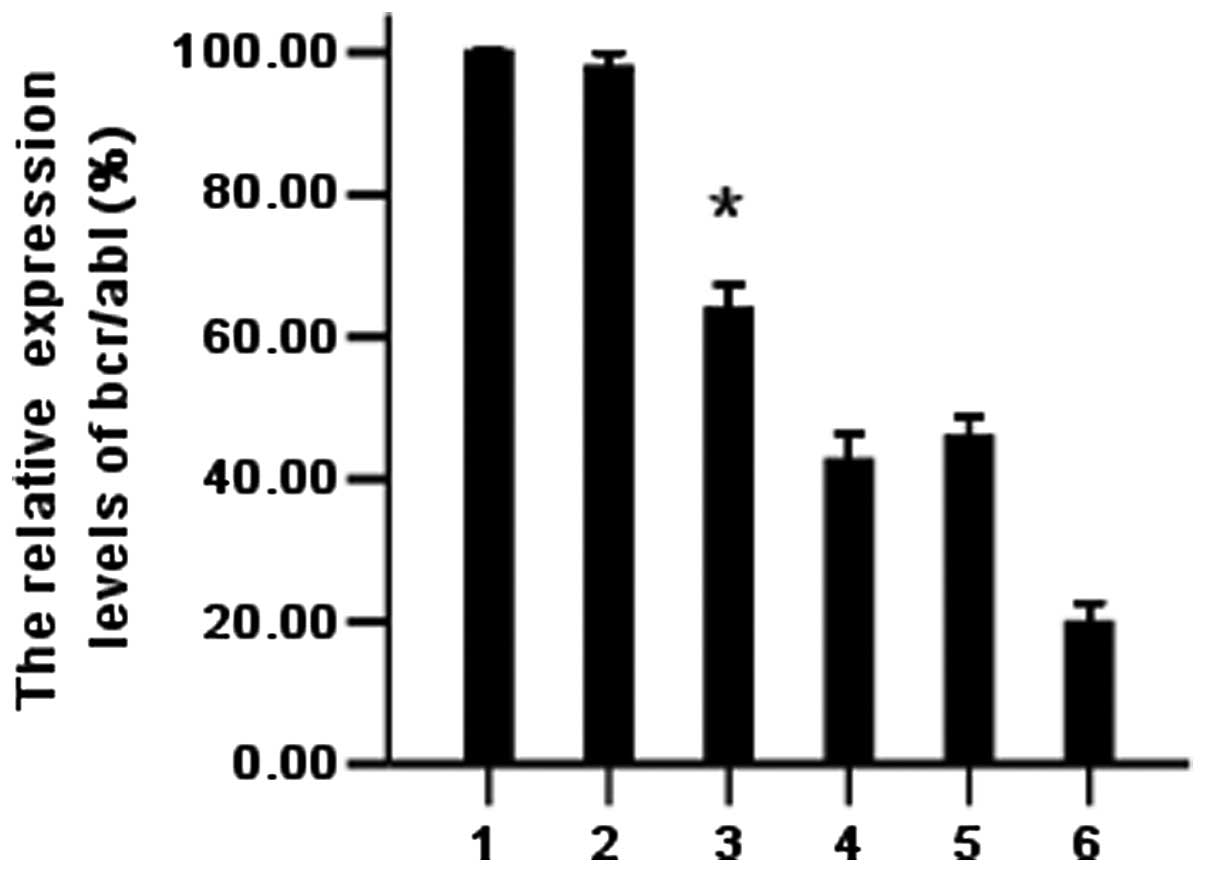
PmiR-203 downregulates the bcr/abl mRNA
level as detected by real-time PCR
To evaluate the effects of PmiR-203 on the bcr/abl
mRNA level of the cells, K562 cells transfected with PmiR-203 were
processed and analyzed for mRNAs. The bcr/abl expression was
determined by quantitative real-time PCR. GAPDH was used as the
internal control. The fold-change of the expression level was
calculated using 2−ΔΔCt, as described in Materials and
methods. The 2−ΔΔCt value of K562 cells treated with
PmiR-203 was only 0.189 (18.9%). The results indicate that PmiR-203
effectively downregulates the bcr/abl level (Fig. 7).
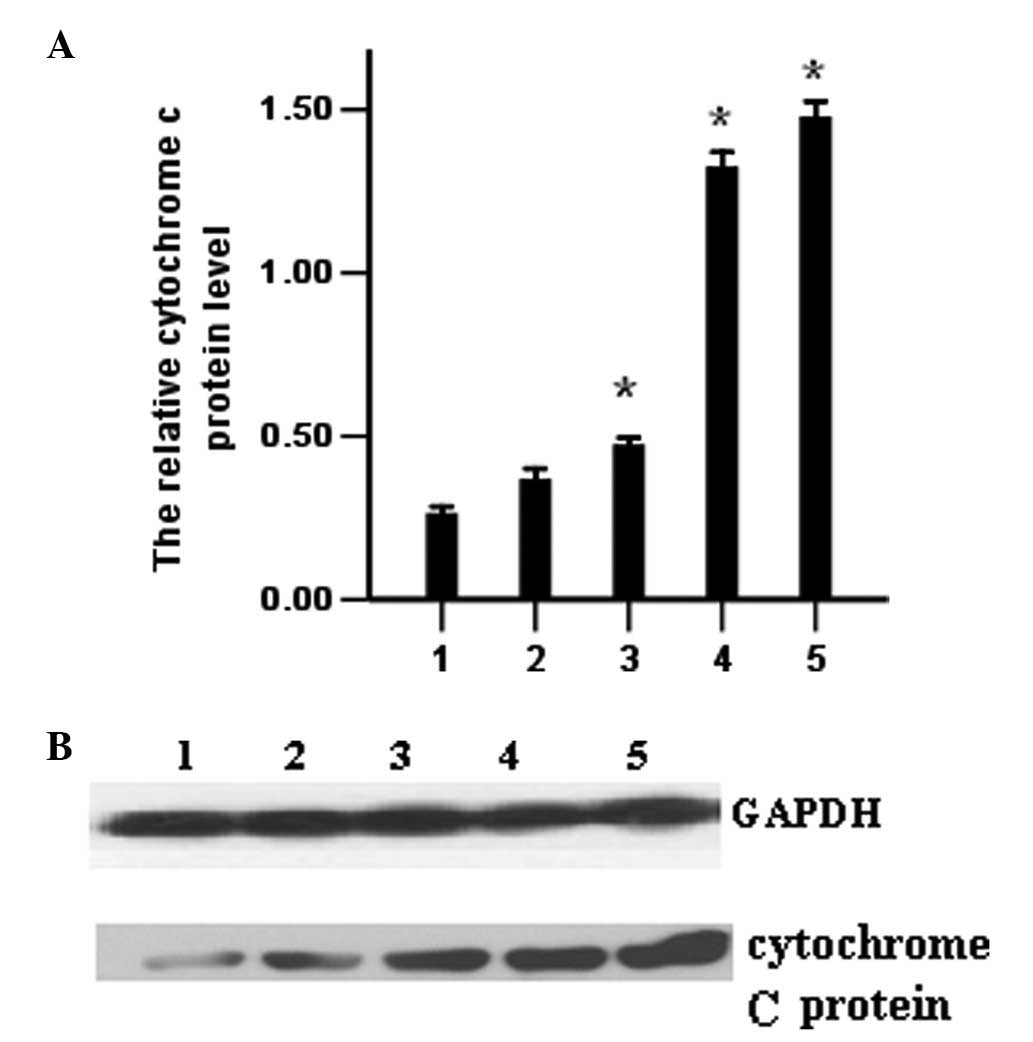
Release of cytochrome c protein detected
by western blotting
To evaluate the levels of cytochrome c
protein, K562 cells transfected with PmiR-203 were processed and
analyzed for cytochrome c protein by western blotting as
described in Materials and methods. The results indicate that the
release of cytochrome c protein increased in the PmiR-203
and the ATO groups (Fig. 8).
Discussion
Increasing evidence suggests that significantly
low-expressed miRNAs in tumors may be considered to be tumor
suppressor genes. These miRNAs usually suppress tumor development
by negatively regulating oncogenes that control biological
processes. Therefore, increasing the expression of tumor suppressor
genes may be a valuable strategy for cancer treatment (9).
A number of studies have shown that miR-203 plays a
role in carcinogenesis. Melar-New and Laimins (10) reported that high levels of miR-203
inhibit human papillomavirus (HPV) amplification and miR-203 has
been proposed to be a new prognostic marker for pancreatic
adenocarcinoma (11). miR-203 has
also been proposed to be a tumor-suppressive miRNA in
hepatocellular carcinoma and hematopoietic malignancies (7,12).
In a previous study, we successfully constructed a eukaryotic
expression vector expressing the hsa-miR-203 plasmid (PmiR-203) and
then transfected it into K562 cells using Lipofectamine 2000.
miR-203 presented a gain-of-function phenotype in K562 cells, which
inhibits the proliferation of K562 cells (8).
The identification of target genes is a key step in
assessing the role of aberrantly expressed miRNA in human cancer
and is used for the further development of miRNA-based gene
therapy. Yi et al(12)
confirmed p63 as a target of miR-203. The authors demonstrated that
miR-203 expression is conspicuous in terminally differentiating
epithelial cells. abl1 is activated in hematopoietic malignancies
in certain cases as a bcr-abl1 fusion protein (Philadelphia
chromosome). bcr-abl1 fusion is a landmark of CML (13). The current study indicated that the
bcr/abl mRNA expression level was downregulated by PmiR-203
(Fig. 7) and the results
demonstrated that increasing the expression of PmiR-203
specifically reduces the expression of the reporter gene that
carries two tandem bcr/abl-3′UTRs without changing the expression
level of the mutations’ reporter gene in the seed sites (Fig. 1). The results demonstrate that
bcr/abl is directly targeted by miR-203 in K562 cells.
The current study also indicated that ATO alone
inhibits cell growth, increases the G1 phase population and induces
early apoptosis in K562 cells (Figs.
2, 5 and 6). This is consistent with the study by
Li et al(9). It has been
reported that cytochrome c release from the mitochondria
precedes dissipation of the voltage gradient (mitochondrial
trans-membrane potential) across the inner membrane, which supports
the specific channel hypothesis, suggesting that the escape of
cytochrome c from the mitochondria occurs prior to
permeability transition pore opening (loss of mitochondrial
transmembrane potential) (14). In
the current study, we demonstrated that ATO and PmiR-203 induce
apoptosis of K562 cells. The induction of apoptosis may involve the
loss of mitochondrial transmembrane potential, cytochrome c
release and caspase-9 and caspase-3 activation, as shown in
Figs. 3 and 8 and Table
I. PmiR-203 significantly sensitized K562 cells to ATO by
inducing apoptosis. When PmiR-203 and ATO were used in combination,
PmiR-203 reduced the therapeutic dose of ATO; this may lead to low
non-specific effects and high tolerance and also possibly reverse
drug resistance in leukemia. Theoretically, this strategy may be
feasible. Therefore, exploiting the synergistic effects between
PmiR-203 and ATO may be an effective clinical strategy for leukemia
chemotherapy and miR-203 may be a potential drug target in K562
cells.
Acknowledgements
This study was supported by grants
from the Guangdong Science and Technology Department social
development projects (No. 2011B080702011).
References
|
1.
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Salim H, Akbar NS, Zong D, et al:
miRNA-214 modulates radio-therapy response of non-small cell lung
cancer cells through regulation of p38MAPK, apoptosis and
senescence. Br J Cancer. 107:1361–1373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Niederer F, Trenkmann M, Ospelt C, et al:
Down-regulation of microRNA-34a* in rheumatoid arthritis synovial
fibroblasts promotes apoptosis resistance. Arthritis Rheum.
64:1771–1779. 2012.
|
|
4.
|
Chen Z, Chen GQ, Shen ZX, Sun GL, Tong JH,
Wang ZY and Chen SJ: Expanding the use of arsenic trioxide:
leukemias and beyond. Semin Hematol. 39(Suppl 1): 22–26. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Wu W: MicroRNA: potential targets for the
development of novel drugs? Drugs R D. 10:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Greither T, Grochola LF, Udelnow A,
Lautenschläger C, Würl P and Taubert H: Elevated expression of
microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated
with poorer survival. Int J Cancer. 26:73–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Furuta M, Kozaki KI, Tanaka S, Arii S,
Imoto I and Inazawa J: miR-124 and miR-203 are epigenetically
silenced tumor-suppressive microRNAs in hepatocellular carcinoma.
Carcinogenesis. 31:766–776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
He JH, Li YG, Chen SY and Wang L:
Construction of the eukaryotic expression vector targeting
hsa-miR-203 and its effects on proliferation and apoptosis of K562
cell lines. Chin J Clin Lab Sci. 30:595–598. 2012.(In Chinese).
|
|
9.
|
Li Y, Zhu XJ, Gu JY, et al: Anti-miR-21
oligonucleotide sensitizes leukemic K562 cells to arsenic trioxide
by inducing apoptosis. Cancer Sci. 101:948–954. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Melar-New M and Laimins LA: Human
papillomaviruses modulate expression of microRNA 203 upon
epithelial differentiation to control levels of p63 proteins. J
Virol. 84:5212–5221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ikenaga N, Ohuchida K, Mizumoto K, Yu J,
Kayashima T, Sakai H, Fujita H, Nakata K and Tanaka M: MicroRNA-203
expression as a new prognostic marker of pancreatic adenocarcinoma.
Ann Surg Oncol. 17:3120–3128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Yi R, Poy MN, Stoffel M and Fuchs E: A
skin microRNA promotes differentiation by repressing ‘stemness’.
Nature. 452:225–229. 2008.
|
|
13.
|
Bueno MJ, Pérez de Castro I, Gómez de
Cedrón M, Santos J, Calin GA, Cigudosa JC, Croce CM,
Fernández-Piqueras J and Malumbres M: Genetic and epigenetic
silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene
expression. Cancer Cell. 13:496–506. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Riedl SJ, Renatus M, Schwarzenbacher R,
Zhou Q, Sun C, Fesik SW, Liddington RC and Salvesen GS: Structural
basis for the inhibition of caspase-3 by XIAP. Cell. 9:791–800.
2001. View Article : Google Scholar : PubMed/NCBI
|