Introduction
The incidence of myocardial infarction (MI) is
higher in patients with diabetes than that in patients without
diabetes, and recovery is often slower, resulting in higher
mortality rates (1). The etiology of
diabetes-associated impaired myocardial healing and cardiac
dysfunction is multifaceted (2–7) and
remains to be completely elucidated.
It has previously been demonstrated that the sonic
hedgehog (Shh) pathway is critical to the growth and repair of
cardiovascular systems during embryonic and postnatal development
and adult life (8). Shh signaling
occurs via interaction of the Shh protein with its receptor,
patched-1 (Ptc1), which terminates the inhibition of the smoothened
receptor. This subsequently leads to activation of the
transcription factor Gli, which induces the expression of
downstream target genes, including Ptc1 itself (9–11).
Numerous studies have suggested that the Shh pathway
is endogenously upregulated in MI, and Shh gene transfer following
an MI resulted in the preservation of left ventricular function
(12,13). Our previous study reported that the
Shh pathway is impaired in diabetic mice following MI, which
contributes to diminished myocardial healing and the exacerbation
of cardiac dysfunction (14);
however, the mechanisms underlying the impaired Shh pathway in
diabetes remain unclear.
Oxidative stress is well documented in the diabetic
state and leads to diabetic complications, particularly diabetic
cardiovascular diseases. Reactive oxygen species (ROS) have been
confirmed as the major source of oxidative stress (15). Previous studies have demonstrated
that tissue and blood levels of ROS are higher in numerous human
diseases, including atherosclerosis, chronic inflammation,
neurodegenerative disease, diabetes and various types of cancer
(16–20). In addition, it has been suggested
that excessive ROS production may interfere with normal signaling
pathways and play a key pathogenic role in disease (21).
The present study aimed to test the hypothesis that
oxidative stress in type 1 diabetic mice contributes to the
impaired Shh pathway that leads to exacerbated cardiac dysfunction,
and that this may be rescued by protecting the Shh pathway using
the antioxidant Tempol.
Materials and methods
Animals and experimental design
Adult male C57BL/6 mice (Guangdong Medical
Laboratory Animal Center, Foshan, China), weighing ~20 g and aged
6–7 weeks, were used in the present study. The mice were handled in
accordance with the principles of the Animal Management Rule of the
Ministry of Health, China (acceptance no. 2011-62), and the Guide
for the Care and Use of Laboratory Animals published by the US
National Institutes of Health (NIH publication no. 85–23, revised,
1996). Mice were maintained at 21–23°C, with a humidity of 40–55%
and a 12-h light cycle (lights on 06:00–18:00), with free access to
food and water.
The mice were divided into six groups: i) Control
(CON), ii) diabetes (DM), iii) diabetes plus Tempol (DM + Tempol),
iv) control plus MI (CMI), v) diabetes plus MI (DMI), and vi) DM
plus Tempol and MI (DTMI). The control mice were administrated
drinking water, which is a solvent of Tempol in vivo.
The rat neonatal cardiomyocytes were divided into
seven groups: i) Control (CON), ii) xanthine oxidase (XO, 1
U/l)/xanthine (X, 0.5 mM), iii) XO (1.5 U/l)/X (0.5 mM), iv) XO (2
U/l)/X (0.5 mM), v) XO (2 U/l)/X (0.5 mM) + Tempol (0.1 mM), vi) XO
(2 U/l)/X (0.5 mM) + Tempol (0.5 mM), and vii) Tempol (0.5 mM). The
control cells were treated with sterilized and purified water,
which is the solvent of Tempol in vitro.
The superoxide scavenger Tempol
(4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy; 3 mM; Sigma-Aldrich,
St. Louis, MO, USA) was administered in the drinking water at the
onset of diabetes until the end of the experiment. Mice were
sacrificed using sodium pentobarbital (100 mg/kg; Guangzhou Qihua
Medical Equipment Co., Ltd., Guangzhou, China). The hearts of the
mice were harvested at various time-points of diabetes (7, 10, 14
and 18 weeks), in order to analyze the protein expression levels of
the Shh pathway and the oxidative stress levels. The surgery to
induce MI was performed 7 weeks after the induction of diabetes.
The heart tissue was harvested on day 7 after MI in order to detect
the expression levels of the Shh pathway-associated proteins using
western blot analysis. A total of 21 days after MI,
echocardiography was performed to assess ventricular function, and
immunohistochemical analysis was conducted to evaluate capillary
density. In addition, Masson's trichrome staining was used to
assess the percentage area of myocardial infarct.
Induction of type 1 diabetes
Adult male C57/BL6 mice (6–7 weeks of age) received
an intraperitoneal (i.p.) injection of streptozotocin (STZ;
Sigma-Aldrich) dissolved in sterile citrate buffer (0.05 mol/l
sodium citrate, pH 4.5, 45 mg/kg). The mice were administered STZ
or citrate buffer (control) for 5 days consecutively during the
first week of the study. Blood samples were collected from the vena
caudalis. Whole blood glucose levels were measured using a glucose
analyzer [OneTouch® UltraMini® Blood Glucose Monitoring system;
Johnson & Johnson Medical (China) Ltd., Shanghai, China]. Mice
with a blood glucose level ≥16.7 mmol/l were considered diabetic
and were used for subsequent MI experiments.
Induction of MI
Adult male C57/BL6 mice were anesthetized with
sodium pentobarbital (50 mg/kg, i.p.). The mice were orally
intubated with a 22G intravenous catheter (Guangzhou Qihua Medical
Equipment Co., Ltd.) and subjected to artificial ventilation using
a respirator (Tai Meng Technology Co., Ltd., Chengdu, China). A
small, oblique thoracotomy was performed lateral to the left
intercostal line in the third costal space, in order to expose the
heart. Following the opening of the pericardium, the proximal left
anterior descending artery branch of the left coronary artery was
ligated using 8-0 polypropylene sutures (Guangzhou Qihua Medical
Equipment Co., Ltd.) under a dissecting microscope (Carl Zeiss AG,
Oberkochen, Germany). The sham-operated animals had an untied left
anterior descending artery; with suture material in place but the
ligature not tightened.
Echocardiography
Transthoracic echocardiography was performed using a
VisualSonics system (Vevo® 2100; Fujifilm VisualSonics, Inc.,
Toronto, ON, Canada) equipped with a 25-MHz imaging transducer. The
anesthesia of the mice was maintained with 2% isoflurane gas (RWD
Life Science Co., Ltd., Shenzhen, China) with an inflow rate of
0.5–1.5 ml/min during the echocardiographic examination. The left
ventricle (LV) was analyzed under parasternal long- and short-axis
views, with Doppler images for LV systolic function, LV cavity
diameter, wall thickness, diastolic function and LV end-systolic
and end-diastolic volume determination.
Assessment of the percentage of
myocardial infarct (Masson's trichrome staining)
The hearts of the mice were perfusion-fixed with 10%
buffered formalin (Sigma-Aldrich), horizontally sectioned between
the point of ligation and the apex and then embedded in paraffin.
The paraffin sections were stained with Masson's trichrome staining
solution (Beyotime Institute of Biotechnology, Shanghai, China).
The percentage area of myocardial infarct in the Masson's
trichrome-stained tissue sections was determined using Image
Pro-Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD,
USA). The levels of fibrosis and total LV area were measured, and
the area of myocardial infarct was expressed as a final
percentage.
Immunohistochemistry for the
determination of capillary density
The hearts of the mice were horizontally sectioned
between the point of ligation and the apex and embedded in optimal
cutting temperature compound (OCT; Sakura Finetek Japan Co., Ltd.,
Tokyo, Japan). Briefly, for the assessment of capillary density,
the OCT sections were stained with rat polyclonal
anti-CD31/platelet endothelial cell adhesion molecule-1 antibody
(1:100; 553708; BD Pharmingen, San Diego, CA, USA), followed by
incubation with a secondary anti-rat antibody (1:5,000; sc-2006;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Sections were
staining using a 3,3-diaminobenzidine kit (K3468; Dako, Glostrup,
Denmark) according to the manufacturer's instructions. The
antigen-antibody interaction was visualized using
3,3′-diaminobenzidine (DAB) substrate using a DAB Substrate kit
(Dako). Counts of capillary density per square millimeter of the
border zone of infarcted myocardium (MI group) or the free wall of
the LV (Sham group) were conducted using Image Pro-Plus software
(Media Cybernetics, Inc.).
ROS detection in the myocardium
At each time-point of diabetes, the mice were
sacrificed and heart tissue sections were harvested and directly
embedded in OCT, as stated previously. Superoxide production in the
heart was detected using dihydroethidium (DHE) staining
(Sigma-Aldrich). Frozen heart sections (10-µm) were incubated with
10 µM DHE for 45 min at 37°C in a humidified chamber protected from
the light. The average fluorescence intensity of the nuclei was
then analyzed using Image Pro-Plus software (Media Cybernetics,
Inc.).
Cell culture
The primary culture of cardiomyocytes was performed
according to previously described methods (22). Briefly, rats were sacrificed by
cervical dislocation and sterilized with 75% alcohol. The chests of
new-born rats were opened using ophthalmic scissors and the hearts
were dissected with ophthalmic forceps. Hearts from the 1-day-old
Sprague Dawley rats (Guangdong Medical Laboratory Animal Center)
were dissected, minced and placed in a petri dish. The tissue was
trypsinized at 37°C in D-Hank's Balanced Salt Solution (HBSS; Gibco
Life Technologies, Carlsbad, CA, USA). Following centrifugation
(1,200 × g, 10 min), the cells were collected and suspended in
Dulbecco's modified Eagle's medium (Gibco Life Technologies),
supplemented with 15% fetal bovine serum (Gibco Life Technologies),
100 U/ml penicillin (Sigma-Aldrich) and 100 µg/ml streptomycin
(Sigma-Aldrich). The cells were then incubated at 37°C for 1 h. The
cells were diluted to 5×106 cells/ml, plated in a 60-mm
petri dish (Corning Incorporated, Corning, NY, USA) and cultured
for 48 to 72 h in medium supplemented with 0.1 mmol/l
bromodeoxyuridine (Sigma-Aldrich), in order to prevent the
proliferation of non-myocytes. Subsequently, a series of chemicals
(XO/X and the superoxide dismutase homolog Tempol), all purchased
from Sigma-Aldrich, were used to treat the cells. The rat neonatal
cardiomyocytes were treated with the mixture of XO/X for 4 or 24 h,
and Tempol was administered at 1 h prior to XO/X.
ROS detection in rat neonatal
cardiomyocytes
Intracellular ROS levels were assessed using the
ROS-specific probe 2′,7′-dichlorofluorescein diacetate (DCF-DA;
Molecular Probes Life Technologies, Carlsbad, CA, USA). On culture
day 4, the cultured cardiomyocytes were washed with HBSS (Gibco
Life Technologies) and then incubated with DCF-DA (5 µmol/l) in
HBSS at 37°C. Following a 1-h incubation, the cardiomyocytes were
further washed with HBSS. In each case, five randomly selected
fields in each well were selected for examination. Data were
collected using a fluorescence reader (Carl Zeiss AG) at excitation
and emission wavelengths of 485 and 530 nm, respectively. The
average fluorescence intensity was then analyzed using Image
Pro-Plus 6.0 software (Media Cybernetics, Inc.).
Western blot analysis
Western blot analysis was used to detect the protein
expression levels of Shh and Ptc1. Cells were washed with
phosphate-buffered three times, then collected using sodium dodecyl
sulfate (SDS) sample buffer containing 62.5 mmol/l Tris-HCl (pH
6.8), 2% SDS, 10% glycerol, 50 mmol/l dithiothreitol and 0.1%
bromphenol blue. Cells were incubated for 5 min at 95°C, cooled on
ice for 5 min and stored at −20°C until required. Cell lysates were
quantified using a Bicinchonininic Acid Protein Assay Kit (Beyotime
Institute of Biotechnology) and 40-µg protein samples were
separated on 10% SDS-PAGE and transferred to PVDF membranes using a
semi-dry electroblot chamber. Proteins in the gel were visualized
with Coomassie brilliant blue staining. Membranes were blocked in
Tris-buffered saline (pH 7.4) containing 0.1% Tween-20 and 5%
non-fat dry milk for 1 h at room temperature. The membranes were
incubated with anti-Shh (1:1,000; 06–1106; EMD Millipore,
Billerica, MA, USA), anti-Ptc1 (1:1,000; 06–1102; EMD Millipore)
and anti-β-actin (1:1,000; sc-130657; Santa Cruz Biotechnology,
Inc.) primary antibodies at 4°C overnight, as recommended by the
manufacturer. Subsequently, the membranes underwent incubation with
goat anti-rabbit IgG horse radish peroxidase-conjugated secondary
antibodies (1:5,000; sc-2004; Santa Cruz Biotechnology, Inc.) for 1
h at room temperature. The blots were visualized using a
SuperSignal® West Pico Chemiluminescent substrate (Pierce
Biotechnology, Inc., Rockville, MD, USA), and molecular band
intensity was determined via densitometry (Quantity One 1-D
Analysis Software; Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Statistical analysis
All data were analyzed using the statistical
software GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA,
USA), and all values are expressed as the mean ± standard error of
the mean. The differences between two groups were analyzed using
the Student's unpaired t-test, and differences between three or
more groups were evaluated via one-way analysis of variance with
Bonferroni correction. P≤0.05 was considered to indicate a
statistically significant difference.
Results
Treatment with the antioxidant Tempol
improves cardiac function in type 1 diabetic mice
Five days of low-dose STZ treatment resulted in 18
weeks of sustained hyperglycemia and body weight loss throughout
the present study (Fig. 1A). An
echocardiographic assessment of cardiac function was performed from
7 to 18 weeks after STZ treatment. The superoxide scavenger Tempol
(3 mM) was administered in the drinking water at the onset of
diabetes and was continued for 18 weeks. Cardiac dysfunction was
detected in the diabetic mice between 10 and 18 weeks. Treatment
with Tempol between 0 and 18 weeks significantly increased E-wave
velocity (31.92%), the E- to A-wave ratio (34.83%) and the E-wave
deceleration rate (40.02%), and decreased isovolumetric relaxation
time (19.32%) and ejection time (14.55%) in diabetic mice at 18
weeks, as compared with the untreated diabetic mice (Fig. 1B and C).
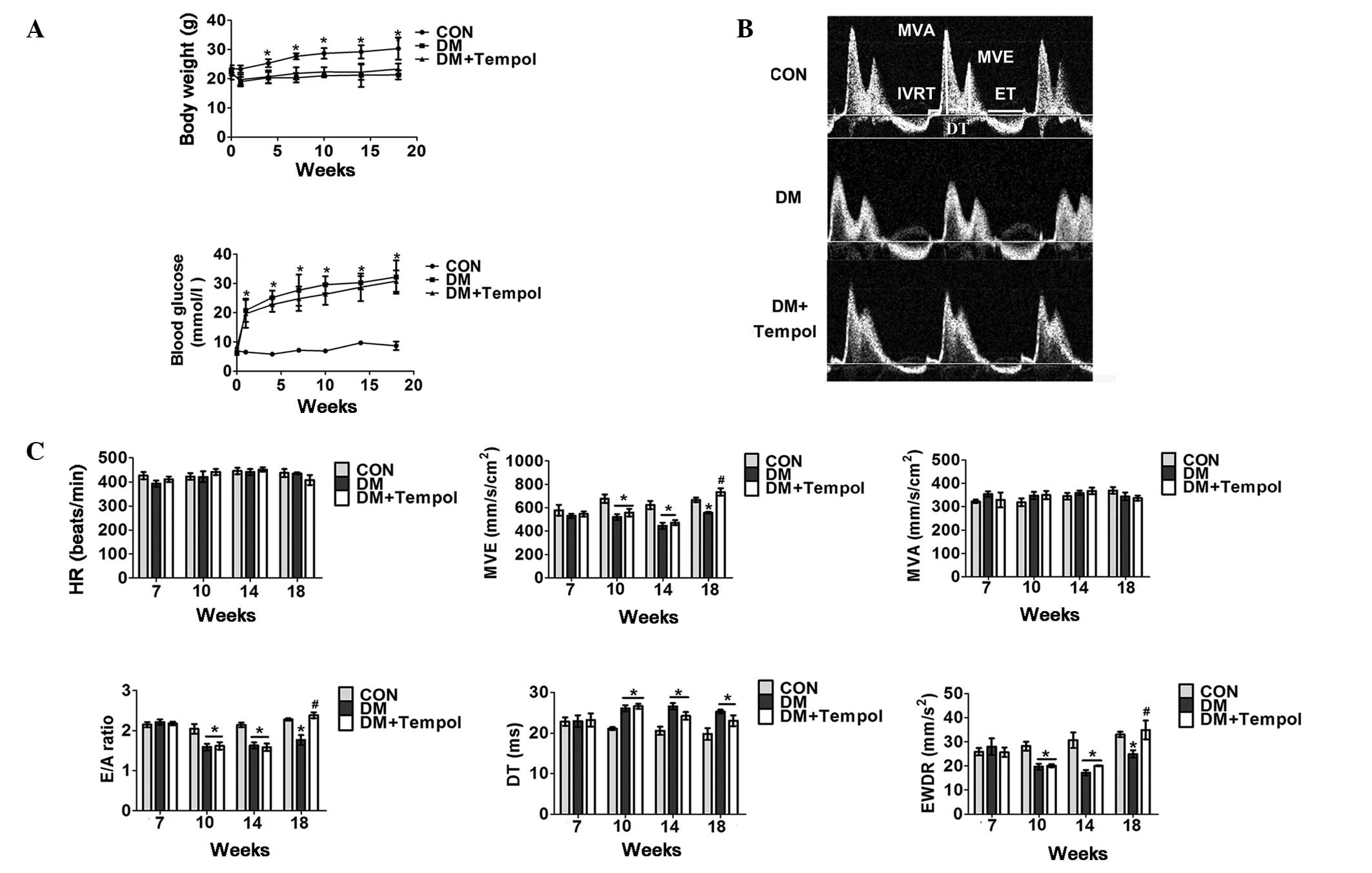 | Figure 1.Treatment with the antioxidant Tempol
improves cardiac function in type 1 diabetic mice. (A) Quantitative
analysis of body weight (upper panel) and whole blood glucose
concentration (lower panel) in control (CON), diabetic (DM) and DM
+ Tempol groups (n=5). (B) Representative transmittal Doppler flow
profile of the CON and DM groups at 18 weeks. (C) Quantitative
analysis of heart rate (HR), E-wave velocity (MVE), mitral valve
area (MVA), E- to A-wave ratio (E/A), E-wave detection time (DT),
E-wave deceleration rate (EWDR), isovolumetric relaxation time
(IVRT), and ejection time (ET) of the CON and DM groups at the 7-,
10-, 14- and 18-week time-points (n=5). Values are presented as the
mean ± standard error of the mean. *P≤0.05 vs. the CON group;
#P≤0.05 vs. the DM group. |
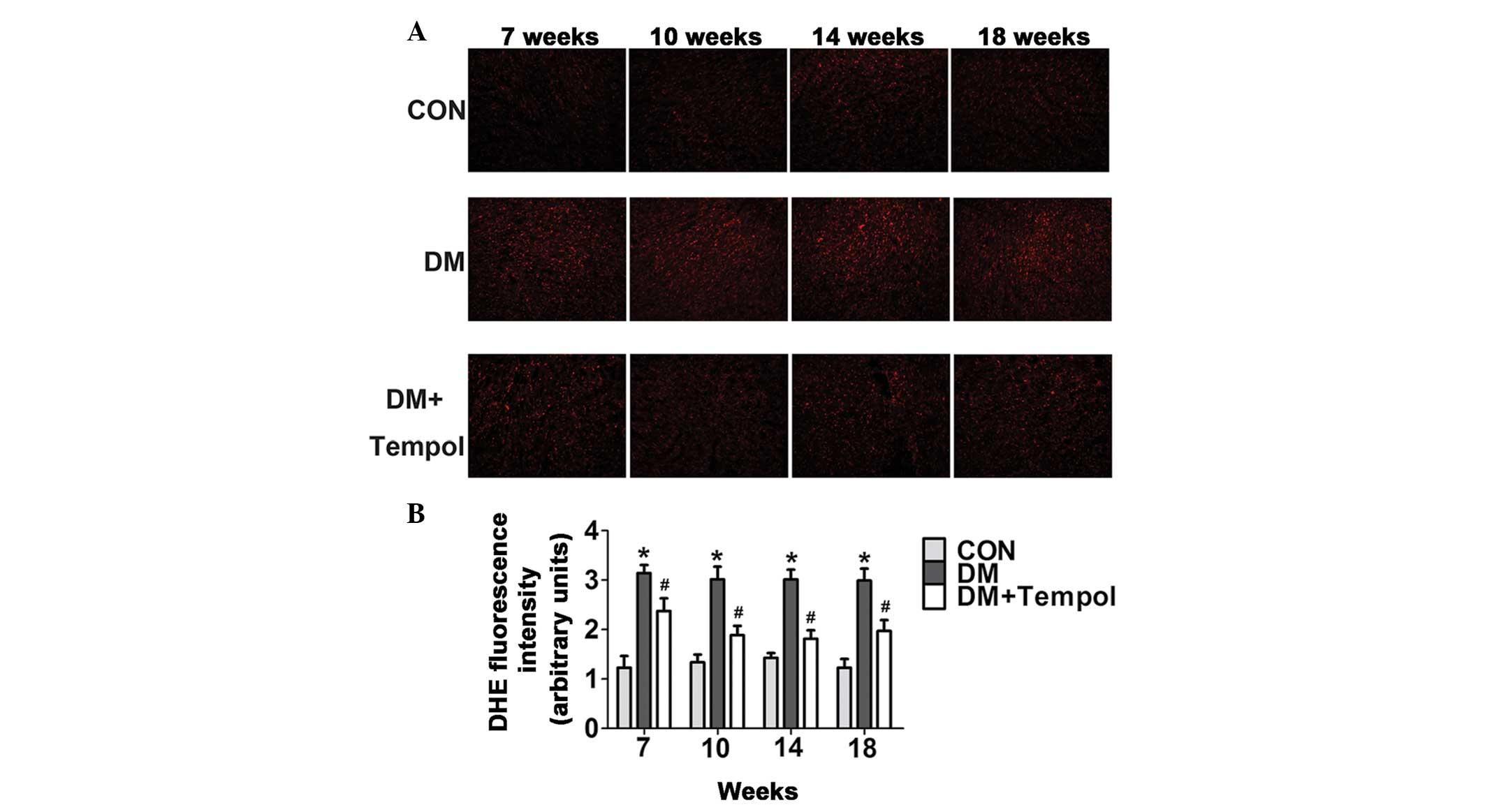
Treatment with the antioxidant Tempol
reduces myocardial ROS levels in type 1 diabetic mice
Myocardial ROS levels were quantified by DHE
staining at the 7-, 10-, 14- and 18-week time-points of diabetes.
The ROS levels were significantly increased in the diabetic mice
during this time. Treatment with Tempol significantly decreased ROS
levels (by 24.42, 37.50, 39.79 and 34.12%, respectively) at 7, 10,
14 and 18 weeks, as compared with the untreated diabetic mice
(Fig. 2).
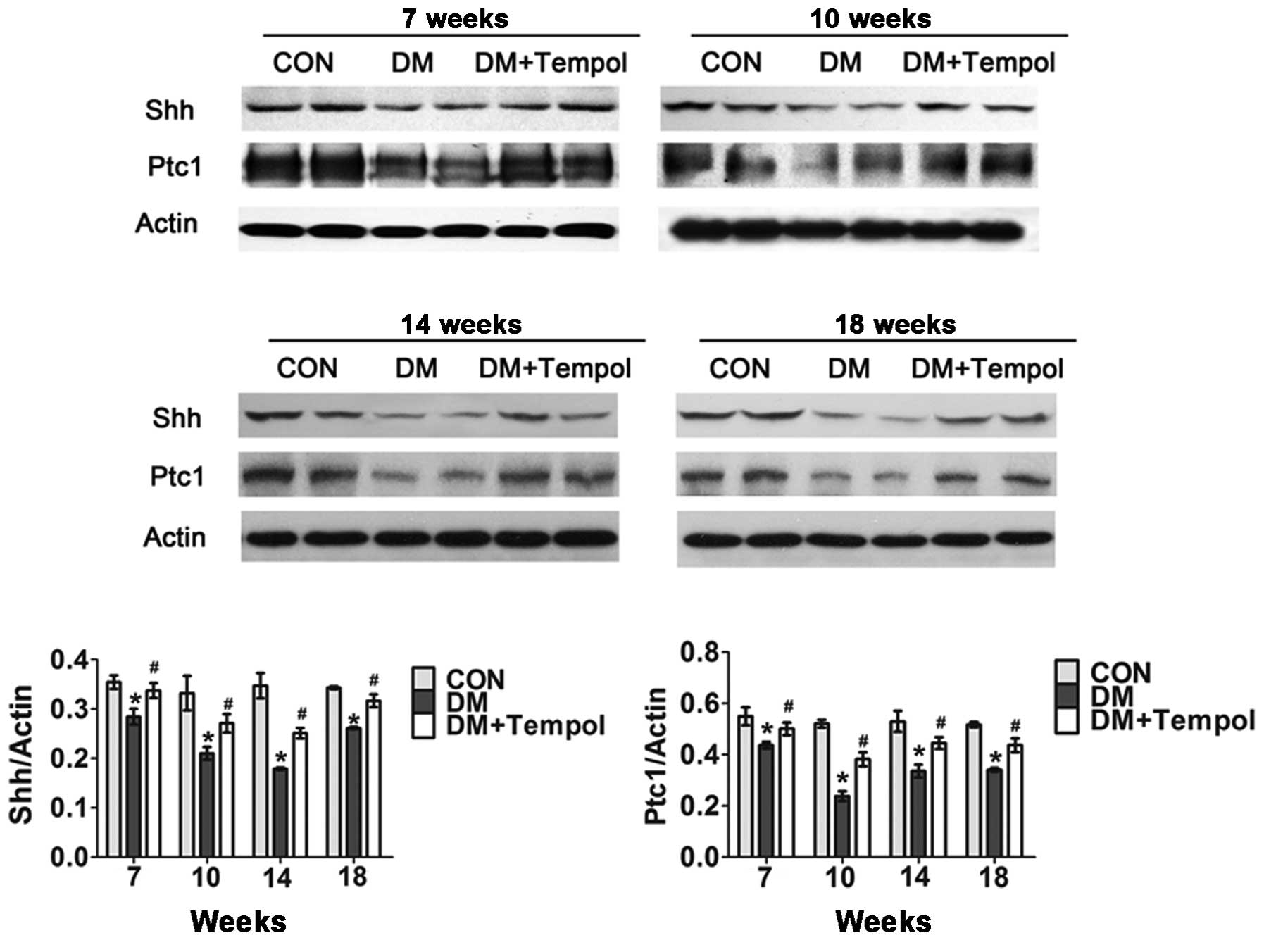
Treatment with the antioxidant Tempol
ameliorates the impaired myocardial Shh pathway in type 1 diabetic
mice
Myocardial Shh and Ptc1 protein expression levels
were detected via western blot analysis at the 7-, 10-, 14- and
18-week time-points of diabetes. The Shh pathway was significantly
impaired in the diabetic mice during this time. Treatment with
Tempol significantly increased the protein expression levels of Shh
(by 15.06, 60.67, 32.57 and 28.18%, respectively) and Ptc1 (by
18.67, 29.12, 40.22 and 21.14%, respectively) at 7, 10, 14 and 18
weeks, as compared with the untreated diabetic mice (Fig. 3).
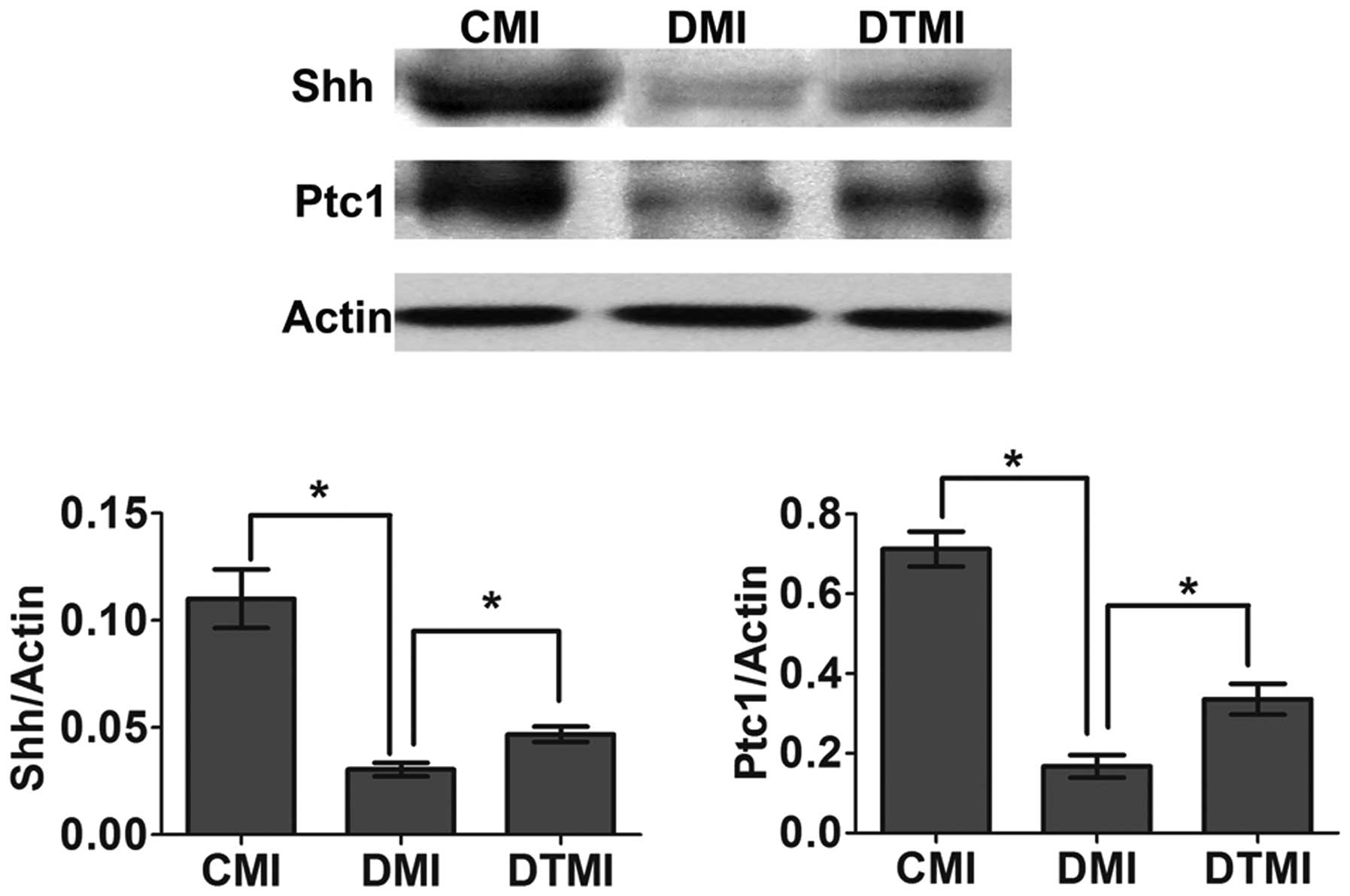
Treatment with the antioxidant Tempol
ameliorates the impaired myocardial Shh pathway in type 1 diabetic
mice with MI
The surgery to induce MI was performed 7 weeks after
the induction of diabetes, and western blot analysis was performed
7 days after MI. Tempol was administered in the drinking water at
the onset of diabetes and continued for 8 weeks (7 weeks for
diabetes and 1 week for diabetes after MI). On day 7 after MI, the
Shh pathway was impaired in the DMI group, whereas treatment with
Tempol significantly increased Shh and Ptc1 protein expression
levels (by 71.04 and 95.07%, respectively) in the DTMI group, as
compared with the DMI group (Fig.
4).
Treatment with the antioxidant Tempol
enhances capillary density and reduces the percentage area of
myocardial infarct in type 1 diabetic mice with MI
Surgery to induce MI was performed 7 weeks after the
induction of diabetes, and histochemical analysis was performed 21
days after MI. Tempol was administered in the drinking water at the
onset of diabetes and continued for 10 weeks (7 weeks for diabetes
and 3 weeks for diabetes after MI). On day 21 after treatment,
Tempol significantly increased capillary density by 25.75% and
reduced the percentage area of myocardial infarct by 20.18% in the
DTMI group, as compared with the DMI group (Fig. 5A-D).
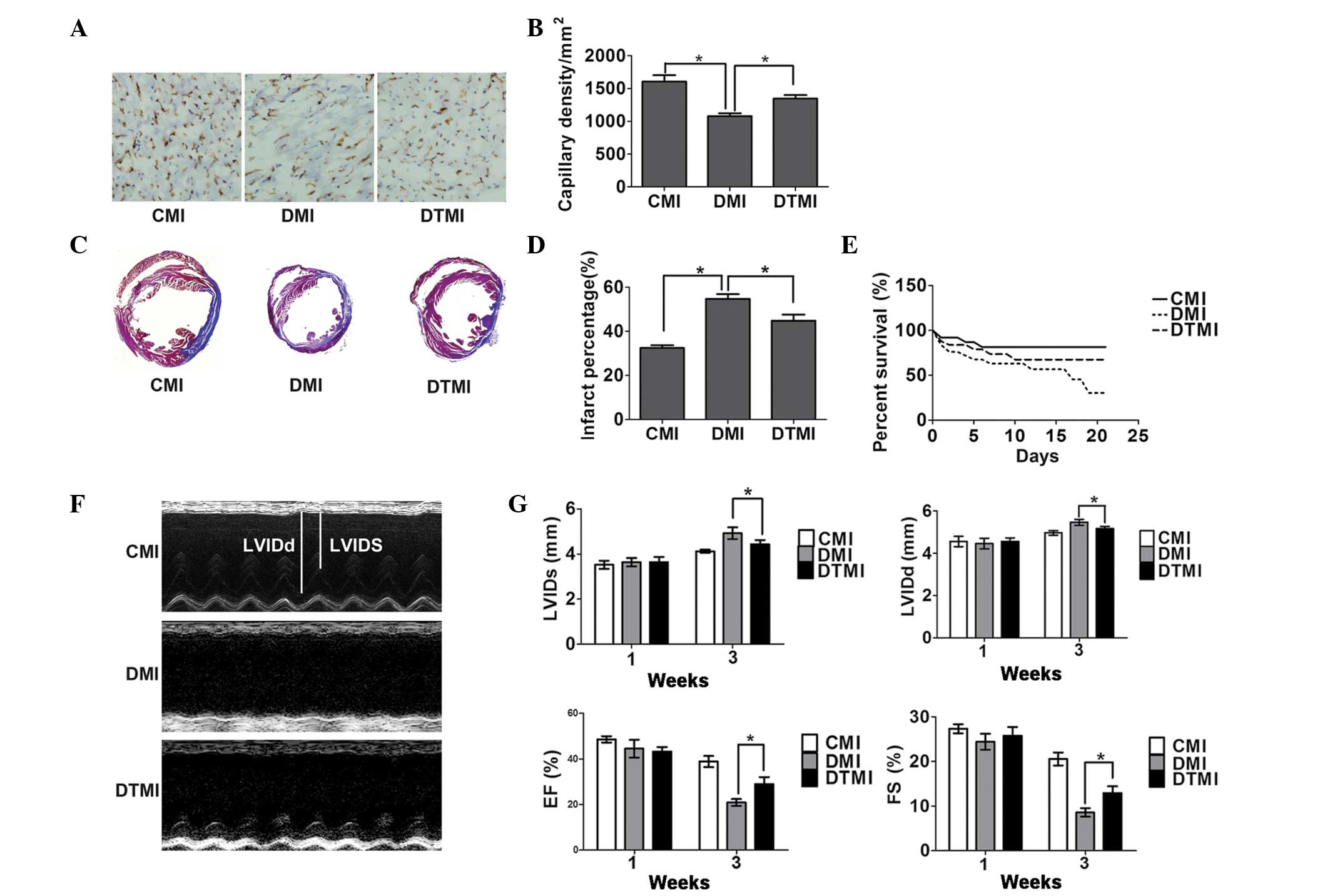 | Figure 5.Treatment with the antioxidant Tempol
ameliorates cardiac status in type 1 diabetic mice following
myocardial infarction (MI). Capillary density, percentage
myocardial infarct area and cardiac function were measured using
immunohistochemistry, Masson's trichrome staining and
echocardiography, respectively, on day 21 after MI. (A)
Immunostaining for CD31 (brown) and the cell nucleus (blue) in the
border zone of the infarcted myocardium. Scale bar, 100 µm. The
number of vessels was measured using Image Pro-Plus. (B)
Quantitative analysis of capillary density (n=5). (C) Masson's
trichrome staining of the mouse hearts. Scale bar, 3 mm. (D)
Percentage infarct area (n=5). (E) Survival rate (n=20). (F)
Representative M-mode images. (G) Quantitative analysis of left
ventricular internal dimension-systole (LVIDs), left ventricular
internal dimension-diastole (LVIDd), fractional shortening (FS) and
ejection fraction (EF) (n=5). Data are presented as the mean ±
standard error of the mean. *P≤0.05. CMI, control MI group; DMI,
diabetic mice with MI; DTMI, DMI group treated with Tempol. |
Treatment with the antioxidant Tempol
protects against cardiac dysfunction and enhances the survival rate
in type 1 diabetic mice with MI
The cardiac function and survival rate were examined
21 days after MI. Tempol significantly enhanced the survival of DMI
mice by 87.5% (Fig. 5E) and
significantly prevented the enlargement of the LV end-systolic and
LV end-diastolic dimensions (by 20.1 and 16.75%, respectively). In
addition, Tempol reduced the fractional shortening and ejection
fraction (by 38.33 and 50.40%, respectively) in the DTMI group, as
compared with the DMI group (Fig. 5F and
G).
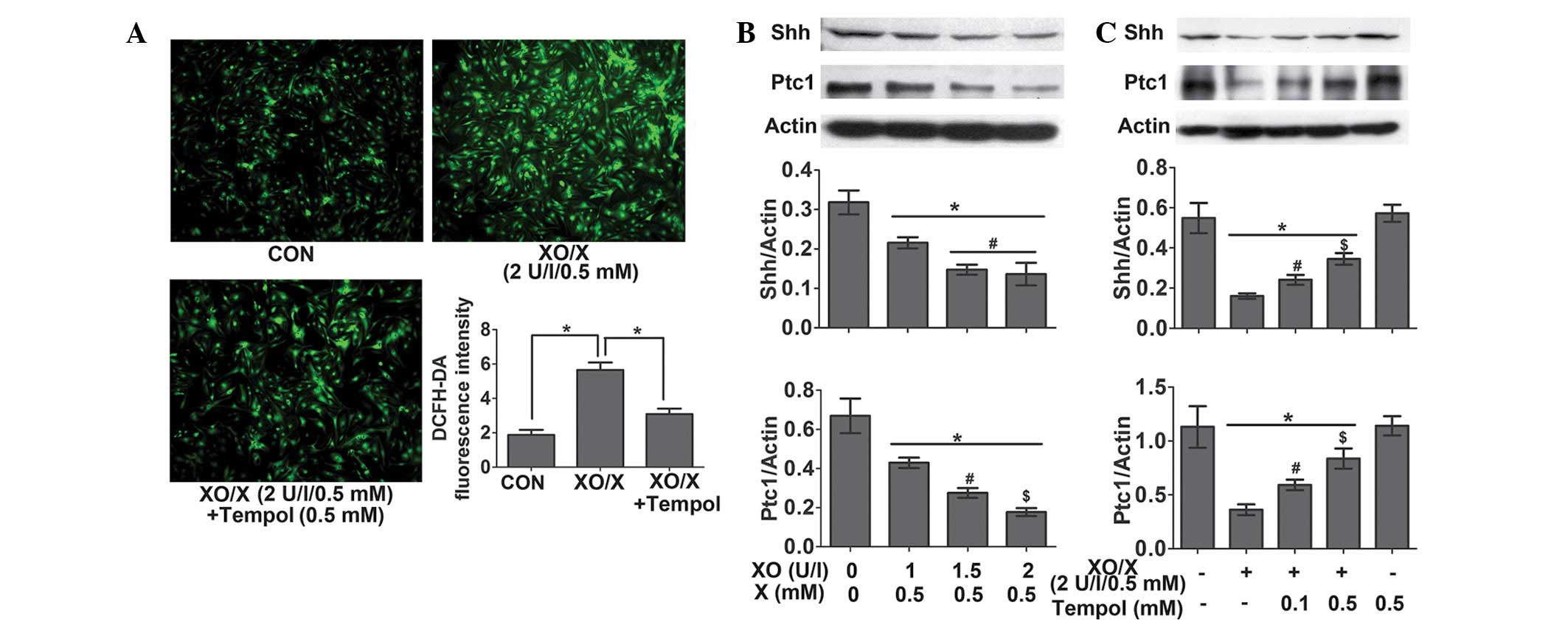
The oxidative-stress system XO/X
inhibits the Shh pathway in rat neonatal cardiomyocytes
Following the 4-h treatment of rat neonatal
cardiomyocytes, XO (2 U/l)/X (0.5 mM) significantly increased ROS
levels by 201.26%. This effect was reversed by 45.28% following the
administration of Tempol (0.5 mM) (Fig.
6A). Following the 24-h treatment of rat neonatal
cardiomyocytes, XO (1–2 U/l)/X (0.5 mM) significantly decreased the
protein expression levels of Shh and Ptc1 in a
concentration-dependent manner (Fig.
6B); however, this effect was attenuated by treatment with
Tempol (0.1 and 0.5 mM) (Fig.
6C).
Discussion
The present study is the first, to the best of our
knowledge, to demonstrate that: i) In type 1 diabetic mice,
treatment with the antioxidant Tempol can improve the impaired
myocardial Shh pathway following MI and eventually ameliorate
cardiac dysfunction; ii) in neonatal cardiomyocytes, administration
of XO/X significantly increases ROS levels and reduces the protein
expression levels of Shh pathway-associated proteins. These
findings suggest that oxidative stress may contribute to the
impaired Shh pathway in type 1 diabetic mice.
The subsequent generation of ROS and accompanying
oxidative stress in diabetes are hallmarks of the molecular
mechanisms underlying diabetic cardiovascular disease. In diabetic
cardiomyopathy, the production of ROS induces inflammation,
endothelial dysfunction, cell apoptosis and myocardial remodeling
(23). ROS from the hexosamine and
polyol pathways impair the expression and activity of nitric oxide
synthase and decrease the function of nitric oxide, which results
in the induction of endothelial dysfunction. In addition, increased
ROS levels may lead to cell apoptosis, via the aggregation of
apoptosis signal-regulating kinase 1 and ceramide, and induce
myocardial remodeling, cardiac dysfunction and arrhythmia (24). ROS also have a critical role in
diabetes with cardiac ischemia. A previous study demonstrated that
exogenous glutathione (an endogenous ROS-scavenging agent) was able
to normalize Rac1 and hypoxia inducible factor-1α expression and
ameliorate infarct size in isolated high-glucose-perfused ischemic
hearts (7). In addition,
administration of thioredoxin-1, an endogenous ROS-scavenging
protein that is required for the maintenance of redox environments,
immediately following MI reversed the reduction of vascular
endothelial growth factor and heme oxygenase-1 and ameliorated
myocardial healing and cardiac dysfunction in diabetic rats
(5). In the present study,
therefore, it was hypothesized that the oxidative stress-induced
impairment of the Shh pathway in diabetes and diabetic MI may have
also been associated with cardiac dysfunction.
Tempol is a redox cycling nitroxide that promotes
the metabolism of numerous ROS and improves nitric oxide
bioavailability. Tempol is effective in preventing various adverse
consequences of oxidative stress, which is associated with numerous
diseases including shock, hypertension, aging, metabolic syndrome
and diabetes (25). In the present
study, the effects of Tempol on oxidative stress in the Shh pathway
were analyzed. Myocardial and cardiomyocyte ROS levels were
subsequently detected, and the results indicated that Tempol was
able to act as an effective and efficient antioxidant.
The results of the present study suggested that the
antioxidant Tempol was able to improve the impaired Shh pathway and
cardiac function in diabetic mice. Since previous studies have yet
to establish a causal link between an impaired Shh pathway and
cardiac dysfunction in diabetes, the present study was only able to
report a preliminary association between the oxidative
stress-induced Shh pathway inhibition and oxidative stress-induced
cardiac dysfunction. Therefore, the significance of the oxidative
stress-induced Shh pathway inhibition in diabetes required further
exploration.
As our previous study demonstrated (14), activation of the Shh pathway in
diabetic mice was impaired on day 7 after MI. Notably, the impaired
Shh pathway contributed to cardiac dysfunction at 3 weeks after MI
in diabetic mice. In the present study, MI was induced in order to
analyze the effect and significance of diabetes-stimulated
oxidative stress on the inhibition of the Shh pathway in diabetic
mice with MI. MI was induced after 7 weeks of diabetes, when a
markedly impaired Shh pathway was detected.
The results of the present study suggested that
Tempol was able to significantly upregulate the Shh pathway in
diabetic mice with MI, which was beneficial for the enhancement of
capillary density, the reduction of the percentage area of
myocardial infarct and the improvement in cardiac function and
survival rate. These results are consistent with those observed
following treatment with the Shh pathway agonist SAG1.3 in our
previous study (14).
In order to eliminate the interference of various
factors that may occur in vivo, the XO/X oxidative stress
producer system was used in neonatal cardiomyocytes, in order to
investigate the effect of oxidative stress on the Shh pathway in
vitro. The results demonstrated that treatment with XO/X
significantly induced the production of ROS and inhibited the Shh
pathway in a concentration-dependent manner; however, Tempol
attenuated these effects.
The results of the present study collectively
suggest that oxidative stress can impair the endogenous Shh pathway
in diabetes and weaken its ability when combined with MI, which may
subsequently exacerbate cardiac dysfunction in diabetic MI.
Antioxidants used in preclinical animal studies have
provided promising results (26–29);
however, the explanation for why antioxidative drugs, including
vitamin C and vitamin E, do not lead to satisfactory results when
applied clinically is complex. Numerous studies have demonstrated
that antioxidants appear promising as substrate-modifying ancillary
agents for use with the major therapy (e.g. injection of angiogenic
factors) in the treatment of various diseases, including diabetes,
hypercholesterolemia and ischemia, since the molecular and cellular
environments of these diseases are not suitable for regular therapy
(30). Preliminary evidence from
rodent models of hindlimb ischemia has suggested that
supplementation with vitamins C and E, which prevents the excessive
generation of ischemia-induced oxygen radicals, enhances the
angiogenic effect of L-arginine on bone marrow cell infusion
(31). Our previous study also
revealed that administration of a sufficient Shh agonist
inadequately activated the Shh pathway in diabetic MI (14). Therefore, further studies should
consider combining long-time applied antioxidants with Shh pathway
receptor agonists, Shh protein or Shh-adenoviral vector gene
therapy, in order to improve the effectiveness of the
treatment.
In conclusion, the present study has demonstrated
that oxidative stress may contribute to the impaired Shh pathway in
type 1 diabetic mice, leading to diminished myocardial healing and
cardiac dysfunction. Antioxidative strategies that are aimed at
restoring the endogenous Shh pathway may offer a useful means for
improving cardiac function.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81173062 and
81302767), the Science and Technology Planning Project of Guangdong
Province (grant no. 2014A020212361) and the Medical Scientific
Research Foundation of Guangdong Province (grant no. A2015444).
References
|
1
|
Krum H and Gilbert RE: Demographics and
concomitant disorders in heart failure. Lancet. 362:147–158. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marfella R, Filippo CD, Portoghese M,
Siniscalchi M, Martis S, Ferraraccio F, Gustafierro S, Nicoletti G,
Barbieri M, Coppola A, et al: The ubiquitin-proteasome system
contributes to the inflammatory injury in ischemic diabetic
myocardium: The role of glycemic control. Cardiovasc Pathol.
18:332–345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shishido T, Woo CH, Ding B, McClain C,
Molina CA, Yan C, Yang J and Abe J: Effects of MEK5/ERK5
association on small ubiquitin-related modification of ERK5:
Implications for diabetic ventricular dysfunction after myocardial
infarction. Circ Res. 102:1416–1425. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bucciarelli LG, Ananthakrishnan R, Hwang
YC, Kaneko M, Song F, Sell DR, Strauch C, Monnier VM, Yan SF,
Schmidt AM and Ramasamy R: RAGE and modulation of ischemic injury
in the diabetic myocardium. Diabetes. 57:1941–1951. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Samuel SM, Thirunavukkarasu M, Penumathsa
SV, Koneru S, Zhan L, Maulik G, Sudhakaran PR and Maulik N:
Thioredoxin-1 gene therapy enhances angiogenic signaling and
reduces ventricular remodeling in infarcted myocardium of diabetic
rats. Circulation. 121:1244–1255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Di Filippo C, Marfella R, Cuzzocrea S,
Piegari E, Petronella P, Giugliano D, Rossi F and D'Amico M:
Hyperglycemia in streptozotocin-induced diabetic rat increases
infarct size associated with low levels of myocardial HO-1 during
ischemia/reperfusion. Diabetes. 54:803–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marfella R, D'Amico M, Di Filippo C,
Piegari E, Nappo F, Esposito K, Berrino L, Rossi F and Giugliano D:
Myocardial infarction in diabetic rats: Role of hyperglycaemia on
infarct size and early expression of hypoxia-inducible factor 1.
Diabetologia. 45:1172–1181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mimeault M and Batra SK: Frequent
deregulations in the hedgehog signaling network and cross-talks
with the epidermal growth factor receptor pathway involved in
cancer progression and targeted therapies. Pharmacol Rev.
62:497–524. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lewis PM, Dunn MP, McMahon JA, Logan M,
Martin JF, St-Jacques B and McMahon AP: Cholesterol modification of
sonic hedgehog is required for long-range signaling activity and
effective modulation of signaling by Ptc1. Cell. 105:599–612. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chari NS and McDonnell TJ: The sonic
hedgehog signaling network in development and neoplasia. Adv Anat
Pathol. 14:344–352. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Varjosalo M and Taipale J: Hedgehog
signaling. J Cell Sci. 120:3–6. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kusano KF, Pola R, Murayama T, Curry C,
Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, et
al: Sonic hedgehog myocardial gene therapy: Tissue repair through
transient reconstitution of embryonic signaling. Nat Med.
11:1197–1204. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ueda K, Takano H, Niitsuma Y, Hasegawa H,
Uchiyama R, Oka T, Miyazaki M, Nakaya H and Komuro I: Sonic
hedgehog is a critical mediator of erythropoietin-induced cardiac
protection in mice. J Clin Invest. 120:2016–2029. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiao Q, Hou N, Wang YP, He LS, He YH,
Zhang GP, Yi Q, Liu SM, Chen MS and Luo JD: Impaired sonic hedgehog
pathway contributes to cardiac dysfunction in type 1 diabetic mice
with myocardial infarction. Cardiovas Res. 95:507–516. 2012.
View Article : Google Scholar
|
|
15
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Newsholme P, Haber EP, Hirabara SM,
Rebelato EL, Procopio J, Morgan D, Oliviera-Emilio HC, Carpinelli
AR and Curi R: Diabetes associated cell stress and dysfunction:
Role of mitochondrial and non-mitochondrial ROS production and
activity. J Physiol. 583:9–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martinon F: Signaling by ROS drives
inflammasome activation. Eur J Immunol. 40:616–619. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Y and Tabas I: Emerging roles of
mitochondria ROS in atherosclerotic lesions: causation or
association? J Atheroscler Thromb. 21:381–390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sabharwal SS and Schumacker PT:
Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles
heel? Nat Rev Cancer. 14:709–721. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
de Vries HE, Witte M, Hondius D,
Rozemuller AJ, Drukarch B, Hoozemans J and van Horssen J:
Nrf2-induced antioxidant protection: A promising target to
counteract ROS-mediated damage in neurodegenerative disease? Free
Radic Biol Med. 45:1375–1383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pennathur S and Heinecke JW: Mechanisms
for oxidative stress in diabetic cardiovascular disease. Antioxid
Redox Signal. 9:955–969. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hou N, Luo MS, Liu SM, Zhang HN, Xiao Q,
Sun P, Zhang GS, Luo JD and Chen MS: Leptin induces hypertrophy
through activating the peroxisome proliferator-activated receptor α
pathway in cultured neonatal rat cardiomyocytes. Clin Exp Pharmacol
Physiol. 37:1087–1095. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Khullar M, Al-Shudiefat AA, Ludke A,
Binepal G and Singal PK: Oxidative stress: A key contributor to
diabetic cardiomyopathy. Can J Physiol Pharmacol. 88:233–240. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pitocco D, Zaccardi F, Di Stasio E,
Romitelli F, Santini SA, Zuppi C and Ghirlanda G: Oxidative stress,
nitric oxide, and diabetes. Rev Diabet Stud. 7:15–25. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wilcox CS: Effects of tempol and
redox-cycling nitroxides in models of oxidative stress. Pharmacol
Ther. 126:119–145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang Q: Natural forms of vitamin E:
Metabolism, antioxidant, and anti-inflammatory activities and their
role in disease prevention and therapy. Free Radic Biol Med.
72:76–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Danta CC and Piplani P: The discovery and
development of new potential antioxidant agents for the treatment
of neurodegenerative diseases. Expert Opin Drug Discov.
9:1205–1222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Feng Y and Wang X: Antioxidant therapies
for Alzheimers disease. Oxid Med Cell Longev. 2012:2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kamat CD, Gadal S, Mhatre M, Williamson
KS, Pye QN and Hensley K: Antioxidants in central nervous system
diseases: Preclinical promise and translational challenges. J
Alzheimers Dis. 15:473–493. 2008.PubMed/NCBI
|
|
30
|
Boodhwani M and Sellke FW: Therapeutic
angiogenesis in diabetes and hypercholesterolemia: Influence of
oxidative stress. Antioxid Redox Signal. 11:1945–1959. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Napoli C, Williams-Ignarro S, de Nigris F,
de Rosa G, Lerman LO, Farzati B, Matarazzo A, Sica G, Botti C,
Fiore A, et al: Beneficial effects of concurrent autologous bone
marrow cell therapy and metabolic intervention in ischemia-induced
angiogenesis in the mouse hindlimb. Proc Natl Acad Sci USA.
102:17202–17206. 2005. View Article : Google Scholar : PubMed/NCBI
|