Introduction
Epidermolysis bullosa (EB) is a term that describes
a group of inherited skin diseases that are characterized by
blistering of the skin caused by mechanical trauma (1). EB has been classified into three major
subtypes on the basis of the level of the dermoepidermal separation
within the basement membrane zone (1). Epidermolysis bullosa simplex (EBS)
comprises the most common subtype of EB, which results from tissue
separation within the epidermal basal keratinocytes. The worldwide
prevalence of EBS has been estimated to be between 1 in 30,000 and
1 in 50,000 (1).
EBS can be subdivided into three major subtypes: i)
The mildest subtype, localized EBS (EBS-loc, OMIM 131800),
characterized by blistering confined to the hands and feet; ii) the
moderately severe variant, generalized EBS (EBS-gen; OMIM 131900),
characterized by more generalized blistering, and iii) the most
severe variant, the Dowling-Meara type (EBS-DM; OMIM 131760)
characterized by severe herpetiform blistering (2).
EBS is mostly caused by a single mutation in either
of the keratin genes, keratin 5 (KRT5) or KRT14,
inherited in an autosomal dominant fashion (3,4). The
majority of mutations are nucleotide substitutions that lead to
missense mutations. The KRT5 and KRT14 genes encode
the intermediate filament cytoskeleton proteins in basal
keratinocytes, which maintain the mechanical integrity of the
epidermis (5). Each of these two
keratin proteins comprises a central α-helical rod domain of 310
amino acids, which consists of four segments (1A, 1B, 2A and 2B)
and is interrupted by three nonhelical linkers (L1, L12 and L2)
(5).
Mutation screening of KRT5 and KRT14
in an EBS proband and their family members could potentially help
to improve the understanding of the pathogenesis of EBS and the
mutation spectrum of Chinese patients with EBS. In the present
study, Chinese patients with EBS were examined for KRT5
germline mutations.
Patients and methods
Patients and pedigree
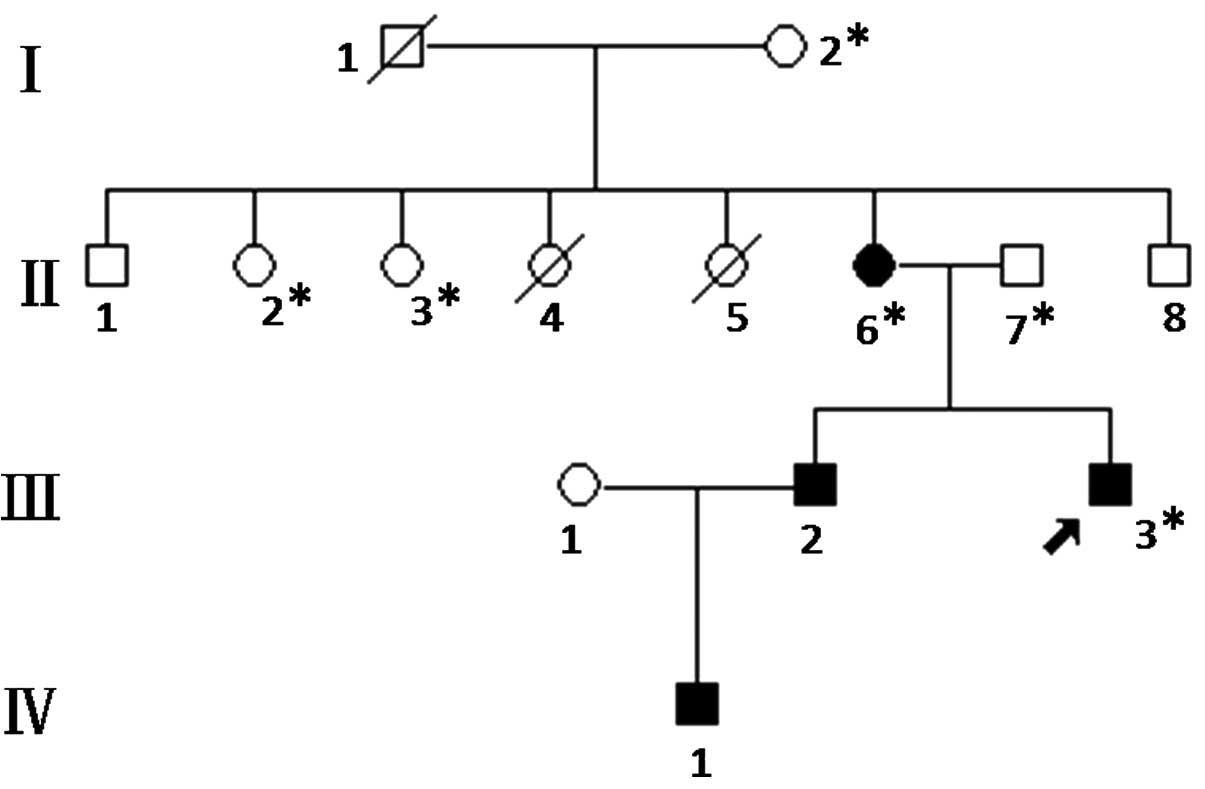
In total, 6 members of the EBS pedigree (Fig. 1) were enrolled in the study in
October 15th, 2013. The members II:6 (female, 46-years-old) and
III:3 (male, 23-years-old) were diagnosed with EBS and treated in
the Zhongnan Hospital of Wuhan University (Wuhan, China). The
diagnosis was based on the clinical features, family history and
histopathological examination of the skin of the patients. The
clinical records, family history and results from the
histopathological examination of the patients were collected.
The proband presented with blistering after
mechanical trauma, which occurred mainly on the feet or hands
following exercise and the symptoms deteriorated in the summer. The
bullae healed without scarring. All four affected members in the
pedigree had the same clinical manifestations as the proband.
Analyses also included 100 unaffected control subjects.
The study was approved by the Zhongnan Hospital
Research Ethics Committee and was conducted in accordance with the
Declaration of Helsinki. Informed consent was obtained from all
individuals prior to their participation in the study.
Mutational screening
Approximately 4–5 ml peripheral venous blood was
drawn from the family members and 100 population controls. Genomic
DNA was extracted using the standard sodium dodecyl
sulfate-proteinase K method (6),
quantified using a spectrophotometer (Nanodrop 2000; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 260 nm and stored at −20°C
until use. The total RNA sample of the proband was extracted using
the ComWin RNA extraction kit (Beijing ComWin Biotech Co., Ltd.,
Beijing, China) according to the manufacturer's instructions.
Reverse transcription (RT) was performed with 1 µg total RNA using
a Revert First Strand cDNA Synthesis kit (Thermo Scientific, Inc.,
Waltham, MA, USA). Specific primers for KRT5 were designed using
Primer 3, version 0.4.0 (http://frodo.wi.mit.edu/primer3/) (sequences listed in
Table I) and synthesized by Qingke
Biotechnology Co., Ltd. (Wuhan, China). DNA or cDNA fragments were
amplified using PCR tubes containing 10 pmol of each primer, 1.5
mmol/l Mg2+, 2.0 mmol/l dNTP, 1.0 units Taq DNA
polymerase (Fermentas; Thermo Fisher Scientific, Inc.) and 100 ng
DNA. The amplification was performed as follows: Initial
denaturation at 95°C for 10 min followed by 35 cycles of
denaturation at 95°C for 30 sec, annealing at 56, 57 or 59°C (as
specified in Table I) for 30 sec,
extension at 72°C for 1 min and final extension at 72°C for 10 min.
Amplifications were performed in a Hema 9600 PCR thermocycler (Hema
Medica Instrument Co., Ltd., Zhuhai, China). PCR-amplified
fragments were purified and sequenced by Qingke Biotechnology Co.,
Ltd. using an Applied Biosystems Genetic Analyzer 3730×l (Thermo
Fisher Scientific, Inc.).
 | Table I.Primers and conditions for keratin 5
amplification by reverse transcription-polymerase chain
reaction. |
Table I.
Primers and conditions for keratin 5
amplification by reverse transcription-polymerase chain
reaction.
|
| Primers (5′→3′) |
|
|
|---|
|
|
|
|
|
|---|
| Exon | Forward | Reverse | Product size
(bp) | Annealing temperature
(°C) |
|---|
| 2, 3, 4, 5, 6 (for
cDNA) |
CGGTAGTGGATTTGGTTTCG |
TGGTGTTGCGGAGGTCAT | 845 | 56 |
| 7, 8 (for cDNA) |
CGATGACCTCCGCAACA |
CTCCTGGGAACCAAAGAAT | 798 | 57 |
| 2 (for DNA) |
AGAGGGACGGAAAGAGG |
CAAAGGAAGGCATGGTAG | 401 | 59 |
Mutation analysis was conducted using the
PCR-restriction fragment length polymorphism (PCR-RFLP) method in 6
family members and 100 population controls. The PCR products (8 µl)
were digested with EcoNI restriction enzyme (New England
Biolabs Inc., Ipswich, MA, USA) at 37°C overnight. The resulting
fragments were separated by electrophoresis on a 2% agarose gel
(Biowest, Madrid, Spain) for 20 min with 100 V in 0.5X
Tris/borate/EDTA buffer (Sheng Gong Biotechnology Co., Ltd.,
Shanghai, China). Subsequently, the fragments were stained by
goldview (Sai Baisheng Biotechnology Co., Ltd., Shanghai, China)
were visualized under a UV transilluminator (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Protein-protein Basic Local Alignment
Search Tool (BLAST) of KRT5
The protein BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to
align the proteins of different species. The protein sequences of
keratin were inserted in the pblast frame, and the proteins of
different species (such as human, dog, mouse and rat) were
determined.
Prediction of the biophysical
properties of the mutant protein
Biophysical predictions of the altered protein were
made using bioinformatics tools. In particular, Antheprot 2000
version 5.0 (Institut de Biologie et Chimie des Protéines, Lyon,
France) was used for the prediction of secondary structures and
hydrophobicity.
Results
Autosomal dominant pedigree of
EBS
Six Chinese patients with EBS were enrolled into the
present study, of which two patients (III:3 and II:6) were
classified into the EBS-loc subtype based on previously established
diagnostic criteria (4,7). The patients had a history suggestive of
autosomal dominance. The diagnosis of EBS-loc was confirmed by
histopathological examination of the patients' skin, which revealed
basal cell cytolysis.
Novel mutation of L202Q in KRT5
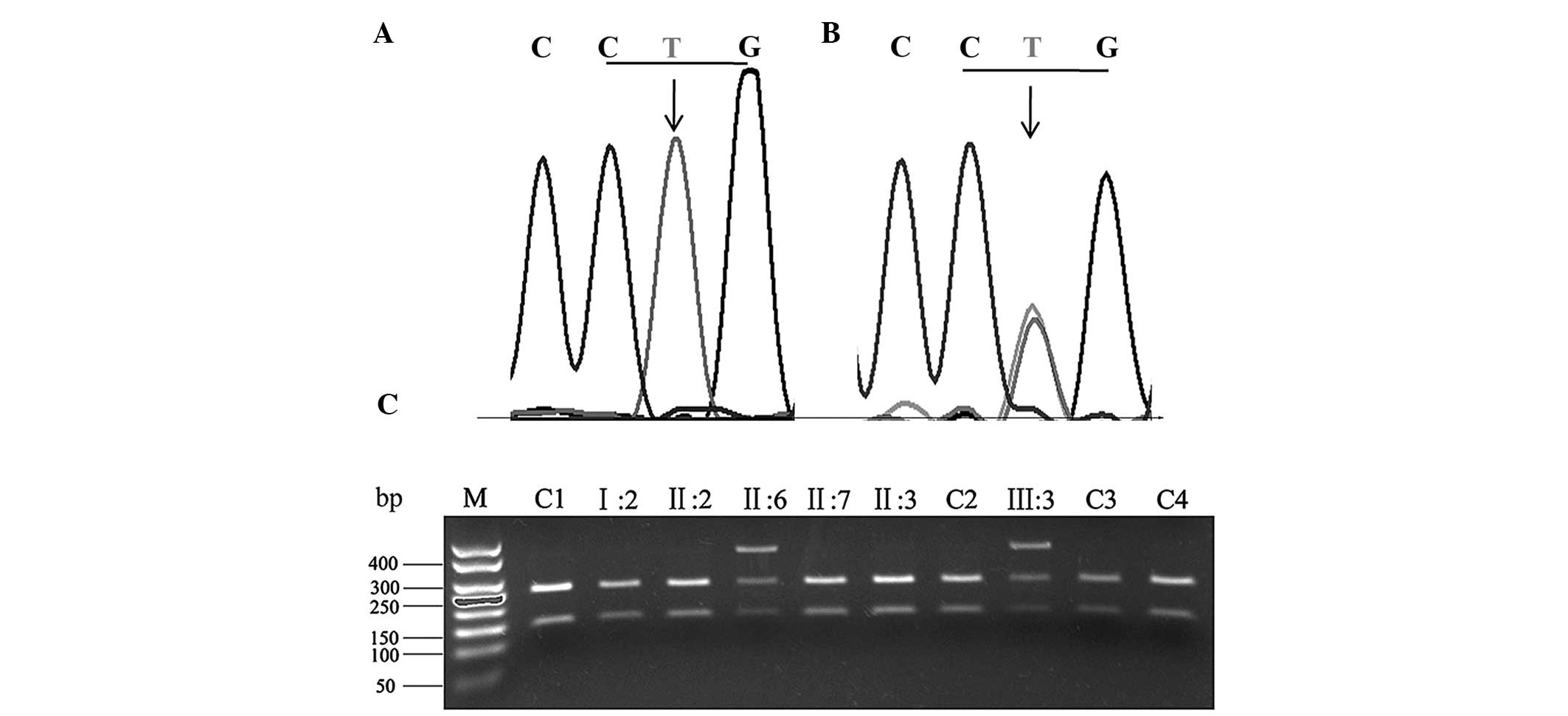
In this particular pedigree, the results of direct
PCR sequencing in the two affected individuals (III:3 and II:6), in
comparison with the wild-type allele (Fig. 2A), revealed a heterozygous T→A
mutation at nucleotide 605 in KRT5 (Fig.
2B). At the protein level, this mutation leads to an amino acid
change from leucine to glutamine in codon 202 of exon 2. The
results of the PCR-RFLP analysis indicated that this mutation was
not present in the unaffected family members or the 100 population
controls (Fig. 2C).
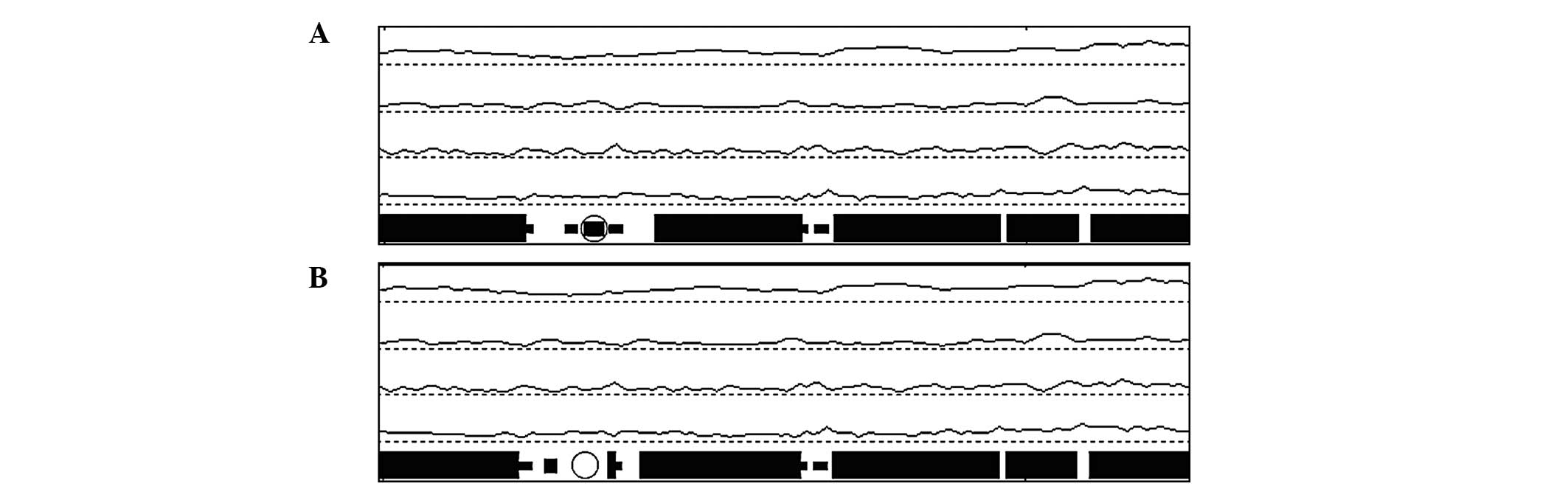
Structure predictions
Computer-assisted prediction of the human KRT5
protein was performed in order to obtain a better understanding of
the effects of the mutation on its biochemical properties and
structure. The results showed that the L202Q mutation caused a
variation of the secondary structure with the omission of a β sheet
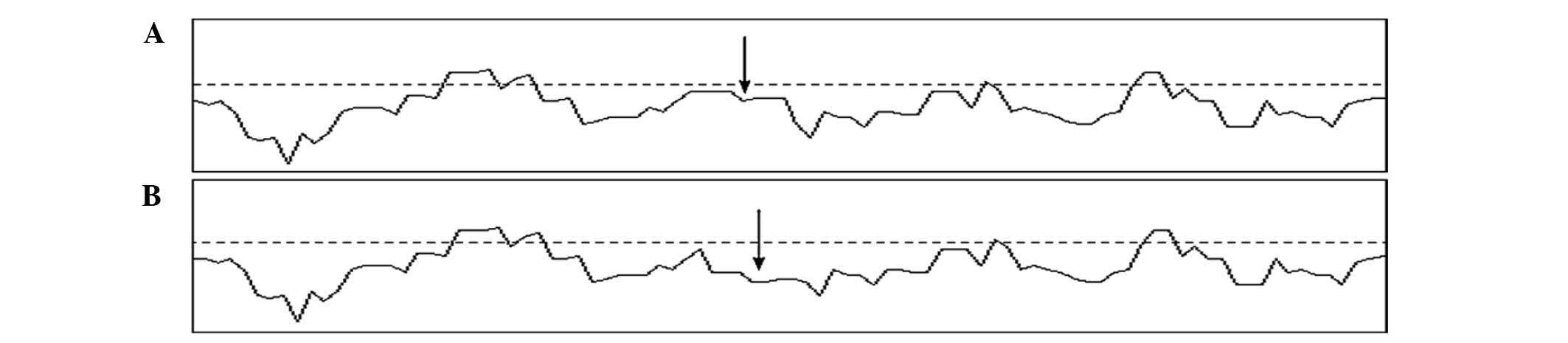
(Fig. 3). In addition, the
hydrophobicity of the corresponding region (Fig. 4) was decreased.
Discussion
In this pedigree, the diagnosis of EBS-loc with an
autosomal dominant transmission mode was made on the basis of the
clinical manifestations, family history and histopathological
examination of the patients' skin.
Subsequently mutation analysis of the commonest
candidate gene KRT5 was conducted using direct PCR
sequencing and PCR-RFLP, and a missense mutation (c.605T>A,
p.L202Q) was identified in KRT5. Since this mutation
cosegregated with the disease in the family and was absent from the
population controls, the possibility of a rare polymorphism was
excluded, and it was considered as a novel mutation. The mutation
replaces leucine with glutamine at the 202th amino acid on position
34 of the 1A helical domain of KRT5, which is highly conserved
among other type II keratins (data not shown) and different species
including human, dog, rat, mouse, chicken and zebrafish (Table II).
| Table II.Sequences producing specific alignment
of amino acids in keratin 5. |
Table II.
Sequences producing specific alignment
of amino acids in keratin 5.
| Species | 605 T>A
(L202Q) |
|---|
| Dog |
VRFLEQQNKVLDTKWTLLQEQGTKTVRQN |
| Mouse |
VRFLEQQNKVLDTKWALLQEQGTKTIKQN |
| Rat |
VRFLEQQNKVLDTKWALLQEQGTKTVRQN |
| Rabbit |
VRFLEQQNKVLDTKWTLLQEQGTKTVRQS |
| Chicken |
VRFLEQQNKVLETKWSLLQEQGMKTVRNN |
| Pig |
VRFLEQQNKVLDTKWTLLQEQGIKTVRQN |
| Cattle |
VRFLEQQNKVLDTKWTLLQEQGTKTVRQN |
| Zebrafish |
VRFLEQQNKVLETKWSLLQEQT-TTRSNI |
| Human
(wild-type) |
VRFLEQQNKVLDTKWTLLQEQGTKTVRQN |
| Human (III:3) |
VRFLEQQNKVLDTKWTQLQEQGTKTVRQN |
In general, the most severe phenotype of EBS,
EBS-DM, has been associated with mutations in more highly conserved
helix boundary motifs, including the head of segment 1A and end of
segment 2B in the rod domains of KRT5, which induce greater keratin
aggregation in basal keratinocytes. By contrast, milder phenotypes,
such as EBS-loc and EBS-gen, exhibit keratin monofilaments that
appear relatively normal ultrastructurally and have been linked to
mutations in linker domains and elsewhere in the rod domain of KRT5
(8–10). Consistent with previous reports, the
p.L202Q mutation in this pedigree was identified at the end of the
1A domain, a non-helix boundary motif of KRT5, which resulted in
the EBS-loc phenotype.
Computer-assisted prediction results indicated that
the L202Q mutation would have a significant effect on the secondary
structure and hydrophobicity of the protein. As shown in Fig. 4, the mutant appears to be missing a β
sheet at the mutation site, which may due to the alteration of the
polarity of the affected amino acid by the replacement of a
non-polar hydrophobic leucine by a polar neutral glutamine. It has
been demonstrated that a mutation in the coiled-coil motif of 1A
domain of KRT5 often results in a significant distortion of the
backbone over a turn, increasing the likelihood of impaired chain
aggregation, and hence molecular assembly (11). It was therefore speculated that the
omission of a β sheet could result in the reduction of
hydrophobicity; however, further functional experiments are
necessary to explore the underlying mechanisms in detail.
In this pedigree, the individual II:6 suffering from
EBS had unaffected parents. There are two possible causes for that:
Either the mutation is a de novo mutation or the penetrance
of EBS was incomplete.
The present study reported a novel mutation of EBS
in a Chinese pedigree. A previous report has revealed EBS types in
Israel that have a unique mutation spectrum and different patterns
of inheritance, including a higher incidence of recessive cases
than in families from Europe or the USA (12). In addition, the proportion of
Japanese patients with EBS with KRT5 mutations is 3-fold
higher than those with KRT14 mutations, despite the fact
that mutations in these two genes have been reported to be equally
prevalent (13,14). Whether the mutation spectrum in
Chinese patients with EBS is similar or unique to other ethnic
groups remains unclear; therefore, further accumulation of clinical
data is required, in order to confirm the spectrum of EBS mutations
in China.
Acknowledgements
The authors would like to thank all the members of
the pedigree for their kind cooperation. This study was supported
by the National Natural Science Foundation of China (grant no.
81270365).
References
|
1
|
Fine JD, Eady RA, Bauer EA, Briggaman RA,
Tuderman Bruckner L, Christiano A, Heagerty A, Hintner H, Jonkman
MF, McGrath J, et al: Revised classification system for inherited
epidermolysis bullosa: Report of the Second International Consensus
Meeting on diagnosis and classification of epidermolysis bullosa. J
Am Acad Dermatol. 42:1051–1066. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Coulombe PA, Kerns ML and Fuchs E:
Epidermolysis bullosa simplex: A paradigm for disorders of tissue
fragility. J Clin Invest. 119:1784–1793. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fuchs EV: The molecular biology of
epidermolysis bullosa simplex. Clinical. Bauer EA, McGuire J and
Moshell A: (Baltimore). Johns Hopkins University Press. 280–299.
1999.
|
|
4
|
Horn HM and Tidman MJ: The clinical
spectrum of epidermolysis bullosa simplex. Br J Dermatol.
142:468–472. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Steinert PM: The two-chain coiled-coil
molecule of native epidermal keratin intermediate filaments is a
type I-type II heterodimer. J Biol Chem. 265:8766–8774.
1990.PubMed/NCBI
|
|
6
|
Soetens O, Vauloup-Fellous C, Foulon I,
Dubreuil P, De Saeger B, Grangeot-Keros L and Naessens A:
Evaluation of different cytomegalovirus (CMV) DNA PCR protocols for
analysis of dried blood spots from consecutive cases of neonates
with congenital CMV infections. J Clin Microbiol. 46:943–946. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fine JD, Eady RA, Bauer EA, Bauer JW,
Bruckner-Tuderman L, Heagerty A, Hintner H, Hovnanian A, Jonkman
MF, Leigh I, et al: The classification of inherited epidermolysis
bullosa (EB): Report of the third international consensus meeting
on diagnosis and classification of EB. J Am Acad Dermatol.
58:931–950. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liovic M, Stojan J, Bowden PE, Gibbs D,
Vahlquist A, Lane EB and Komel R: A novel keratin 5 mutation
(K5V186L) in a family with EBS-K: A conservative substitution can
lead to development of different disease phenotypes. J Invest
Dermatol. 116:964–969. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Murrell DF, Trisnowati N, Miyakis S and
Paller AS: The yin and the yang of keratin amino acid substitutions
and epidermolysis bullosa simplex. J Invest Dermatol.
131:1787–1790. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bolling MC, Lemmink HH, Jansen GH and
Jonkman MF: Mutations in KRT5 and KRT14 cause epidermolysis bullosa
simplex in 75% of the patients. Br J Dermatol. 164:637–644.
2011.PubMed/NCBI
|
|
11
|
Smith TA, Steinert PM and Parry DA:
Modeling effects of mutations in coiled-coil structures: Case study
using epidermolysis bullosa simplex mutations in segment 1a of
K5/K14 intermediate filaments. Proteins. 55:1043–1052. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ciubotaru D, Bergman R, Baty D, Indelman
M, Pfendner E, Petronius D, Moualem H, Kanaan M, Ben Amitai D,
McLean WH, et al: Epidermolysis bullosa simplex in Israel: Clinical
and genetic features. Arch Dermatol. 139:498–505. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yasukawa K, Sawamura D, Goto M, Nakamura
H, Jung SY, Kim SC and Shimizu H: Epidermolysis bullosa simplex in
Japanese and Korean patients: Genetic studies in 19 cases. Br J
Dermatol. 155:313–317. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shinkuma S, Natsuga K, Nishie W and
Shimizu H: Epidermolysis bullosa in Japan. Dermatol Clin.
28:431–432. 2010. View Article : Google Scholar : PubMed/NCBI
|