Stem cells: Genetic integrity
Stem cells (SCs) have high potential and hold great
promise in the rapid development of regenerative medicine.
Pluripotent SCs are able to self-renew unrestrictedly. They can
differentiate in vitro and in vivo in all cell types
deriving from the three germ layers (1,2). This
ability makes them useful in cell replacement therapy and the
treatment of numerous diseases (3),
including diabetes (4),
neurodegenerative (5), retinal
(6) and cardiac (7) diseases, as well as muscular dystrophy
(8). SC therapy raises questions
concerning the consequences of their influence on an organism.
In vivo studies constitute only a small proportion of all
research on SCs (9).
Despite the clearly demonstrated effectiveness of
SC-derived therapies, this approach has a number of impediments.
The response of SCs and stem-derived cells to ionizing radiation
(IR) and chemotherapeutics is a questionable issue, particularly
with regard to the increase of cancer morbidity in patients >50
years old (10,11). Tumor diseases are frequently
diagnosed, particularly in elderly patients often burdened with
other diseases. How SC therapies affect the organism during cancer
treatment (radiotherapy and/or chemotherapy) remains unknown.
Exposure to gamma radiation and cisplatin is known to cause DNA
damage in cancer cells. These treatments are intended to deprive
cancer cells of multiplication potential, and trigger irreparable
DNA damage leading to their death (12). However, knowledge concerning the
effects of anticancer therapies on healthy cells, including SCs is
limited. The exposure of SCs to IR will be unavoidable during
treatment and routine diagnosis using computed tomography, positron
emission tomography and single-photon emission computed tomography
(13).
An additional difficulty in the application of SCs
is the evidence that human-induced pluripotent SCs (hiPSCs) and
human embryonic SCs (hESCs) are prone to genetic instability during
in vitro culture. Frequently chromosomal rearrangements,
aneuploidy or defective DNA methylation in both cell types are
observed. This results in decreased differentiation capacity and
increased proliferation rate (14).
Cellular stress, such as freeze-thaw cycles, causes them to be more
prone to gene mutation. Manipulation of culture conditions in
vitro may contribute to epigenetic instability. Although the
majority of cell lines retain a normal karyotype during multiple
passages, long-term culture increases the risk of anomalies
(15). It has also been reported
that the process of reprogramming leads to the creation of
genetically unstable induced pluripotent SCs (iPSCs). Chromosomal
abnormalities in those cells occur at the very early passages
(16). The first reports involving
abnormal karyotypes of hESCs concerned trisomy of chromosome 12.
Chromosomal aberrations may apply to all chromosomes or occur at
subchromosomal level. Many of them are also observed in iPSCs
(17). Trisomy of chromosome 8
occurs more frequently in hiPSCs than in hESCs. In turn, trisomy 17
was not identified in hiPSCs, but was present in hESCs (18).
Inzunza et al investigated the karyotypes of
three hESC lines. The karyotypes of two of the cell lines did not
differ, but in the third a monosomy × was demonstrated (19). Genomic and phenotypic changes may be
associated with abnormal functioning of SCs, both in the
undifferentiated and differentiated stages. Thus, the issue of
genetic stability of SCs and cells differentiated from them is
crucial in the context of the application of these cells in
clinical trials. Further studies are required to demonstrate that
iPSCs have no deleterious effect for patients. A high level of DNA
damage disrupts the normal functioning of cells. Changes occurring
in DNA play an important role during aging, disease conditions and
cancer development (20).
Specialized repair mechanisms, checkpoints of the cell cycle and
tolerance to certain DNA damage protect the integrity of the cell
genome, which is required for the normal functioning of cells and
their progeny (21). DNA damage is
caused by numerous factors, which can arise during replication and
transcription, or in response to endogenous and exogenous factors,
such as UV radiation, reactive oxygen species, IR and chemical
agents (22). However, the nature of
the cellular response of SCs to damaging agents and the repair
mechanisms remain poorly understood.
The present article provides an overview of the stem
and stem-derived cell DNA-damage response to cytotoxic and
genotoxic agents during anticancer therapies. Although some
research has been carried out on the DNA repair mechanisms of SCs,
the complicated mechanisms in undifferentiated, partially
differentiated and differentiated cells require elucidation in
further studies. The current literature data on DNA repair
mechanisms in SCs are explored and discussed in the present
review.
Cell cycle of stem cells and DNA damage
recognition
SCs are required to constantly deal with potential
damage to their DNA (23). When
severe DNA damage occurs, the cell cycle is arrested to prevent
aberrant replication, transcription and translation, as well as to
preserve energy. However, when DNA damage repair is impossible,
cell death mechanisms are activated (24). In response to DNA double-strand
breaks (DSBs), ESCs undergo similar events to those of
differentiated cells: The kinase ataxia telangiectasia mutated
(ATM) becomes phosphorylated at serine 1981 and relocates to DSBs,
where it phosphorylates the histone variant protein H2AX within a
few minutes. In mouse embryonic stem cells (mESCs) following
exposure to IR, the ATM and ataxia talengiectasia and Rad3-related
protein (ATR) kinases phosphorylate >900 sites in ~700 proteins
(25). The phosphorylation of
histone H2AX is visible as γH2AX foci. The level of endogenous foci
can be also detected in the presence of unrepaired or misrepaired
DNA DSBs, the dysfunction of telomeres, genomic instability or
senescence. The size of γH2AX depends on the chromatin structure.
During the condensation of chromatin regions, smaller structures
can be observed. In turn, when the hyperacetylation of chromatin
proceeds or a deficiency of histone H1 occurs, the γH2AX foci
create bigger structures (26).
Hundreds of much smaller γH2AX foci appear in differentiated cells,
although these small foci probably are unrelated to the DNA damage
response (27). Moreover, mESCs
reveal increased basal levels of γH2AX, even in the absence of DNA
DSBs (28). Another important factor
is replication protein A, which is single-stranded DNA-binding
protein playing a major role in DNA repair pathways (including
nucleotide excision, base excision and double-strand break repairs)
(29).
The majority of accomplished experiments have been
carried out on mESCs. mESCs and hESCs are not equivalent, and
differences between them must be taken into consideration. mESCs do
not possess the G1 checkpoint and readily undergo tumor protein 53
(p53)-independent apoptosis, as well as demonstrating good repair
mechanisms in response to oxidative damage (30). The lack of a functional G1 checkpoint
is caused by sequestration of p53 into the cytoplasm. The cells
with DNA damage transition from G1 into the S phase, where the
lesion can be intensified by a round of replication. mESCs spend
~75% of their time in S phase, which favors homologous
recombination as a main DNA repair mechanism. The p53 protein has
an ability to inhibit the activity of homeobox protein Nanog
throughout the association with its promoter, which facilitates the
differentiation of mESCs. It allows the maintenance of an unchanged
population of cells (31). mESCs
tolerate only a low level of DNA damage, and for that reason they
readily undergo apoptosis or differentiation. As a result of these
defensive mechanisms, mESCs generate fewer mutations than somatic
cells (32).
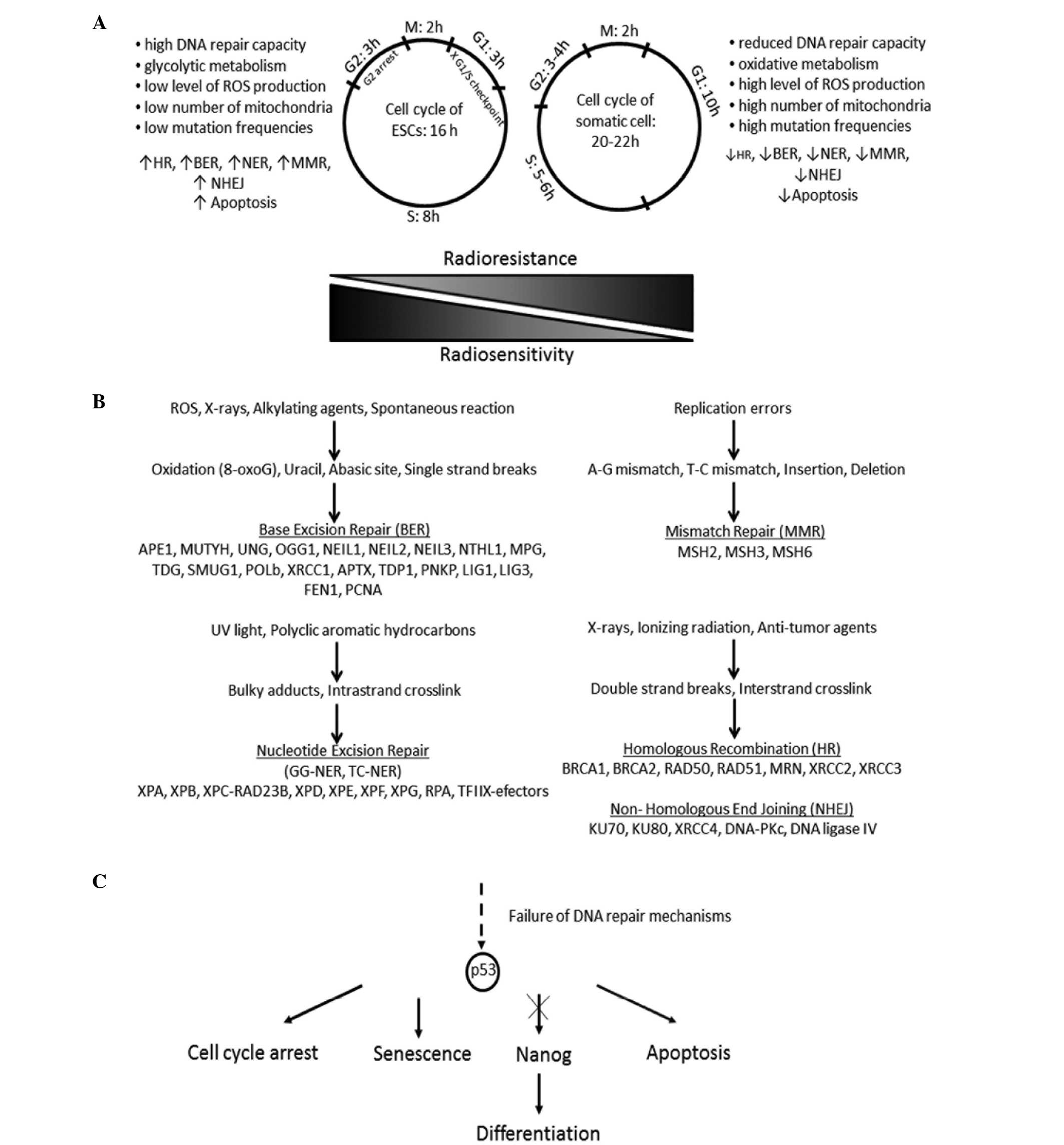
The cell cycle in hESCs and mESCs has been found to
be shorter in duration than that in somatic cells (Fig. 1A). This is caused by the
significantly shorter G1 phase and the enhanced expression of
cyclin-dependent kinase 4 (CDK4) and cyclin D2 facilitating
transition to the S phase. The defective G1/S checkpoint leads to
the accumulation of DNA damage in the S phase, where DNA damage
repair is activated or cell death proceeds (33). hESCs activate cell cycle arrest in G2
and, in contrast to mESCs, p53-dependent apoptosis. Their DNA
repair mechanisms are enhanced, which has improved genome
protection effects, such as a higher level of DNA-dependent protein
kinase catalytic subunit (DNA-PKcs) following irradiation and
ultimately more effective repair of DSBs (34). p53 prevents the accumulation of
unrepaired DNA lesions throughout cell arrest and DNA repair; in
addition, it inhibits the expression of genes responsible for
pluripotency, such as Nanog. Studies have indicated that the
suppression of p53 may contribute to successful reprogramming.
However, in this case the tumorigenicity of iPSCs and their
derivatives requires intensive examination (35).
Desmarais et al disclosed that hESCs fail to
activate checkpoint kinase (CHK1) and ATM following exposure to
cisplatin. Consequently, these cells undergo apoptosis rather than
DNA repair. Furthermore, hESCs are able to activate CHK1 or ATM
following IR treatment. hESCs and mESCs reveal less oxidative
damage than do differentiated cells (36). This phenomenon may be explained by
two concepts. First, ESCs have higher levels of antioxidants that
decrease with the progression of differentiation; the
downregulation of antioxidant genes results in an increase in the
levels of reactive oxygen species. Secondly, mESCs and hESCs
demonstrate better DNA repair capacity than differentiated cells
such as fibroblasts (37).
DNA repair systems in stem cells
In accordance with the law of Bergoniè and
Tribondeau, cells undergoing intensive division, that are
mitotically active or characterized by a high mitotic index, as
well as undifferentiated cells, are much more radiosensitive in
contrast to other cells. This statement has been confirmed in
vitro numerous times, with the exception of mature lymphocytes,
that although are differentiated, reveal considerably decreased
radioresistance (38).
Wilson et al conducted investigations that
concerned the irradiation of hESCs cells with low and high doses of
gamma radiation. Unsurprisingly, high doses of radiation brought
about massive cell death. However, all samples of irradiated hESCs
continued to be able to form teratomas, which is a clear test in
the determination of pluripotency. It is likely that, although
high-dose radiation alters some developmental pathways, it does not
influence the expression of pluripotency genes (39). Moreover, hESCs and human mesenchymal
SCs (MSCs) are less susceptible to radiation-induced bystander
effect signaling than fully differentiated cells in in vitro
experiments, which has a direct relevance to cancer therapy
(40).
DNA repair pathways
DNA repair is essential for genetic integrity. DNA
repair mechanisms can be divided into two main groups: Direct and
indirect DNA damage response (DDR). The former includes
O6-methylguanine-DNA methyl-transferase (MGMT) activity, which
removes methyl from newly created O6methylguanine adducts; this
compound is harmful because it forms complementary base pairs with
thymine instead of cytosine, and thus is potentially mutagenic
(41). Indirect DNA repair pathways
are composed of nucleotide excision repair (NER), base excision
repair (BER), mismatch repair (MMR) and DSB recombination repair.
DSBs are repaired by error-free homologous recombination (HRR) and
error-prone nonhomologous end-joining (NHEJ) (Fig. 1B). The majority of damage triggered
by chemo- and radiotherapy is repaired by HRR and NHEJ (42). Numerous proteins involved in the five
basic DNA repair mechanisms take part also in apoptosis. In HHR:
Breast cancer 1, early onset (BRCA1), ATM, ATR and p53; in NHEJ:
DNA-PK; in NER, excision repair cross-complementation group 2
(XPD), p53 and excision repair cross-complementation group 3 (XPB);
in BER, poly (ADP-ribose) polymerase 1 (PARP-1),
apurinic/apyrimidinic endonuclease/redox effector factor-1
(Ref-1/Ape) and p53; in MMR, mut S homolog 2 (MSH2), Mut S homolog
6 (MSH6) and Mut L homolog 1 (MLH1) (43). This process is discussed later in
this review.
DSB repair mechanisms
DSBs cause a massive loss of genetic information,
genomic rearrangements and/or cell death. The DSBs are repaired by
homologous recombination (HR) and NHEJ. During HR, the undamaged
DNA template on the sister chromatid is necessary in order to
recover the original sequence. In NHEJ, homology is not required
due to modification and ligation of the DNA ends. It often results
in deletion or insertions. HR is active in S/G2/M phases and NHEJ
in G1/S/G2/M phases (44,45). RAD51 and BRCA1 are the most common
biomarkers for HR, and DNA-PKcs for NHEJ (46). The RAD51 gene encodes a recombinase
protein that induces DNA strand exchange during HR. It plays a
pivotal role in DSBs during replication, because RAD51 associates
with chromatin during the S phase and interacts with components of
the DNA replication apparatus (47).
RAD51 is active in three phases of HR: Presynapsis, synapsis and
post-synapsis. During the first stage, the presynaptic filament
consisting of six RAD51 molecules and a single-strand DNA (ssDNA)
filament is formed. In the synapsis phase, RAD51 creates a
heteroduplex DNA (D-loop) created by connection of invading
substrate and homologous duplex DNA template. During the last
phase, RAD51 exposes the 3-OH group required for DNA synthesis
throughout dissociation from double-strand DNA (48). DSB repair using a homologous template
can involve HR or single-strand annealing (SSA). SSA is always
characterized by mutagenic alterations, as it involves the
annealing of sequence repeats located near the DSB and as a result,
a deletion between these repeats is observed (49). The participation of very precise HR
decreases with the progress of differentiation, whereas the
contribution of error-prone NHEJ increases. Oliver et al
demonstrated that even during the early stages of differentiation,
hMSCs are more prone to cell death. This phenomenon may be
explained through the commitment of caspases both in apoptosis and
cell differentiation (50).
NHEJ repairs exogenous DSBs triggered by radiation
exposure or variable diversity joining (VDJ) recombination. The
proteins engaged in this process involve formation of a complex
from DNA-PK, Ku70 and Ku80 at the lesion, the alignment of short
homologies in overhand regions and recruitment of the ligase
IV/X-ray repair cross-complementing protein 4 (XRCC4) complex
(20). The Ku complex, due to its
toroidal shape, is able to detect DNA damage, maintain the two DNA
ends in proximity, inhibit or regulate the activity of exonuclease,
and also recruit DNA-PKcs polymerase and ligase (51). DNA-PKcs is the largest known protein
with serine/threonine kinase activity. DNA-PKcs and Ku bind
together to form a DNA duplex. Then, autophosphorylation occurs at
>15 sites. This activated protein induces the ligase activity of
ligase IV/XRCC4. Positive feedback loops are evident: The presence
of DNA ligase IV/XRCC4 evokes the autophosphorylation activity of
DNA-PKcs. The DNA-PKcs takes part in the regulation of the
endonucleolytic activities of artemis, which is another relevant
factor for NHEJ (52). Artemis is
hyperphosphorylated by the ATM kinase in response to IR. The
artemis-DNA-PKcs complex excises damaged DNA overhangs due to its
5′ and 3′ endonuclease activity. Moreover, artemis possesses an
apparent 5′ exonuclease activity (53).
DSB repair is more precise in hESCs than it is in
somatic human cells. The differentiation of hESCs into astrocytes
results in a reduction in the efficiency and fidelity of DSB repair
mechanisms. hESCs use homologous recombinational repair rather than
NHEJ, which is contrary to the situation in other types of human
cells. In addition, a reduction in the accuracy of NHEJ is observed
during differentiation. Hence, NHEJ in hESCs is likely to be
independent on ATM, DNA-PKcs and PARP as well as dependent on XRCC4
with considerable effectiveness (13). Zou et al examined the response
of hESCs and hESC-derived neuronal stem cells (NSCs) to IR
(54). They reported that hESCs have
DNA repair mechanisms comparable with those of somatic cells, which
was not in accordance with other literature data, and revealed that
following high-dose treatment, 90% of irradiated hESCs underwent
apoptosis. Following differentiation, the DNA repair capacity of
stem-derived cells was found to decline and a higher level of
reactive oxygen species was shown. In contrast to hESCs, a sizeable
fraction of the NSCs survived high doses of IR (54). In conclusion, although hESCs
demonstrate robust DNA repair capacity and tolerate stress, they
seems to be considerably more sensitive to IR than are the cells
differentiated from them.
Sokolov and Neumann investigated the dynamics of
transcriptional changes of IR-responsive genes, such as
cyclin-dependent kinase inhibitor 1A (CDKN1A/p21/Cip1), growth
arrest and DNA-damage-inducible α (GADD45A), proliferating cell
nuclear antigen (PCNA), BTG family member 2 (BTG2), BCL2-binding
component 3 (BBC3), sestrin 1 (SESN1), DNA damage-binding protein 2
(DDB2), immediate early response 5 (IER5), polo-like kinase 3
(PLK3) and growth differentiation factor 15 (GDF15) during the
‘early’ and ‘late’ radioresponse of hESCs. The activation of stress
genes exhibited a dose-response correlation, although not in clear
linear manner. This may indicate the existence of a threshold for
changes in gene expression in certain human ESC genomes for low
doses of IR (55). CDKN1A is one of
the principal cyclin-dependent kinase inhibitors necessary for
effective activation of the G1/S checkpoint following IR treatment
in majority of human cells in vitro. However, in response to
IR, hESCs and iPSCs fail to induce the G1/S checkpoint. This
phenomenon may be caused by reduced p21 protein levels in
hESCs/iPSCs following IR (56).
However, the G1/S checkpoint is characteristic for NHEJ not for
HR.
Single-strand break repair
mechanisms
The mismatch repair (MMR) pathway is responsible for
the repair of mismatches created between bases on complementary DNA
strands. The mismatches are formed due to polymerase slippage
during replication that initiates errors and alterations in
nucleotide incorporation, as well as short insertions and deletions
(indels). Deficiencies of MMR may induce the accumulation of
mutations and microsatellite instability, which is characteristic
for hereditary nonpolyposis colon cancer (57). The study carried out by Tichy et
al indicates that mESCs are characterized by elevated levels of
MMR proteins, which correlates with more efficient MMR. Moreover,
in these cells, BER protein expression levels are also elevated and
better able to repair an oligonucleotide template (58).
The BER pathway covers two main types of repair:
Short patch repair dependent upon DNA polymerase β and long patch
repair dependent upon PCNA. In short patch repair, single
nucleotides are first removed and then replaced. The longer
mechanism is based on the removal of nucleotides from the damaged
strand, DNA synthesis and consequently, ligation. Those mechanisms
eliminate base damage caused by oxidation, alkylation or
deamination as well as ssDNA breaks (59).
NER is responsible for eliminating a wide variety of
bulky, helix-distorting lesions from DNA, such as cisplatin-DNA
intrastrand crosslinks. With regard to the mechanical process, BER
and NER are very similar; however, the NER pathway is much more
complex mechanism. NER involves DNA damage recognition, local
opening of the DNA helix around the lesion, excision of a short
single-strand segment of DNA spanning the defect, as well as
sequential repair synthesis and strand ligation (60). The NER pathway consists of two
subgroups, termed global genome NER (GG-NER) and
transcription-coupled NER (TC-NER). The predominant damage
recognition factor of GG-NER is the XP complementation group C
(XPC)/Rad23 homolog B (HR23B)/Centrin (CEN2) system. This system
eliminates or reduces DNA lesions throughout the genome. TC-NER
damage recognition includes the arrest of elongating RNA polymerase
II (RNAPII) and two proteins: Cockayne syndrome A (CSA) and B
(CSB). The XPC complex, CSA and CSB recruit 10 protein complexes
and the multi-functional transcription factor TFIIH to the site of
the lesion (61). Undifferentiated
cells have higher efficacy of NER and BER than non-pluripotent
cells. The GG-NER, but not TC-NER repair mechanism, is superior in
human undifferentiated cells in comparison with fibroblasts
(62).
Failure of DNA repair systems:
Apoptosis
Failure of DNA repair processes directs cells to
undergo apoptosis-programmed cell death (63). Dying cells send signals to
neighboring and living cells, inducing the proliferation of stem
and progenitor cells. This phenomenon is crucial for the
regenerative process (64). The main
reason for apoptosis is a loss of genomic integrity caused by the
accumulation of DNA damage (65).
Although SCs have very efficient DNA repair mechanisms, the lack of
a G1 checkpoint causes them to be hypersensitive to IR and other
DNA-damaging agents, which facilitates their apoptosis (66). However, the final decision regarding
the induction of apoptosis and/or cell cycle arrest is dependent on
the magnitude and duration of the damage stimuli (67). In mammalian cells, two main pathways
initiating apoptosis exist: Extrinsic and intrinsic
(mitochondrial). The key mechanism in both is based on the
activation of enzymes known as caspases (68). Caspases can be divided into initiator
(caspase-8, −9 and −12) and effector (caspase-3, −6 and −7) types
(69,70). An important role in the process of
apoptosis is played by p53. In physiological conditions, the
concentration of p53 in the cytosol is low. In response to cellular
stress, p53 is accumulated, which leads to the activation of
apoptosis (71).
The extrinsic apoptotic pathway is based mainly on
binding of death receptor ligand to the receptor membrane. These
death receptors include the tumor necrosis factor (TNF) receptor
superfamily of TNF-Fas (Apo-1, CD95), TNF receptor 1 (TNF-R1),
death receptor 3 (DR-3, Apo-3, TRAMP, WSL-1, LARD), death receptor
4 (TRAIL-R1, DR-4), death receptor 5 (TRAIL-R2, DR-5), death
receptor 6 (DR-6), ectodysplasin receptor (EDA-R) and nerve growth
receptor (NGF-R). All death receptors are composed of an
extracellular domain, transmembrane portion and cytoplasmic death
domain (DD), which is necessary for interaction with the adapter
protein complex. Receptor-ligand binding leads to a conformational
change in the receptor internal domain and formation of the
death-inducing signaling complex (DISC), consisting of a death
receptor adapter protein known as Fas-associated protein with death
domain (FADD) and procaspase-8 or procaspase-10. As a result of
DISC dimerization, the activation of caspase-8 or −10 proceeds.
Activated caspases initiate a cascade of reactions leading to cell
death (72–74).
The intrinsic apoptotic pathway is associated with
the mitochondria and activated by DNA damage or cytotoxic drugs,
which triggers changes within the mitochondria, such as increased
permeability of the mitochondrial membrane and outflow of
cytochrome c to the cytoplasm. Subsequently, cytochrome
c activates apoptosis-inducing protein (Apaf-1) and
procaspase-9. An apoptosome is formed, which induces other effector
caspases (75). Membrane proteins
belonging to the B-cell lymphoma 2 (Bcl-2) family are involved in
the regulation of this process. Some of them: Bcl-2 and B-cell
lymphoma-extra large (Bcl-xL) are anti-apoptotic and increase the
likelihood of cell survival. Others, such as Bcl-2-associated ×
protein (Bax) and Bcl-2 homologous antagonist killer (Bak), direct
the cell to programmed death (76).
Although the intrinsic and extrinsic pathways act in an independent
manner, the receptor pathway can be associated with the internal
pathway via BH3 interacting domain death agonist (Bid) protein.
Caspase-8 is capable of proteolysis of the pro-apoptotic protein,
Bid, which is translocated to the mitochondrial surface. This
results in release of cytochrome c and activation of the
intrinsic pathway of apoptosis (77).
Mechanism of apoptosis in pluripotent
SCs
The mechanism of apoptosis varies, according to cell
type and the nature of the stimulus. Filion et al showed
that in the majority of hESCs undergoing apoptosis due to DNA
damage, apoptosis was directed through the mitochondrial pathway.
In stress conditions, the accumulation of p53 contributes to the
inhibition of octamer-binding transcription factor 4 (Oct-4) and
Nanog expression, and the induction of spontaneous differentiation
(Fig. 1C). This suggests that the
utilization of this mechanism ensures the genomic integrity of
hESCs (78). In comparison with
hESCs, mESCs have lower levels of apoptosis and a reduced capacity
for differentiation (79). However,
mESCs exposed to DNA damage-inducing UV radiation also reveal a
certain degree of ability to induce differentiation by suppressing
the expression of Nanog (80). HESCs
have a high capacity for DNA repair and a considerable threshold of
apoptosis induction compared with differentiated cells. iPSCs
exhibit an inferior apoptosis response to reactive oxygen species
in comparison with hESCs (32). SCs
isolated from the small intestine are sensitive to DNA
damage-induced cell death due to their low expression levels of the
anti-apoptotic Bcl-2 protein. By contrast, colon SCs are more
resistant to radiation; they express high levels of Bcl-2. This
demonstrates that the sensitivity to DNA damage and p53-induced
apoptosis differs significantly among different types of SCs
(81). In conclusion, apoptosis
constitutes a defensive mechanism that protects entire populations
of cells against the effects of endo-exogenous factors.
Conclusions
In conclusion, SCs possess a unique cell cycle,
which has a DDR-enhancing effect. The DNA repair mechanisms in
pluripotent SCs are more efficient than those in differentiated
cells. In particular, HR is favored as a main repair mechanism of
DNA DSBs. Unrepaired DNA damage in pluripotent SCs readily directs
them to programmed cell death or differentiation. This phenomenon
prevents the accumulation of mutations and contributes to the
genetic stability of SCs. The genetic integrity of pluripotent SCs
and their derivatives is very relevant due to the unavoidable
exposure of SCs to genotoxic and cytotoxic agents during diagnostic
procedures, as well as during anti-cancer therapies. For that
reason, further studies concerning the safety of stem and
stem-derived cells treated with IR and/or chemotherapeutics are
required.
Acknowledgements
The present study was supported by the National
Science Centre (grant number 2012/07/E/NZ3/01819).
References
|
1
|
Tomizawa M, Shinozaki F, Sugiyama T,
Yamamoto S, Sueishi M and Yoshida T: Activin A maintains
pluripotency markers and proliferative potential of human induced
pluripotent stem cells. Exp Ther Med. 2:405–408. 2011.PubMed/NCBI
|
|
2
|
Lach M, Trzeciak T, Richter M, Pawlicz J
and Suchorska WM: Directed differentiation of induced pluripotent
stem cells into chondrogenic lineages for articular cartilage
treatment. J Tissue Eng. 5:20417314145527012014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fragma AM, de Araújo ÉSS, Vergani N,
Fonseca SAS and Pereira LV: Use of human embryonic stem cells in
therapy. Stem Cells and Cell Therapy. Al-Rubeai M and Naciri M:
(Dordrecht). Springer. 1–19. 2014. View Article : Google Scholar
|
|
4
|
Domínguez-Bendala J, Lanzoni G, Inverardi
L and Ricordi C: Concise review: Mesenchymal stem cells for
diabetes. Stem Cells Transl Med. 1:59–63. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Feng Z and Gao F: Stem cell challenges in
the treatment of neurodegenerative disease. CNS Neurosci Ther.
18:142–148. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin ZB, Okamoto S, Mandai M and Takahashi
M: Induced pluripotent stem cells for retinal degenerative
diseases: A new perspective on the challenges. J Genet. 88:417–424.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bolli R, Chugh AR, D-Amario D, Loughran
JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T,
et al: Cardiac stem cells in patients with ischaemic cardiomyopathy
(SCIPIO): Initial results of a randomised phase 1 trial. Lancet.
378:1847–1857. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abujarour R, Bennett M, Valamehr B, Lee
TT, Robinson M, Robbins D, Le T, Lai K and Flynn P: Myogenic
differentiation of muscular dystrophy-specific induced pluripotent
stem cells for use in drug discovery. Stem Cells Transl Med.
3:149–160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Drummond RJ, Kunath T, Mee PJ and Ross JA:
Induced pluripotent stem cell technology and stem cell therapy for
diabetes. Exp Ther Med. 2:3–7. 2011.PubMed/NCBI
|
|
10
|
de Magalhães JP: How ageing processes
influence cancer. Nat Rev Cancer. 13:357–365. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zaman MH: The role of engineering
approaches in analysing cancer invasion and metastasis. Nat Rev
Cancer. 13:596–603. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baskar R, Lee KA, Yeo R and Yeoh KW:
Cancer and radiation therapy: Current advances and future
directions. Int J Med Sci. 9:193–199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sokolov MV and Neumann RD: Human embryonic
stem cells responses to ionizing radiation exposures: Current state
of knowledge and future challenges. Stem Cells Int.
2012:5791042012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nguyen HT, Geens M and Spits C: Genetic
and epigenetic instability in human pluripotent stem cells. Hum
Reprod Update. 19:187–205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lund RJ, Närvä E and Lahesmaa R: Genetic
and epigenetic stability of human pluripotent stem cells. Nat Rev
Genet. 13:732–744. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martins-Taylor K, Nishler SB, Taapken SM,
Compton T, Crandall L, Montgomery KD, Lalande M and Xu RH:
Recurrent copy number variations in human induced pluripotent stem
cells. Nat Biotechnol. 29:488–491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peterson SE and Loring JF: Genomic
instability in pluripotent stem cells: Implications for clinical
applications. J Biol Chem. 289:4578–4584. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Taapken SM, Nisler BS, Newton MA,
Sampsell-Barron TL, Leonhard KA, Mclntire EM and Montgomery KD:
Karyotypic abnormalities in human induced pluripotent stem cells
and embryonic stem cells. Nat Biotechnol. 29:313–314. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Inzunza J, Sahlén S, Holmberg K, Strömberg
AM, Teerijoki H, Blennow E, Hovatta O and Malmgren H: Comparative
genomic hybridization and karyotyping of human embryonic stem cells
reveals the occurrence of an isodicentric X chromosome after
long-term cultivation. Mol Hum Reprod. 10:461–466. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kenyon J and Gerson SL: The role of DNA
damage repair in aging of adult stem cells. Nucleic Acids Res.
35:7557–7565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Giglia-Mari G, Zotter A and Vermeulen W:
DNA damage response. Cold Spring Harb Perspect Biol. 3:a0007452011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Blanpain C, Mohrin M, Sotiropoulou PA and
Passegué E: DNA- damage response in tissue-specific and cancer stem
cells. Cell Stem Cell. 8:16–29. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
von Stechow L, Ruiz-Aracama A, van de
Water B, Peijnenburg A, Danen E and Lommen A: Identification of
cisplatin-regulated metabolic pathways in pluripotent stem cells.
PLoS One. 8:e764762013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pines A, Kelstrup CD, Vrouwe MG, Puigvert
JC, Typas D, Misovic B, de Groot A, von Stechow L, van de Water B,
Danen EH, et al: Global phopshoproteome profiling reveals
unanticipated networks responsive to cisplatin treatment of
embryonic stem cells. Mol Cell Biol. 31:4964–4977. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Banáth JP, Bañuelos CA, Klokov D, MacPhail
SM, Lansdorp PM and Olive PL: Explanation for excessive DNA
single-strand breaks and endogenous repair foci in pluripotent
mouse embryonic stem cells. Exp Cell Res. 315:1505–1520. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han J, Hendzel MJ and Allalunis-Turner J:
Quantitative analysis reveals asynchronous and more than
DSB-associated histone H2AX phosphorylation after exposure to
ionizing radiation. Radiat Res. 165:283–292. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Middel V and Blattner C: DNA repair in
embryonic stem cells. DNA Repair - On the Pathways to Fixing DNA
Damage and Errors. Storici F: (Rijeka, Croatia). InTech. 357–380.
2011.
|
|
29
|
Prendergast ÁM, Cruet-Hennequart S, Shaw
G, Barry FP and Carty MP: Activation of DNA damage response
pathways in human mesenchymal stem cells exposed to cisplatin or
γ-irradiation. Cell Cycle. 10:3768–3777. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo LZ, Gopalakrishna-Pillai S, Nay SL,
Park SW, Bates SE, Zeng X, Iverson LE and O-Connor TR: DNA repair
in human pluripotent stem cells is distinct from that in
non-pluripotent human cells. PLoS One. 7:e305412012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tichy ED and Stambrook PJ: DNA repair in
murine embryonic stem cells and differentiated cells. Exp Cell Res.
314:1929–1936. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fan J, Robert C, Jang YY, Liu H, Sharkis
S, Byalin SB and Rassool FV: Human induced pluripotent cells
resemble embryonic stem cells demonstrating enhanced levels of DNA
repair and efficacy of nonhomologous end-joining. Mutat Res.
713:8–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rocha CRR, Lerner LK, Okamoto OK,
Marchetto MC and Menck CFM: The role of DNA repair in the
pluripotency and differentiation of human stem cells. Mutat Res.
752:25–35. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harfouche G and Martin MT: Response of
normal stem cells to ionizing radiation: A balance between
homeostasis and genomic stability. Muat Res. 704:167–174. 2010.
View Article : Google Scholar
|
|
35
|
Zhao T and Xu Y: p53 and stem cells: New
developments and new concerns. Trends Cell Biol. 20:170–175. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Desmarais JA, Hoffmann MJ, Bingham G,
Gagou ME, Meuth M and Andrews PW: Human embryonic stem cells fail
to activate CHK1 and commit to apoptosis in response to DNA
replication stress. Stem Cells. 30:1385–1393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maynard S, Swistowska AM, Lee JW, Liu Y,
Liu ST, Da Cruz AB, Rao M, de Souza-Pinto NC, Zeng X and Bohr VA:
Human embryonic stem cells have enhanced repair of multiple forms
of DNA damage. Stem Cells. 26:2266–2274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Christensen DM, Iddins CJ and Sugarman SL:
Ionizing radiation injuries and illnesses. Emerg Med Clin North Am.
32:245–265. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wilson KD, Sun H, Huang M, Zhang WY, Lee
AS, Li Z, Wang SX and Wu JC: Effects of ionizing radiation on
self-renewal and pluripotency of human embryonic stem cells. Cancer
Res. 70:5539–5548. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sokolov MV and Neumann RD:
Radiation-induced bystander effects in cultured human stem cells.
PLoS One. 5:e141952010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Park Y and Gerson SL: DNA repair defects
in stem cell function and aging. Annu Rev Med. 56:495–508. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Maugeri-Saccà M, Bartucci M and De Maria
R: DNA damage repair pathways in cancer stem cells. Mol Cancer
Ther. 11:1627–1636. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bernstein C, Bernstein H, Payne CM and
Garewal H: DNA repair/pro-apoptotic dual-role proteins in five
major DNA repair pathways: Fail-safe protection against
carcinogenesis. Mutat Res. 511:145–178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lundin C, Erixon K, Arnaudeau C, Schultz
N, Jenssen D, Meuth M and Helleday T: Different roles for
nonhomologous end joining and homologous recombination following
replication arrest in mammalian cells. Mol Cell Biol. 22:5869–5878.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mao Z, Bozzella M, Seluanov A and
Gorbunova V: DNA repair by nonhomologous end joining and homologous
recombination during cell cycle in human cells. Cell Cycle.
7:2902–2906. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lund PK: Fixing the breaks in intestinal
stem cells after radiation: A matter of DNA damage and death or DNA
repair and regeneration. Gastroenterology. 143:1144–1147. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Serrano L, Liang L, Chang Y, Deng L,
Maulion C, Nguyen S and Tischfield JA: Homologous recombination
conserves DNA sequence integrity throughout the cell cycle in
embryonic stem cells. Stem Cells Dev. 20:363–374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Krejci L, Altmannova V, Spirek M and Zhao
X: Homologous recombination and its regulation. Nucleic Acids Res.
40:5795–5818. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fung H and Weinstock DM: Repair at single
targeted DNA double-strand breaks in pluripotent and differentiated
human cells. PLoS One. 6:e205142011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Oliver L, Hue E, Séry Q, Lafargue A,
Pecqueur C, Paris F and Vallette FM: Differentiation-related
response to DNA breaks in human mesenchymal stem cells. Stem Cells.
31:800–807. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Downs JA and Jackson SP: A means to a DNA
end: The many roles of Ku. Nat Rev Mol Cell Biol. 5:367–378. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lieber MR: The mechanism of double-strand
DNA break repair by the nonhomologous DNA end joining pathway. Annu
Rev Biochem. 79:181–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wood RD, Mitchell M and Lindahl T: Human
DNA repair genes 2005. Mutat Res. 577:275–283. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zou Y, Zhang N, Ellerby LM, Davalos AR,
Zeng X, Campisi J and Desprez DY: Responses of human embryonic stem
cells and their differentiated progeny to ionizing radiation.
Biochem Biophys Res Commun. 426:100–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sokolov M and Neumann R: Effects of low
doses of ionizing radiation exposures on stress-responsive gene
expression in human embryonic stem cells. Int J Mol Sci.
15:588–604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Momcilović O, Choi S, Varum S, Bakkenist
C, Schatten G and Navara C: Ionizing radiation induces ataxia
telangiectasia mutated-dependent checkpoint signaling and G (2) but
not G (1) cell cycle arrest in pluripotent human embryonic stem
cells. Stem Cells. 27:1822–1835. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kunkel TA and Erie DA: DNA mismatch
repair. Annu Rev Biochem. 74:681–710. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tichy ED, Liang L, Deng L, Tischfield J,
Schwemberger S, Babcock G and Stambrook PJ: Mismatch and base
excision repair proficiency in murine embryonic stem cells. DNA
Repair (Amst). 10:445–451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Robertson AB, Klungland A, Rognes T and
Leiros I: DNA repair in mammalian cells: Base excision repair: The
long and short of it. Cell Mol Life Sci. 66:981–993. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shuck SC, Short EA and Turchi JJ:
Eukaryotic nucleotide excision repair: From understanding
mechanisms to influencing biology. Cell Res. 18:64–72. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dexheimer TS: DNA repair pathways and
mechanisms. DNA Repair of Cancer Stem Cells. Mathews LA, Carbarcas
SM and Hurt E: (Dordrecht). Springer International Publishing.
19–32. 2013. View Article : Google Scholar
|
|
62
|
de Waard H, Sonneveld E, de Wit J, van
Esvaldt Lange R, Hoeijmakers JH, Vrieling H and van der Horst GT:
Cell-type-specific consequences of nucleotide excision repair
deficiencies: Embryonic stem cells versus fibroblasts. DNA Repair
(Amst). 7:1659–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ryoo HD and Bergmann A: The role of
apoptosis-induced proliferation for regeneration and cancer. Cold
Spring Harb Perspect Biol. 4:a0087972012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li F, Huang Q, Chen J, Peng Y, Roop DR,
Bedford JS and Li CY: Apoptotic cells activate the ‘phoenix rising’
pathway to promote wound healing and tissue regeneration. Sci
Signal. 3:ra132010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ferdousi Vahidi L, Rocheteau P, Chayot R,
Montagne B, Chaker Z, Flament P, Tajbakhsh S and Ricchetti M: More
efficient repair of DNA double-strand breaks in skeletal muscle
stem cells compared to their committed progeny. Stem Cell Res.
13:492–507. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hong Y and Stambrook PJ: Restoration of an
absent G1 arrest and protection from apoptosis in embryonic stem
cells after ionizing radiation. Proc Natl Acad Sci USA.
101:14443–14448. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Rich T, Allen RL and Wyllie AH: Defying
death after DNA damage. Nature. 407:777–783. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
He YC, Zhou FL, Shen Y, Liao DF and Cao D:
Apoptotic death of cancer stem cells for cancer therapy. Int J Mol
Sci. 15:8335–8351. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fuchs Y and Steller H: Programmed cell
death in animal development and disease. Cell. 147:742–758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Creagh EM and Martin SJ: Caspases:
Cellular demolition experts. Biochem Soc Trans. 29:696–702. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Xu X, Cowley S, Flaim CJ, James W, Seymour
L and Cui Z: The roles of apoptotic pathways in the low recovery
rate after cryopreservation of dissociated human embryonic stem
cells. Biotechnol Prog. 26:827–837. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yang JK: FLIP as an anti-cancer
therapeutic target. Yonsei Med J. 49:19–27. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Würstle ML, Laussmann MA and Rehm M: The
central role of initiator caspase-9 in apoptosis signal
transduction and the regulation of its activation and activity on
the apoptosome. Exp Cell Res. 318:1213–1220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mcllwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 5:a0086562013.PubMed/NCBI
|
|
75
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Roos WP and Kaina B: DNA damage-induced
cell death: From specific DNA lesions to the DNA damage response
and apoptosis. Cancer Lett. 332:237–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Igney FH and Krammer PH: Death and
anti-death: Tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
78
|
Filion TM, Qiao M, Ghule PN, Mandeville M,
van Wijnen AJ, Stein JL, Lian JB, Altieri DC and Stein GS: Survival
responses of human embryonic stem cells to DNA damage. J Cell
Physiol. 220:586–592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J,
Li J, Song Z, Qu X, Zhou P, et al: Regulation of apoptosis and
differentiation by p53 in human embryonic stem cells. J Biol Chem.
282:5842–5852. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Neganova I, Vilella F, Atkinson SP, Lloret
M, Passos JF, von Zglinicki T, O-Connor JE, Burks D, Jones R,
Armstrong L and Lako M: An important role for CDK2 in G1 to S
checkpoint activation and DNA damage response in human embryonic
stem cells. Stem Cells. 29:651–659. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Mandal PK, Blanpain C and Rossi DJ: DNA
damage response in adult stem cells: Pathways and consequences. Nat
Rev Mol Cell Biol. 12:198–202. 2011. View Article : Google Scholar : PubMed/NCBI
|