Introduction
Anaplastic lymphoma kinase-positive large B-cell
lymphoma (ALK-positive LBCL) is a rare but distinct lymphoma with a
consistent absence of CD20 expression and acquisition of plasma
cell markers, including CD38 and CD138. The World Health
Organization (WHO) classification of tumors of hematopoietic and
lymphoid tissues defined ALK-LBCL as a separate entity to
non-Hodgkin's lymphoma, which consists of ALK-positive monomorphic
large immunoblast-like B-cells, sometimes with plasmablastic
differentiation (1). ALK-positive
LBCL appears to be a distinct disease entity, with an aggressive
development cycle that is associated with a poor prognosis; the
overall median survival of patients with advanced stage III/IV
ALK-positive LBCL was 11 months. Previous studies have suggested
that this distinct subtype of diffuse large B-cell lymphoma (DLBCL)
should belong to the group of pediatric lymphomas (2,3);
however, we report an example of ALK-positive LBCL in a 90-year-old
male patient, with rapid clinical course. To the best of our
knowledge, this is the eldest patient with this type of lymphoma to
be reported in the literature.
Case report
A 90-year-old Chinese male patient presented a
1-week history of right neck pain and enlargement of the cervical
lymph nodes at the Third Affiliated Hospital of Guangzhou Medical
University (Guangzhou, China). Physical examination revealed that
his general condition was stable with a normal body temperature and
typical laboratory results, including blood count, and
differential, renal and liver functions. Numerous enlarged right
cervical lymph nodes measuring 1.0–4.0 cm in diameter were detected
via palpation of the superficial lymph nodes. A computed tomography
scan of the chest and abdomen detected several enlarged lymph
nodes, which were located in the mediastinum and right
supraclavicular fossa. The patient was considered to have stage III
non-Hodgkin's lymphoma and one of the right cervical lymph nodes
was surgically removed for histopathological examination. The
enlarged lymph node was grey-reddish in color and
well-circumscribed to the surrounding tissues. Written informed
consent was obtained from the patient prior to the publication of
this case report and the accompanying images.
Surgically removed tissue specimens were formalin
fixed and paraffin embedded for subsequent analyses. Hematoxylin
and eosin-stained sections were prepared according to the standard
methods. For cytogenetic analysis, DNA was extracted from the
paraffinized tissue samples using a DNeasy Blood & Tissue Kit
(Qiagen, Inc., Valencia, CA, USA). T-cell receptor and
immunoglobulin gene rearrangement studies were performed. Two sets
of primers (tube A, 145–255 bp; tube B, 80–220 bp) were used to
amplify the rearranged T-cell receptor-γ gene. A T-cell lymphoma
case with a known monoclonal rearrangement was used as the positive
control, a non-lymphoid and hematopoietic tumor was used as the
negative control, and a reaction without template DNA was
simultaneously run as the blank control. β-actin was amplified as
an internal control. FRIII-J segmentation were conducted for IgH
gene rearrangements. RAJI cells (Sigma-Aldrich, St. Louis, MO, USA)
were used as positive controls and a previous negative sample was
used as a negative control.
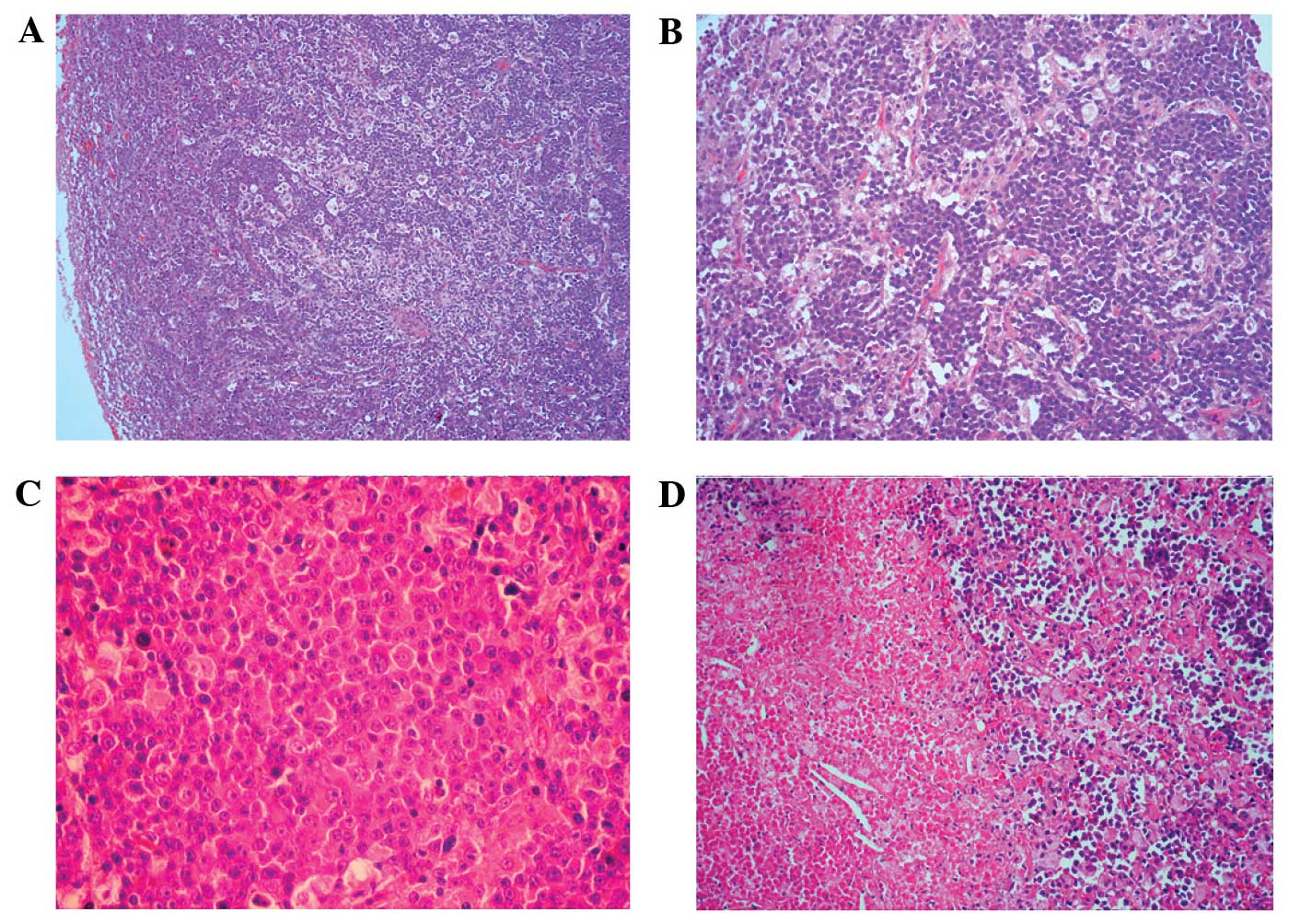
Histologically, the removed specimen resembled a
cervical lymph node with effacement of the normal architecture
(Fig. 1A), and a diffuse
infiltration of large tumor cells with a sinusoidal growth pattern
(Fig. 1B). The tumor cells were
large with round nuclei, dispersed chromatin, a prominent nucleolus
and a moderate eosinophilic or amphophilic cytoplasm. In addition,
some of the large tumor cells were immunoblastic-like in
appearance, with a single central nucleolus and an abundant
cytoplasm, whereas others had a plasmablastic-like appearance, with
eccentrically located nuclei. Furthermore, atypical multinucleated
neoplastic giant cells, and focal necrotic areas, were observed in
the tissue (Fig. 1C and D).
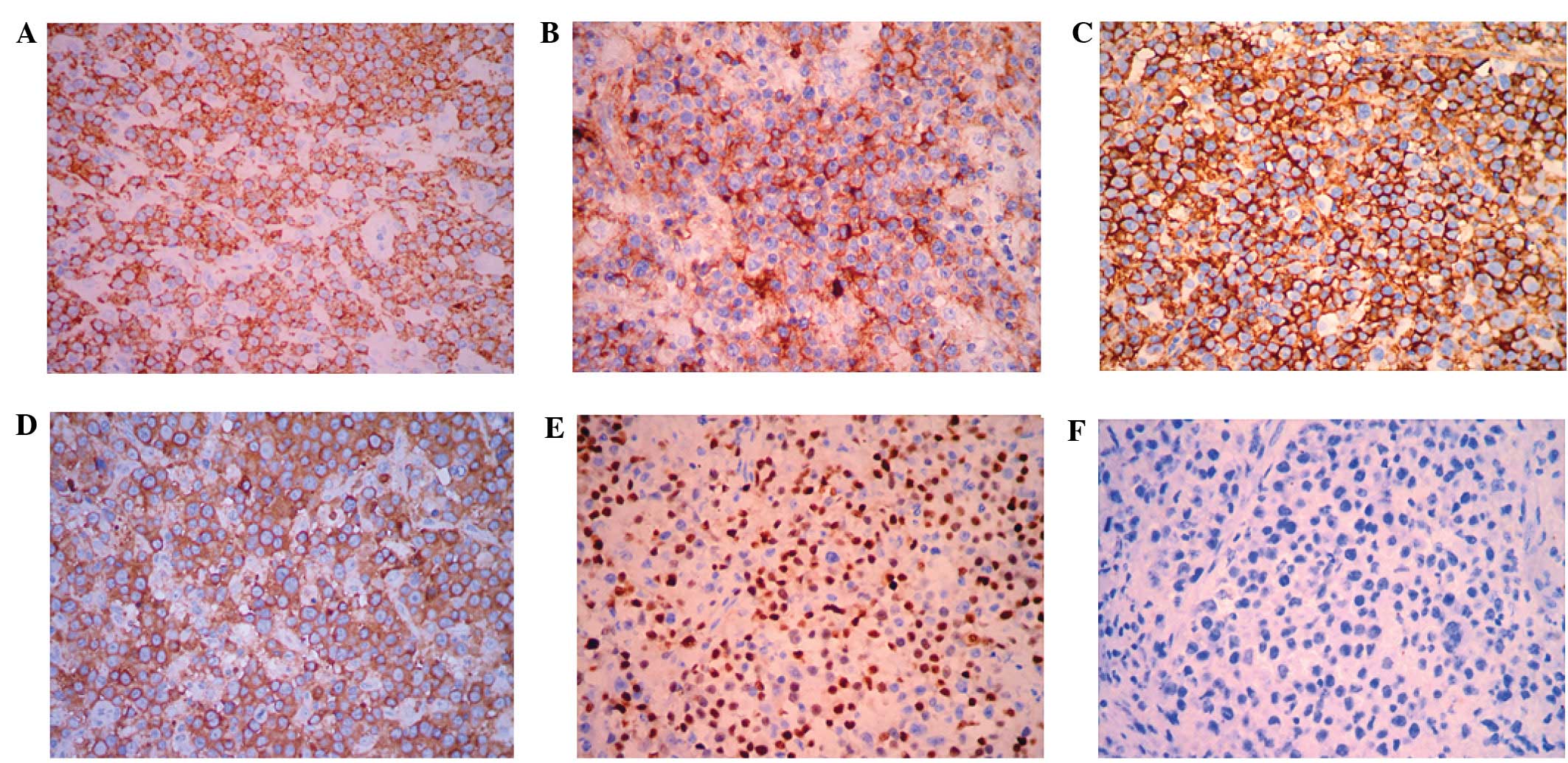
Immunohistochemical analyses demonstrated that the large tumor
cells were strongly positive for ALK in a granular cytoplasmic
distribution, which has previously been described in cases of
clathrin/ALK-fusion (Fig. 2A). In
addition, the tumor cells demonstrated high levels of expression of
CD45, CD138, epithelial membrane antigen (EMA), MUM-1 and lambda
(Fig. 2B–E). Conversely, the tumor
cells were negative for the expression of the B-cell markers CD20
(Fig. 2F) and CD79a. Furthermore,
assays for CD2, CD3, CD7, CD8, CD30, CD38, CD43, CD56, CD68, kappa,
granzyme B, terminal deoxynucleotidyl transferase, Pan-cytokeratin,
S-100, HMB-45 and Melan-A were all negative. The
immunohistochemical analyses were performed using a Dako
Autostainer with Envision (+) Detection Kit (Dako, Glostrup,
Denmark). Polymerase chain reaction assays were unable to detect
clonal rearrangements in the T cell receptor-γ and immunoglobulin
heavy chain genes (IgH) in the tumor lesion. Furthermore, in
situ hybridization was unable to detect an EBV infection in the
lesion. For detection of EBV infection in the tissues, in
situ hybridization for EBV-encoded RNAs (EBERs) was performed
on biopsy samples using an EBER detection kit purchased from Dako,
according to the manufacturer's instructions.
On the basis of the present histopathological and
molecular results, the patient was diagnosed with ALK-positive LBCL
with plasmablastic differentiation, according to the WHO
classification criteria (1).
Subsequently, the patient received treatment with the following
21-day chemotherapy regimen: Cyclophosphamide, 750 mg/m2
d1; doxorubicin, 500 mg/m2 d1; vincristine, 1.4
mg/m2 d1; prednisone, 60 mg/m2 d1-d5; and
etoposide, 100 mg/day d3-d5. However, clinical deterioration was
associated with rapid enlargement of the retroperitoneal lymph node
(diameter, 10 cm). During the initial cycle of chemotherapy, the
patient developed pleural effusions with respiratory distress. A
bone marrow examination was performed following the initial cycle
of chemotherapy and no abnormality was detected. However, the
clinical status of the patient continued to deteriorate rapidly,
and he succumbed to the disease within 4 months of initial
presentation.
Discussion
ALK-positive LBCL is a rare subtype of DLBCL, which
was recognized by the WHO classification of tumors of hematopoietic
and lymphoid tissue in 2001 (4). The
diagnosis of ALK-LBCL is typically based on the detection of
aberrant ALK protein expression levels in the tumor, which may also
exhibit an immunoblastic or plasmablastic morphology, and a lack of
CD20, CD79a and PAX5 expression. Since its initial recognition, 50
well-characterized cases of ALK-positive LBCL have been reported in
the literature, including a total of twelve pediatric cases
(5,6–10). In
adult patients, the majority of ALK-positive LBCL tumors occur in
males (male/female ratio, 4.3:1), whereas it affects the genders
equally in pediatric patients (male/female ratio, 1.4:1) (8). In addition, no significant difference
between the mortality rate of male and female patients with
ALK-LBCL has been reported (9).
Plasmacytic differentiation markers, including CD38, CD138 and
VS38c, have been detected in all previously reported cases of
ALK-LBCL, and EMA expression has been detected in the majority of
the cases. The patient in the present case tested negative for
clonal rearrangement in the IgH gene, and this is consistent with
the finding that some B-cell lymphomas do not demonstrate clonal
IgH amplification, due to of a lack of consensus target sequences
or target site alterations as a result of somatic hypermutation
(11). This result suggests that
cytogenetic analysis of immunoglobulin genes may not be reliable in
the diagnosis of ALK-positive LBCL. Furthermore, consistent with
previous studies (1), EBV infection
was absent in the present case.
The key histological differential diagnoses include
ALK-positive anaplastic large cell lymphoma (ALCL), anaplastic
variants of DLBCL, plasmablastic lymphoma, and primary effusion
lymphoma, all of which exhibit an ALK-positive immunophenotypic
profile or plasmacytic differentiation with CD20-negativity, which
may confound the diagnosis of ALK-positive DLBCL. ALK-positive ALCL
is typically associated with large tumor cells that exhibit an
abundant cytoplasm and pleomorphic nuclei, and elevated expression
levels of CD30 on the cell membrane and in the Golgi region
(11). Conversely, CD30 expression
is typically negative in ALK-positive LBCL, although focal and weak
staining has been reported in a few cases of ALK-positive LBCL
(11). The expression of ALK and
other plasmacytic markers, including CD138, VS38c and MUM-1, and
the lack of viral infection, including infections caused by the
human immunodeficiency virus, EBV, and human herpesvirus 8, should
aid in the diagnosis of ALK-positive LBCL.
ALK-positive LBCL is generally considered a distinct
disease entity with an aggressive progression that is associated
with a poor prognosis; the overall median survival of patients with
advanced stage III/IV ALK-positive LBCL was 11 months in a previous
study (9). However, previous studies
have reported a prolonged survival rate (>10 years) for patients
with advanced stage ALK-positive LBCL (5,7). Of the
50 cases reported in the literature, the clinical stage at disease
presentation was the factor most significantly associated with the
survival rate of patients with ALK-positive LBCL. Notably, there
was no significant difference between the survival rates of
pediatric and adult patients, despite reports of more intensive
therapies being used for pediatric cases (9). To date, no clinical trials
investigating an optimal treatment regimen for ALK-positive LBCL
have been reported. As the ALK-positive LBCL tumor cells are
typically negative for the CD20 antigen, this tumor is thus
insensitive to treatment with rituximab. A previous case reported
an example where ALK-positive LBCL was treated with rituximab
therapy; however, the patient involved succumbed to the disease
within 6 months of diagnosis (9). In
pediatric patients with ALK-positive LBCL tumors, the majority of
regimens used, including the lymphoma malignant B-89, pediatric
oncology group 8719, and CHOP protocols, are highly intensive, and
only four patients (33.3%, 4/12) succumbed to relapse or
progressive disease at the time of the reports (3,6,7). In the present case, an elderly male
patient was diagnosed with stage III ALK-positive LBCL and
exhibited rapid clinical decline despite having been treated with
the E-CHOP regimen of chemotherapy. The patient eventually
succumbed to multiple system organ failure within 4 months of the
initial diagnosis, possibly due to chemotherapy intolerance.
In conclusion, we present a rare case of
ALK-positive LBCL occurring in an elderly male patient, with rapid
tumor progression. To the best of our knowledge, this case is the
eldest patient with ALK-positive LBCL to be described in the
literature. Previous studies have suggested that some patients with
advanced stage ALK-positive LBCL may have prolonged survival rates;
however, it is typically associated with high aggressiveness and a
poor prognosis, particularly in elderly patients. Further research
is required in order to develop novel therapeutic strategies for
treating and improving the prognosis of patients with ALK-positive
LBCL.
References
|
1
|
Swerdlow SH, Campo E and Harris NL:
Chapter 6. WHO classification of Tumours of Haematopoietic and
Lymphoid Tissues (4th). IARC Press. (Lyon, France). 254–255.
2008.
|
|
2
|
Gesk S, Gascoyne RD, Schnitzer B, Bakshi
N, Janssen D, Klapper W, Martín-Subero JI, Parwaresch R and Siebert
R: ALK-positive diffuse large B-cell lymphoma with ALK-Clathrin
fusion belongs to the spectrum of pediatric lymphomas. Leukemia.
19:1839–1840. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yin WH, Guo N, Tian XY, Li Y and Li Z:
Pediatric anaplastic lymphoma kinase-positive large B-cell
lymphoma: A case report and review of the literature. Pediatr Dev
Pathol. 15:318–323. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jaffe ES, Harris NL and Stein H: Chapter
6. World Health Organization classification of tumours: tumours of
haematopoietic and lymphoid tissues (3rd). IARC Press. (Lyon,
France). 214–215. 2001.
|
|
5
|
Delsol G, Lamant L, Mariamé B, Pulford K,
Dastugue N, Brousset P, Rigal-Huguet F, al Saati T, Cerretti DP,
Morris SW and Mason DY: A new subtype of large B-cell lymphoma
expressing the ALK kinase and lacking the 2; 5 translocation.
Blood. 89:1483–1490. 1997.PubMed/NCBI
|
|
6
|
Bubała H, Małdyk J, Włodarska I,
Sońta-Jakimczyk D and Szczepański T: ALK-positive diffuse large
B-cell lymphoma. Pediatr Blood Cancer. 46:649–653. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Isimbaldi G, Bandiera L, d'Amore ES,
Conter V, Milani M, Mussolin L and Rosolen A: ALK-positive
plasmablastic B-cell lymphoma with the clathrin-ALK gene
rearrangement. Pediatr Blood Cancer. 46:390–391. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beltran B, Castillo J, Salas R, Quiñones
P, Morales D, Hurtado F, Riva L and Winer E: ALK-positive diffuse
large B-cell lymphoma: Report of four cases and review of the
literature. J Hematol Oncol. 2:112009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gascoyne RD, Lamant L, Martin-Subero JI,
Lestou VS, Harris NL, Müller-Hermelink HK, Seymour JF, Campbell LJ,
Horsman DE, Auvigne I, et al: ALK-positive diffuse large B-cell
lymphoma is associated with Clathrin-ALK rearrangements: Report of
6 cases. Blood. 102:2568–2573. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Adam P, Katzenberger T, Seeberger H,
Gattenlöhner S, Wolf J, Steinlein C, Schmid M, Müller-Hermelink HK
and Ott G: A case of a diffuse large B-cell lymphoma of
plasmablastic type associated with the t(2;5)(p23;q35) chromosome
translocation. Am J Surg Pathol. 27:1473–1476. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stachurski D, Miron PM, Al-Homsi S,
Hutchinson L, Harris NL, Woda B and Wang SA: Anaplastic lymphoma
kinase-positive diffuse large B-cell lymphoma with a complex
karyotype and cryptic 3′ ALK gene insertion to chromosome 4 q22-24.
Hum Pathol. 38:940–945. 2007. View Article : Google Scholar : PubMed/NCBI
|