Introduction
Acute myocardial infarction (AMI) is a disease that
severely affects the health and life quality of patients.
Arrhythmia is a complication of myocardial infarction, which is one
of the most severe cardiovascular diseases, and it is the
predominant cause of myocardial infarction-associated mortality
(1). Since patients with ischemic
heart disease are particularly prone to arrhythmias, they are often
admitted for arrhythmia monitoring (2). Lysophosphatidic acid (LPA), which is an
intermediate product of membrane phospholipid metabolism, is a
lipid mediator with various biological functions that are
predominantly mediated by specific G protein-coupled receptors
(3). As a water-soluble glycerol
phospholipid with a simple structure, LPA is secreted from numerous
cell types, including platelets, fibroblasts and ovarian cancer
cells (4). The concentration of LPA
in regional myocardial tissue and plasma increases during
myocardial infarction, and it can also cause arrhythmia (5).
During myocardial infarction, both the infarcted
area and the non-infarcted area exhibit perivasculitis, and the
infiltration of a large number of inflammatory cells can be
observed (6,7). A variety of inflammatory factors,
including interferon-γ, interleukin (IL)-4, IL-5, IL-6, IL-8 and
IL-10, increase in the peripheral blood, indicating that myocardial
infarction is a non-infectious immune inflammatory process
(8,9). The immune inflammatory response has an
important role in myocardial infarction (10,11).
Numerous studies have focused on the effect of LPA on myocardial
ischemia, but there are no reports on the immune regulatory effect
of LPA on connexin 43 (Cx43) protein expression and arrhythmias
(1,4,12). In
the present study, various experimental methods were used to
observe the effects of LPA on tumor necrosis factor (TNF)-α, Cx43
and potassium channels. The mechanism underlying the LPA-induced
induction of arrhythmia was also investigated.
Materials and methods
Experimental animals
A total of 80 healthy adult male and female Wistar
rats (clean grade), aged 8–10 week and weighing 240–260 g, were
purchased from the Experimental Animal Center of Jilin University
(Changchun, China). Rats were maintained in an animal care facility
at 21–23°C, relative humidity (55±5%) with a 12-h light/dark cycle
and ad libitum access to food and chow for at least three
days prior to the initiation of the experiment. The procedures used
in the present study were approved by the Animal Ethics Committee
of The First Hospital of Jilin University, in accordance with the
Guide for the Care and Use of Laboratory Animals issued by the US
National Institutes of Health (13).
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA)
unless otherwise stated.
Surface electrocardiogram (ECG)
A total of 40 rats were randomly and equally divided
into 4 groups: A sham-operated group, an AMI model group, an
immune-enhanced group (treated with 1.25 mg/kg thymopeptide
(H20003884; Livzon Pharmaceutical Group, Inc., Shanxi, China) by
intraperitoneal injection) and an immune-suppressed group (treated
with 15 mg/kg cyclophosphamide (H32020857; Jiangsu Hengrui Medicine
Co., Ltd., Lianyungang, China) by intraperitoneal injection). For
the 8 days leading up to the experimental procedures, the rats were
maintained and fed, as outlined. The number of leukocytes was
detected in the peripheral blood. Briefly, the rat tail was soaked
at 45–50°C for ~3 min to dilate the blood vessels and a 5 mm
incision was made to harvest 1 ml blood. Full blood count analysis
was performed within 2 h of collection using a Sysmex XE 2100
hematology analyzer (Sysmex UK, Milton Keynes, UK). If the number
of leukocytes in the immune-enhanced group was
≥1.2×109/l and the number of leukocytes in the
immune-suppressed group was ≤0.6×109/l, then immune
intervention was considered successful.
Rats were anesthetized with 20 g urethane dissolved
in 100 ml saline solution (0.9%) (1 g/kg; 5 µl/g body weight;
U2500) by intraperitoneal injection, and a thoracotomy was
performed in the left 4–5 intercostal space. A total of 10 µl LPA
(0.5 g/l dissolved in 0.9% NaCl saline; L7260) was intravenously
injected into the hearts of the AMI group, the immune-enhanced
group and the immune-suppressed group for <5 sec. The
thoracotomy was completed in 30 sec. The left anterior descending
artery of coronar was ligated. The heart was placed back in the
chest prior to being closed with sutures. Rats in the 4 groups were
analyzed by surface lead II ECG (DECG-03A; Mindray Medical
International, Ltd., Nanshan, China). Successful construction of
the acute myocardial infarction model was observed as ST-segment
elevation and the formation of a flag-shaped waveform of the T
wave. ECG recordings were obtained and analyzed using a BL-420S
Biological Function Recording system (Taimeng Technology Co., Ltd.,
Chengdu, China).
Isolated rat heart perfusion
A total of 40 rats were randomly and equally divided
into 4 groups: A sham-operated group, an LPA group, an
immune-enhanced + LPA group and an immune-suppressed + LPA group.
Hearts were isolated and rapidly perfused with improved
Krebs-Henseleit (K-H) solution in a Langendorff Perfusion system
(Radnoti LLC, Monrovia, CA, USA). The perfusion pressure was 10.1
kPa, and the perfusion temperature was 37°C. The improved K-H
solution was saturated with 95% O2 and 5%
CO2, and the composition of the solution was as follows:
118.0 mmol/l NaCl, 4.7 mmol/l KC1, 1.2 mmol/l
KH2PO4, 1.2 mmol/l MgSO4, 25.0
mmol/l NaHCO3, 1.3 mmol/l CaCl2, and 1.1
mmol/l glucose, with pH titrated to 7.2–7.4. After 10 min the
isolated heart was functioning in a stable manner. LPA (5 µmol/l)
was added to the K-H solution in the hearts of the LPA model group,
the immune-enhanced + LPA group and the immune-suppressed + LPA
group. Surface electrocardiograms were analyzed by surface lead II
ECG (the needle electrode was inserted subcutaneously into the
pulmonary cone and the cardiac apex). ECG recordings were obtained
and analyzed using the BL-420S Biological Function Recording
system. Subsequently, the number of premature ventricular
contractions were observed.
ELISA
Jurkat T cells were purchased from the National
Platform of Experimental Cell Resources (Sci-Tech, Beijing, China)
and plated in a 96-well plate (2×106 cells/ml) in 200 µl
RPMI-1640 medium (R8755) supplemented with 10% fetal bovine serum
(F2442) and 100 mg/ml penicillin/streptomycin (V900929), and
incubated at 37°C in atmosphere containing 5% CO2 and
95% humidity. Jurkat T cells were divided into 3 groups: A control
group, an LPA group (5 µmol/l), and a Ki16425 + LPA group (10
µmol/l and 5 µmol/l, respectively). Cell suspensions were obtained
from each group following cell incubation for 1, 2, 4, 8, 12 and 24
h. The cell suspensions were centrifuged at 560 × g for 10 min at
22–23°C, and the supernatants were collected. The concentration of
TNF-α was determined using an ELISA kit (ERT2010-1; Assaypro LLC,
St. Charles, MO, USA).
Ionic current recordings
Jurkat T cells (2×104 cells/ml) were
placed in a bath solution. Patch electrode pipettes were fabricated
using a vertical pipette puller (Narishige PP-83; Narishige
Scientific Instrument Lab., Tokyo, Japan). Pipettes with tip
diameters of 1–2 µm had resistances of 3–6 MΩ when filled with the
pipette solution. The pipette was placed on the cell surface using
a microelectrode propeller (Narishige Scientific Instrument Lab.),
and a high impedance seal (GΩ) was formed by vacuum suction.
Subsequently, the negative pressure was increased in order to cause
the clamp of the electrode tip to rupture, and the whole-cell patch
clamp mode was formed. All experiments were performed at 20–22°C.
The voltage-clamp circuit was provided by a patch/whole-cell clamp
amplifier (Dagan Total Clamp 8800; Dagan Corporation, Minneapolis,
MN, USA). Pulse protocols and data acquisition were performed using
a digital interface (TL-1 DMA interface; Axon Instruments, Foster
City, CA, USA) coupled to an IBM compatible computer with PCLAMP
6.0 software (Molecular Devices LLC).
Ionic currents of Kv were recorded under
various voltage-clamp protocols of the step pulse: Holding
potential −80 mV, test potentials −50 to +50 mV, step 10 mv,
duration 300 ms, and frequency 1 Hz. The bath solution of
Kv contained the following components: 140 mmol/l NaCl,
4 mmol/l KCl, 1 mmol/l MgC12, 1 mmol/l CaC12,
10 mmol/l glucose and 10 mmol/l HEPES, pH 7.2 (filtered). The
pipette solution of Kv contained the following
components: 90 mmol/l KCl, 50 mmol/l KF, 4 mmol/l NaCl, 4
MgCl2, 0.5 mmol/l ethylene glycol tetraacetic acid
(EGTA) and 10 mmol/l HEPES, pH 7.2 (filtered).
The bath solution of KCa contained the
following components: 160 mmol/l NaCl, 4.5 mmol/l KC1, 2 mmol/l
CaC12, 1 mmol/l MgC12, 5 mmol/l HEPES, pH 7.4
(filtered). The pipette solution of KCa contained the
following components: 150 mmol/l K-asparatic acid, 2 mmol/l
MgCl2, 5 mmol/l HEPES, 10 mmol/l EGTA, 8.7 mmol/l
CaCl2, with a 1 µmol/l Ca2+ concentration and
pH 7.2 (filtered).
Immunohistochemical staining
Following the isolation of the hearts of the rats in
the AMI, immune-enhanced and immune-suppressed groups were isolated
(atria and blood vessels were discarded). The ventricles were fixed
in 4% paraformaldehyde solution (252549) for 16–18 h. Fixed
myocardial tissue samples were subsequently dehydrated in a graded
ethanol series, cleared with xylene and embedded with paraffin
(10152636; Guangjing Weiye Import & Export Co., Ltd., Tianjin,
China). Tissue samples were cut into 3-µm sections using a sliding
microtome (VT-1000S; Leica Microsystems GmbH, Wetzlar, Germany).
One section was used for hematoxylin (H9627) and eosin (E4009)
staining in order to observe the morphology of the myocardium, and
the remaining two sections were used for immunohistochemical SP
staining (SP-9000; Zhongshan Golden Bridge Biotechnology Co., Ltd.,
Beijing, China). Cx43 was observed in the tissue sections using an
Olympus CKX41-A32PH inverted microscope (Olympus Corporation,
Tokyo, Japan).
Statistical analysis
All data were analyzed using SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA) and are presented as the means ±
standard error of the mean (n=4). The results obtained were
compared using a t-test. Plots were generated using GraphPad Prism
6.01 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of LPA on the incidence of
arrhythmia
To determine the effects of LPA on the incidence of
arrhythmia in rats that had altered immune status, ECGs of
myocardial infarction rats and isolated rat hearts were obtained;
the results demonstrated the presence of arrhythmias and
ventricular premature beats (VPBs). Endogenous LPA enhanced the
incidence of VPBs in rats of the immune-enhanced group and reduced
the incidence of VPBs in rats of the immune-suppressed group
(Table I).
 | Table I.Effects of lysophosphatidic acid on
the incidence of arrhythmia in myocardial infarction rats. |
Table I.
Effects of lysophosphatidic acid on
the incidence of arrhythmia in myocardial infarction rats.
|
|
| Prior to
ligation | Following
ligation |
|---|
|
|
|
|
|
|---|
| Group | n | HR (beats/min) | Incidence of VPBs
(%) | Initial VPB time
(min) | HR (beats/min) | VPBs per hour |
|---|
| A | 0 | 322±21 | 0 | 0 | 328±15a | 0a |
| B | 7 | 324±15 | 70 | 30.5±6.3 | 422±46 | 10.1±2.4 |
| C | 8 | 321±17 | 80 |
24.2±5.8b | 463±55b |
13.5±3.9b |
| D | 5 | 324±31 |
50a |
41.2±8.9b | 384±57b |
7.2±2.9b |
ECGs of isolated rat hearts demonstrated that,
following the addition of 5 µmol/l LPA to the K-H solution of the
LPA model, immune-enhanced + LPA and immune-suppressed + LPA
groups, the incidence of VPBs in the immune-enhanced + LPA group
(90%) increased compared with that of the LPA model group (80%).
The occurence of VPBs (7.4±3.7 times/5 min) also increased in the
immune-enhanced + LPA group, as compared with the LPA model group
(5.1±3.9 times/5 min). The incidence of VPBs in the
immune-suppressed + LPA group (50%) significantly decreased
compared with the LPA model group (80%; P<0.05), and occurrences
(3.5±3.8 times/5 min) significantly increased compared with the LPA
model group (5.2±3.9 times/5 min; P<0.05; Table II).
| Table II.Effects of LPA on the incidence of
arrhythmia in isolated rat hearts. |
Table II.
Effects of LPA on the incidence of
arrhythmia in isolated rat hearts.
| Group | n | Incidence of VPBs
(%) | VPBs per 5 min |
|---|
| A | 10 | 0 | 0 |
| B | 10 | 80 |
5.2±3.9a |
| C | 10 | 90 |
7.4±3.7a |
| D | 10 | 50 |
3.5±3.8b |
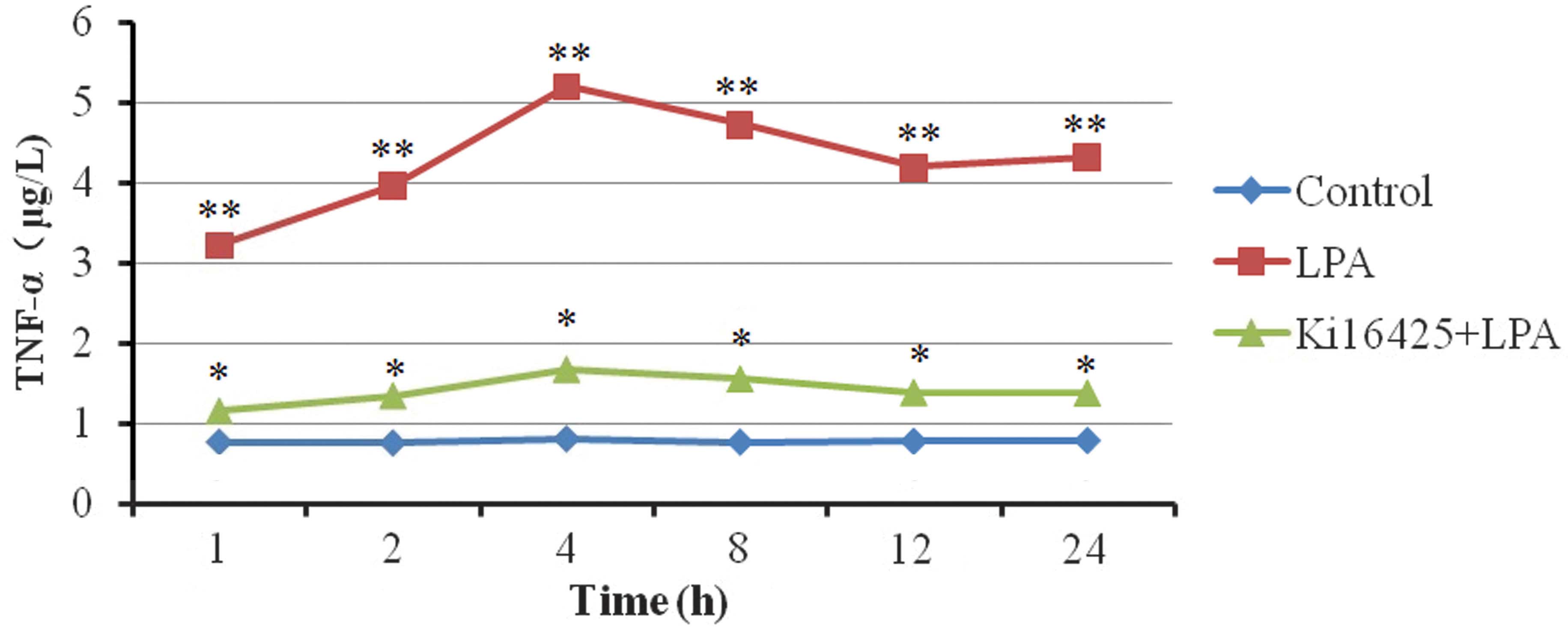
Effects of LPA on TNF-α secretion
The levels of TNF-α secreted by T lymphocytes
significantly increased in the LPA group compared with the control
group (P<0.01). Ki16425, which is a specific inhibitor of LPA,
was able suppress the secretion of TNF-α (Table III). The concentration of TNF-α
increased markedly within a short period of time following the
addition of LPA to the T lymphocytes. The TNF-α concentration
reached its maximum at 4 h and stabilized within 24 h (Table III; Fig.
1).
| Table III.Effects of LPA on tumor necrosis
factor-α secretion in Jurkat T lymphocytes (µg/l). |
Table III.
Effects of LPA on tumor necrosis
factor-α secretion in Jurkat T lymphocytes (µg/l).
| Group | 1 h | 2 h | 4 h | 8 h | 12 h | 24 h |
|---|
| Control |
0.77±0.04a |
0.76±0.05a |
0.81±0.04a |
0.77±0.08a |
0.78±0.04a |
0.79±0.03a |
| LPA | 3.22±0.18 | 3.96±0.19 | 5.20±0.29 | 4.73±0.27 | 4.19±0.30 | 4.32±0.18 |
| Ki16425 + LPA |
1.15±0.18a |
1.34±0.18a |
1.67±0.17a |
1.55±0.22a |
1.38±0.19a |
1.37±0.08a |
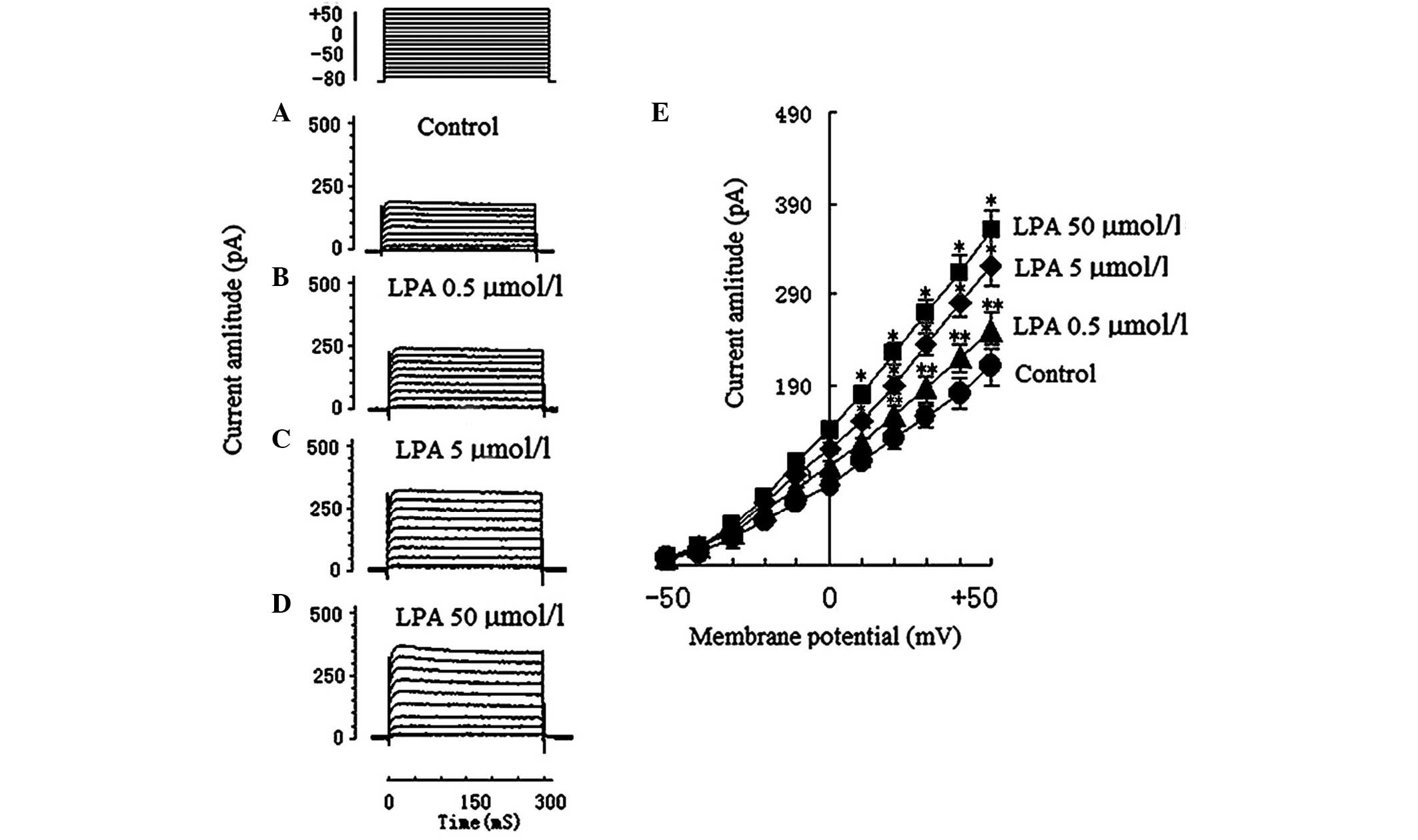
Effects of LPA on voltage-dependent
potassium (Kv) currents
A total of 28 voltage-dependent K+
currents of Jurkat T cells were obtained. To exclude the natural
attenuation of Kv, Kv was continuously
observed for 30 min, which determined that no other current was
significantly attenuated. A total of 7 Jurkat T cells were observed
in the control group (205.5±43.4 pA; Fig. 2A), 7 Jurkat T cells were observed in
the 0.5 µmol/l LPA group (246.65±30.9 pA; Fig. 2B), 7 Jurkat T cells were observed in
the 5 µmol/l LPA group (317.5±32.1 pA; Fig. 2C), and 7 Jurkat T cells were observed
in the 50 µmol/l LPA group (361.8±46.7 pA; Fig. 2D).
The Kv current amplitude in the 5 µmol/l
LPA group (317.5±32.1 pA) and the 50 µmol/l LPA group (361.8±46.7
pA) significantly increased compared with the control group
(205.5±43.4 pA; P<0.01). The Kv current amplitude in
the 0.5 µmol/l LPA group (246.65±30.9 pA) was significantly
increased compared with the control group (P<0.05). The effect
of LPA on Kv currents and the I–V curve of Kv
currents are shown in Fig. 2E. It
was deduced that LPA had a dose-dependent effect on the
Kv current. The electrophysiological characteristics
(current amplitude and activation, and inactivation voltage range)
of Kv in the present study were concordant with those of
previous reports (14,15).
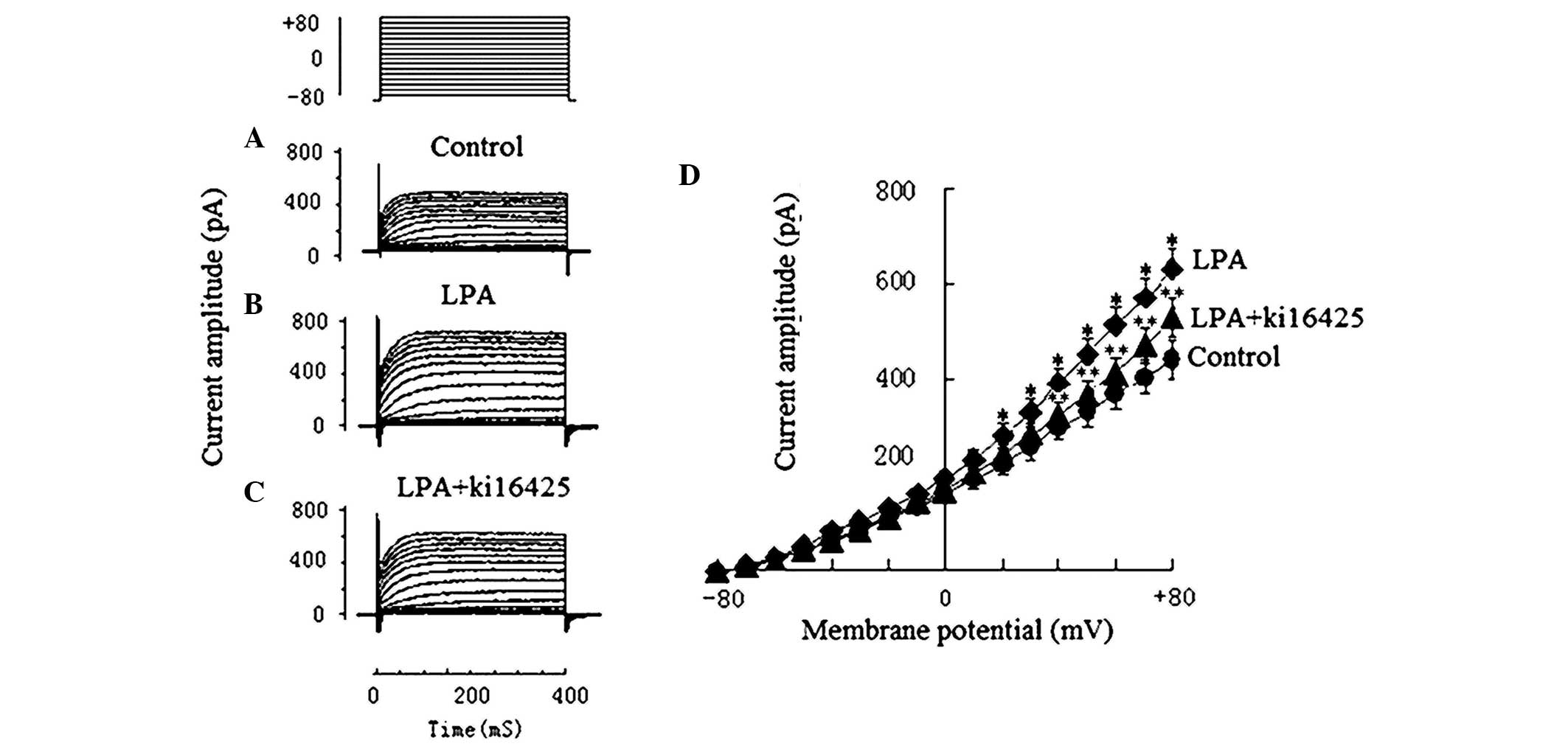
Effects of LPA on
Ca2+-activated potassium (Kca) currents
The KCa of Jurkat T cells in the 5 µmol/l
LPA group and the 10 µmol/l Ki16425 + 5 µmol/l LPA group were
obtained to observe the effects of LPA on KCa and to
examine the inhibitory effect of Ki16425. Ionic currents of
KCa were recorded under the various voltage-clamp
protocols of the step pulse: Holding potential −80 mV, test
potentials −80 to +80 mV, step 10 mv, and duration 400 ms. Compared
with the current amplitude prior to treatment with LPA (439.6±43.7
pA; Fig. 3A), the KCa
current amplitude in the 5 µmol/l LPA group (628.5±46.1 pA;
Fig. 3B) increased significantly
(P<0.01). The KCa current amplitude in the 10 µmol/l
Ki16425 + 5 µmol/l LPA group decreased to 507.5±71.4 pA (Fig. 3C), which suggests that Ki16425 was
able to inhibit the effect of LPA (Fig.
3).
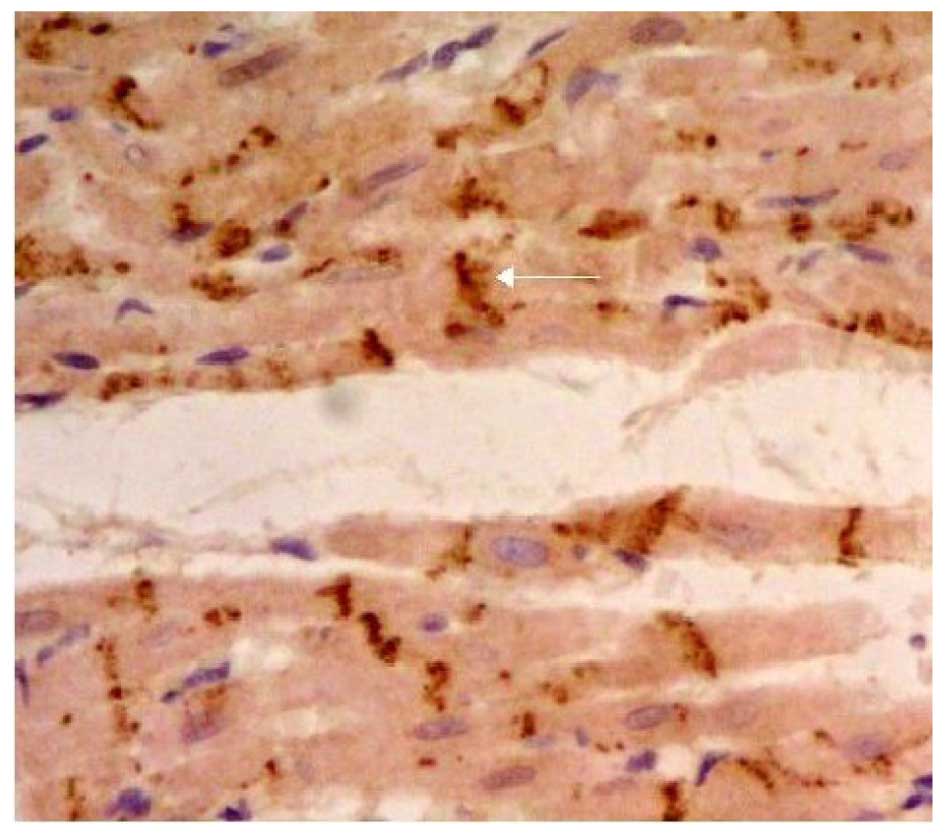
Effects of LPA on Cx43 protein
expression
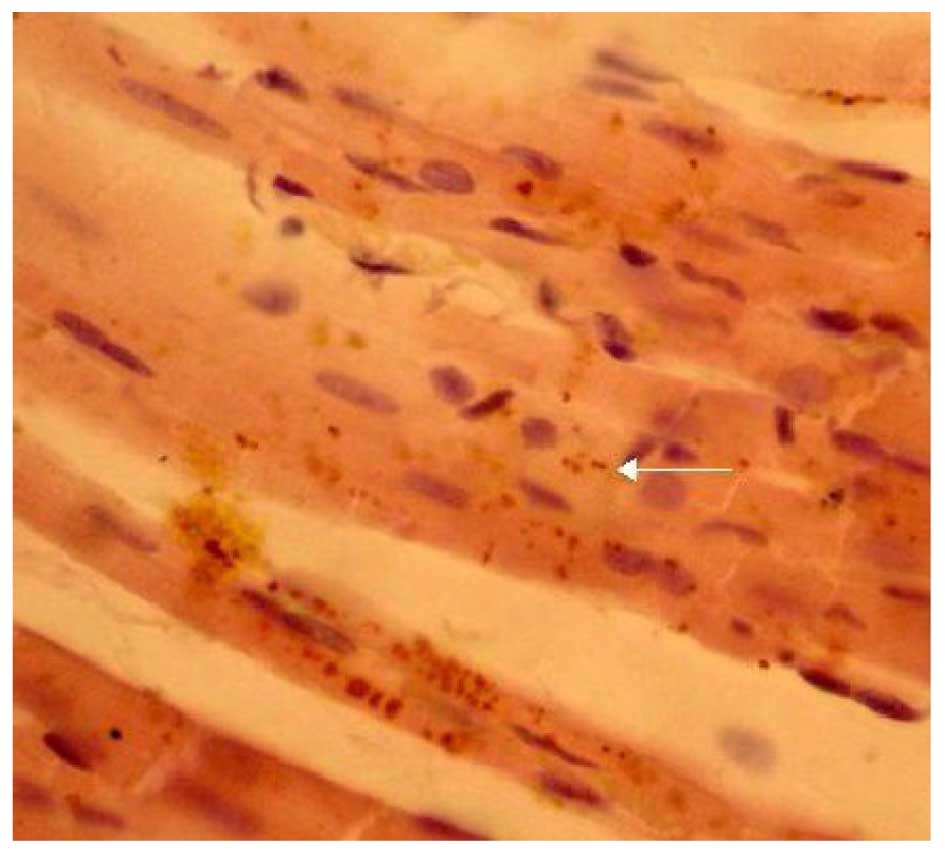
The expression of Cx43 was determined by
immunohistochemical SP staining. Cx43 was abundantly expressed in
the control group and displayed marked positive staining. Cx43 was
uniformly distributed in the intercalated disk between adjacent
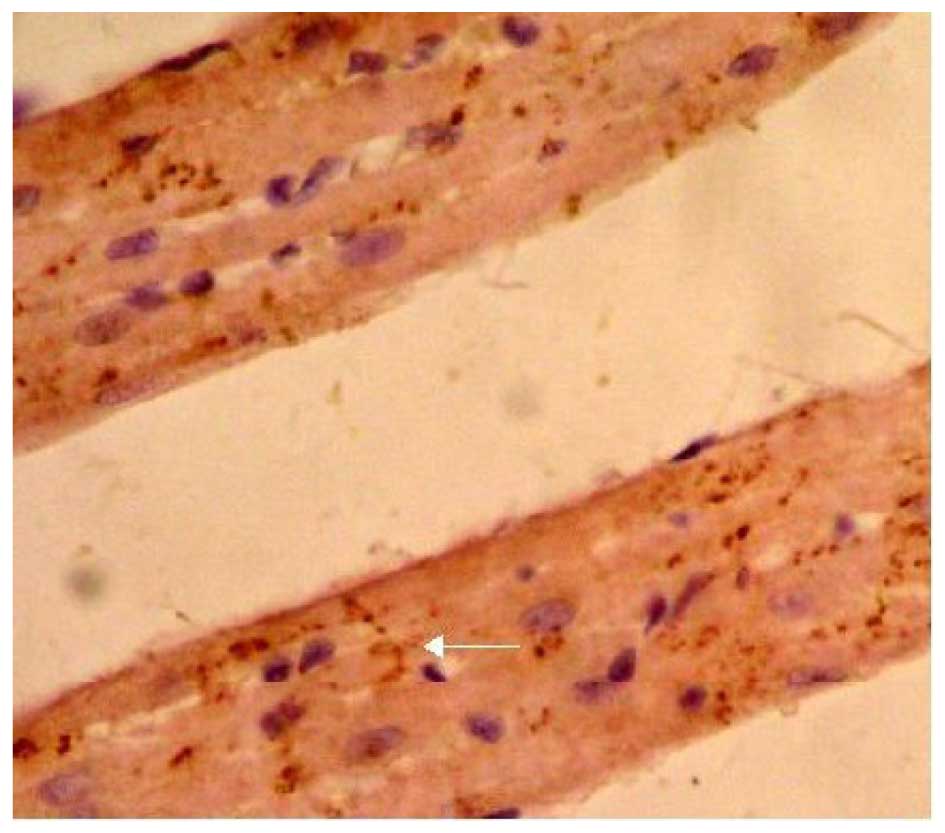
cells and had a clustered distribution (Fig. 4). Cx43 in the LPA + AMI model group
was clearly decreased and had a disordered, uneven and punctate
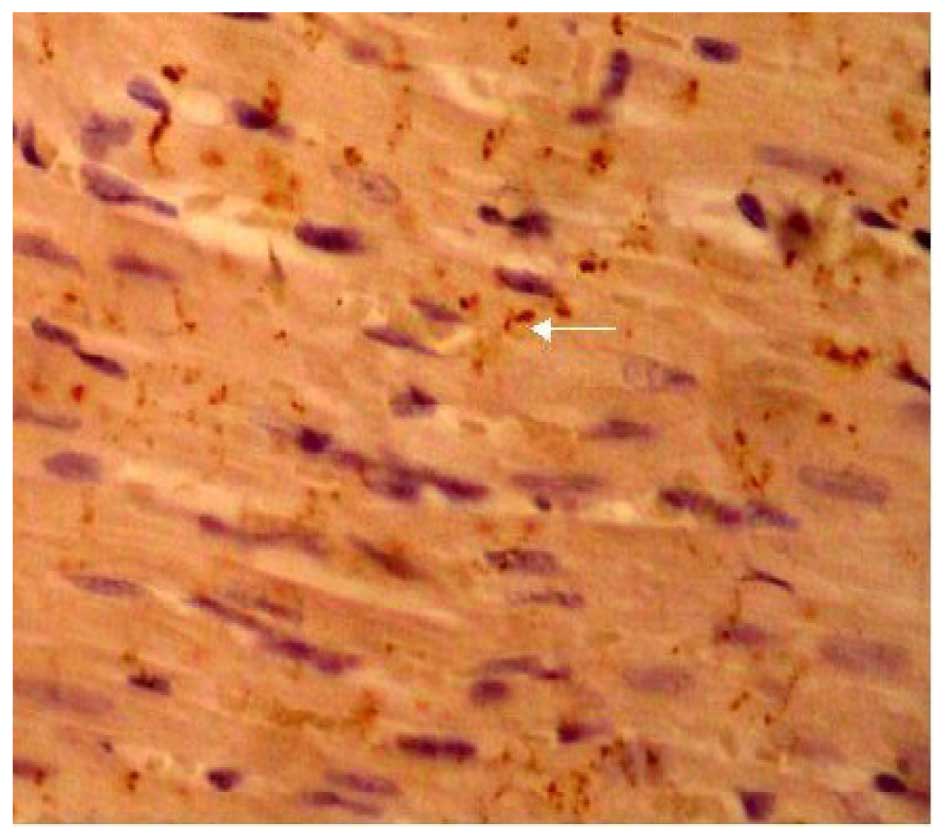
distribution (Fig. 5). The
expression of Cx43 in the Ki16425 + LPA group was very similar to
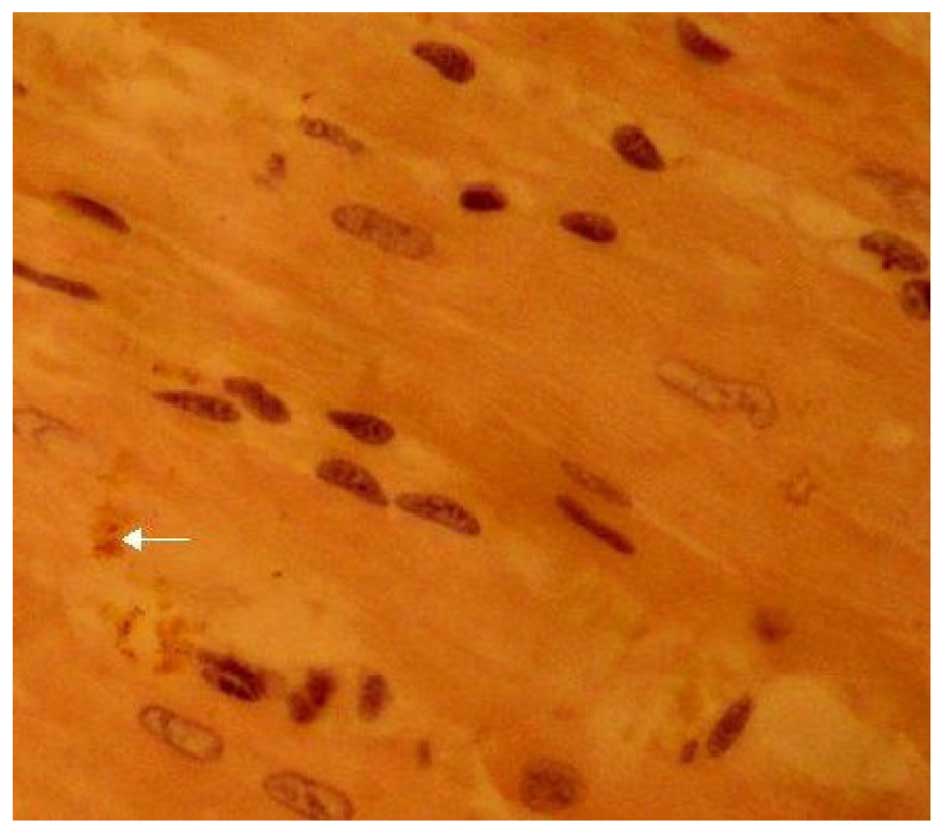
the control group (Fig. 6). The LPA
+ immune-suppressed group expressed decreased Cx43 protein compared
with the LPA + AMI model group. Cx43 was distributed relatively
uniformly in the intercalated disk between adjacent cells and had a
dotted distribution; however, both the density and coloring level
of Cx43 were decreased as compared with the control group (Fig. 7). The expression of Cx43 in the LPA +
immune-enhanced group was marginal (Fig.
8).
Discussion
LPA is an intermediate product of membrane
phospholipid metabolism (1,4,12).
Previous studies have demonstrated that LPA has an important role
in cardiovascular disease. LPA levels increase in infarcted
myocardium following AMI (4,16). In addition, LPA release by platelets
simultaneously increases, resulting in markedly increased LPA
concentration levels in regional myocardial tissue and plasma
(17,18). Leukocyte concentrations increase in
the peripheral blood of patients with AMI, and inflammatory cells
infiltrate the coronary arteries surrounding the tissue (19). These results suggests that AMI is a
process of the immune inflammatory response. Xie et al
(20) and Okudaira et al
(21) hypothesized that LPA may
modulate immune inflammatory responses. In the present study, we
hypothesized that LPA, acting as an important immune regulatory
substance, may be able to induce the release of various cytokines
through the activation of certain immune cells, thereby inducing
arrhythmia.
Immune-enhanced and immune-suppressed rat models
were constructed to validate the hypothesis. By observing the
surface ECGs of AMI rats, the results demonstrated that the
pro-arrhythmic effect of LPA was closely associated with immune
status. The incidence of arrhythmia decreased when the immune
systems of the rats were suppressed. To exclude interference by
in vivo factors and to examine the effect of LPA on
arrhythmia, LPA was added to the perfusate of isolated hearts from
rats of the immune-enhanced and the immune-suppressed groups. The
results demonstrated that the isolated rat hearts of the
immune-enhanced group exhibited increased occurrences of VPBs
compared with normal rats and rats of the immune-suppressed group.
This suggested that LPA has a role in the occurrence of arrhythmia
and is closely associated with the rat immune status.
Various cytokines are closely associated with
arrhythmia, including TNF, IL-6, IL-8 and IL-10 (19,22,23). TNF
is considered to be one of the most important cytokines in
ischemia/reperfusion injury in patients with AMI (24,25).
TNF-α is a multifunctional cytokine; as a key inflammatory mediator
of AMI, TNF-α has been the subject of increased research. Animals
overexpressing TNF suffer from severe heart disease, including
arrhythmia (26–28). This indicates that upregulation of
TNF expression is closely associated with the dysfunction of
cardiac myocytes (29,30). The results of the present study
demonstrated that LPA is able to induce TNF-α release in cultured
Jurkat T cells. These data suggest that LPA induces TNF-α mRNA
expression and promotes the synthesis and release of TNF-α by
activating Jurkat T cells.
Lymphocyte activation is closely associated with
K+ channels on the lymphocyte membrane. K+
channels function in controlling membrane potential, regulating
cell volume and activating lymphocytes (31,32);
therefore, they have an important role in the process of lymphocyte
immunity. The present study investigated the current
characteristics of voltage-dependent K+ channels
(Kv) and Ca2+-activated K+
channels (KCa) in Jurkat T cells. The results
demonstrated that LPA increased the Kv current in Jurkat
T cells and promoted the influx of K+. LPA significantly
increased the current amplitude of KCa. These results
provide further evidence that LPA activates Jurkat T cells by
opening Kv and KCa channels.
The rhythmic contraction of the heart is dependent
on signal transduction between myocardial cells (33,34). It
has been demonstrated that gap junctions (GJs) are the primary mode
of signal transduction between cells (35,36). GJs
are predominantly composed of connexin, and Cx43 is the main
protein of GJs in the heart (37–39). The
incidence of arrhythmia increased significantly and Cx43 decreased
when LPA expression was upregulated (40,41). We
hypothesize that LPA affects signal transduction between myocardial
cells by suppressing the synthesis and expression of Cx43, thereby
causing arrhythmia. The expression pattern of Cx43 was observed in
the myocardium using immunohistochemical staining. The results
indicated that LPA caused the degradation of Cx43 and decreased the
expression of Cx43. This may be one of the most important
mechanisms underlying the regulation of LPA-induced arrhythmias.
The observation that LPA caused the degradation of Cx43 and
decreased its expression is relevant to the immune status of
rats.
In summary, the results of the present investigation
determined that LPA participates in the incidence of arrhythmia
following AMI. To the best of our knowledge, the results provide
the first experimental evidence that LPA causes arrhythmia via the
regulation of immune-inflammatory cells and the release of
cytokines. The experiments demonstrate that LPA is able to induce
TNF-α expression by activating T lymphocytes and suppressing the
synthesis and expression of Cx43.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81170163)
and the Science & Technology Department of Jilin Province
(grant no. 200705367).
References
|
1
|
Sun R, Zhang D, Zhang J, Feng Q, Zhang Y,
Zhao C and Zhang W: Different effects of lysophosphatidic acid on
L-type calcium current in neonatal rat ventricular myocytes with
and without H2O2 treatment. Prostaglandins Other Lipid Mediat.
118(119): 1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sharain K, Vasile VC and Jaffe AS: Does
cardiac rhythm monitoring in patients with elevated troponin levels
lead to changes in management. Eur Heart J Acute Cardiovasc Care.
Jan 27–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aaltonen N, Laitinen JT and Lehtonen M:
Quantification of lysophosphatidic acids in rat brain tissue by
liquid chromatography-electrospray tandem mass spectrometry. J
Chromatogr B Analyt Technol Biomed Life Sci. 878:1145–1152. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei Y, Zhao LQ, Qi BZ, Xiao X, He L, Zhou
GQ, Chen SW, Li HL, Ruan L, Zhang CT and Liu SW: Lysophosphatidic
acid increases the electrophysiological instability of adult rabbit
ventricular myocardium by augmenting L-type calcium current. PLoS
One. 7:e458622012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim Do Y, Song HJ, Jeong JH, Suh JS and
Sohn UD: Regulation of lysophosphatidic acid-induced COX-2
expression by ERK1/2 activation in cultured feline esophageal
epithelial cells. Arch Pharm Res. 31:1331–1338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Latet SC, Hoymans VY, Van Herck PL and
Vrints CJ: The cellular immune system in the post-myocardial
infarction repair process. Int J Cardiol. 179:240–247. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Laskarin G, Zaputovic L, Persic V, Ruzic A
and Tokmadzic Sotosek V: Harmful immune reactions during acute
myocardial infarction. Med Hypotheses. 78:703–706. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hansson GK: Inflammatory mechanisms in
atherosclerosis. J Thromb Haemostasis. 7(Suppl 1): 328–331. 2009.
View Article : Google Scholar
|
|
9
|
Hansson GK, Robertson AK and
Söderberg-Nauclér C: Inflammation and atherosclerosis. Ann Rev
Pathol. 1:297–329. 2006. View Article : Google Scholar
|
|
10
|
Lin CC, Lin CE, Lin YC, Ju TK, Huang YL,
Lee MS, Chen JH and Lee H: Lysophosphatidic acid induces reactive
oxygen species generation by activating protein kinase C in PC-3
human prostate cancer cells. Biochem Biophys Res Commun.
440:564–569. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang CL, Lin ME, Hsu HY, Yao CL, Hwang
SM, Pan CY, Hsu CY and Lee H: Lysophosphatidic acid-induced
interleukin-1 beta expression is mediated through Gi/Rho and the
generation of reactive oxygen species in macrophages. J Biomed Sci.
15:357–363. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karliner JS: Lysophospholipids and the
cardiovascular system. Biochim Biophys Acta. 1582:216–221. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Institute of Laboratory Animal Resources
(US). Committee on Care, Use of Laboratory Animals, and National
Institutes of Health (US). Division of Research Resources: Guide
for the care and use of laboratory animals (8th). National
Academies Press. (Washington, DC). 2011.
|
|
14
|
Bocksteins E, Raes AL, Van de Vijver G,
Bruyns T, Van Bogaert PP and Snyders DJ: Kv2.1 and silent Kv
subunits underlie the delayed rectifier K+ current in
cultured small mouse DRG neurons. Am J Physiol Cell Physiol.
296:C1271–C1278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moreno C, Prieto P, Macías A,
Pimentel-Santillana M, de la Cruz A, Través PG, Boscá L and
Valenzuela C: Modulation of Voltage-Dependent and Inward Rectifier
Potassium Channels by 15-Epi-Lipoxin-A4 in Activated Murine
Macrophages: Implications in Innate Immunity. J Immunol.
191:6136–6146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakasaki T, Tanaka T, Okudaira S, Hirosawa
M, Umemoto E, Otani K, Jin S, Bai Z, Hayasaka H, Fukui Y, et al:
Involvement of the lysophosphatidic acid-generating enzyme
autotaxin in lymphocyte-endothelial cell interactions. Am J Pathol.
173:1566–1576. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng HY, Dong A, Panchatcharam M, Mueller
P, Yang F, Li Z, Mills G, Chun J, Morris AJ and Smyth SS:
Lysophosphatidic acid signaling protects pulmonary vasculature from
hypoxia-induced remodeling. Arterioscler Thromb Vasc Biol.
32:24–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pamuklar Z, Lee JS, Cheng HY,
Panchatcharam M, Steinhubl S, Morris AJ, Charnigo R and Smyth SS:
Individual heterogeneity in platelet response to lysophosphatidic
acid: Evidence for a novel inhibitory pathway. Arterioscler Thromb
Vasc Biol. 28:555–561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kempf K, Haltern G, Füth R, Herder C,
Müller-Scholze S, Gülker H and Martin S: Increased TNF-alpha and
decreased TGF-beta expression in peripheral blood leukocytes after
acute myocardial infarction. Horm Metab Res. 38:346–351. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie Y, Gibbs TC and Meier KE:
Lysophosphatidic acid as an autocrine and paracrine mediator.
Biochim Biophysiolo Acta. 1582:270–281. 2002. View Article : Google Scholar
|
|
21
|
Okudaira S, Yukiura H and Aoki J:
Biological roles of lysophosphatidic acid signaling through its
production by autotaxin. Biochimie. 92:698–706. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Putko BN, Wang Z, Lo J, Anderson T, Becher
H, Dyck JR, Kassiri Z and Oudit GY: Alberta HEART Investigators:
Circulating levels of tumor necrosis factor-alpha receptor 2 are
increased in heart failure with preserved ejection fraction
relative to heart failure with reduced ejection fraction: Evidence
for a divergence in pathophysiology. PLoS One. 9:e994952014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meilleur MA, Akpovi CD, Pelletier RM and
Vitale ML: Tumor necrosis factor-alpha-induced anterior pituitary
folliculostellate TtT/GF cell uncoupling is mediated by connexin 43
dephosphorylation. Endocrinology. 148:5913–5924. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Blancke F, Claeys MJ, Jorens P, Vermeiren
G, Bosmans J, Wuyts FL and Vrints CJ: Systemic inflammation and
reperfusion injury in patients with acute myocardial infarction.
Mediators Inflamm. 2005:385–389. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moreira DM, da Silva RL, Vieira JL, Fattah
T, Lueneberg ME and Gottschall CA: Role of vascular inflammation in
coronary artery disease: Potential of anti-inflammatory drugs in
the prevention of atherothrombosis. Inflammation and
anti-inflammatory drugs in coronary artery disease. Am J Cardiovasc
Drugs. 15:1–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wit AL and Duffy HS: Drug development for
treatment of cardiac arrhythmias: Targeting the gap junctions. Am J
Physiol Heart Circ Physiol. 294:H16–H18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen Y, Zhang Q, Liao YH, Cao Z, Du YM,
Xia JD, Yang H and Chen ZJ: Effect of tumor necrosis factor-α on
neutralization of ventricular fibrillation in rats with acute
myocardial infarction. Mediators Inflamm. 2011:5652382011.
|
|
28
|
Chang WT, Wang YC, Chen CC, Zhang SK, Liu
CH, Chang FH and Hsu LS: The −308G/A of tumor necrosis factor
(TNF)-α and 825C/T of guanidine nucleotide binding protein 3 (GNB3)
are associated with the onset of acute myocardial infarction and
obesity in Taiwan. Int J Mol Sci. 13:1846–1857. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Petkova-Kirova PS, London B, Salama G,
Rasmusson RL and Bondarenko VE: Mathematical modeling mechanisms of
arrhythmias in transgenic mouse heart overexpressing TNF-α. Am J
Physiol Heart Circ Physiol. 302:H934–H952. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lawrence MC, Naziruddin B, Levy MF,
Jackson A and McGlynn K: Calcineurin/nuclear factor of activated T
cells and MAPK signaling induce TNF-{alpha} gene expression in
pancreatic islet endocrine cells. J Biol Chem. 286:1025–1036. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kazama I, Tamada T and Tachi M: Usefulness
of targeting lymphocyte Kv1.3-channels in the treatment of
respiratory diseases. Inflamma Res. 64:753–765. 2015. View Article : Google Scholar
|
|
32
|
Kazama I: Physiological significance of
delayed rectifier K+ channels (Kv1.3) expressed in T
lymphocytes and their pathological significance in chronic kidney
disease. J Physiol Sci. 65:25–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Csordás G, Thomas AP and Hajnóczky G:
Calcium signal transmission between ryanodine receptors and
mitochondria in cardiac muscle. Trends Cardiovasc Med. 11:269–275.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vinogradova TM, Lyashkov AE, Zhu W,
Ruknudin AM, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey
JL, et al: High basal protein kinase A-dependent phosphorylation
drives rhythmic internal Ca2+ store oscillations and
spontaneous beating of cardiac pacemaker cells. Circulation Res.
98:505–514. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hirata N, Kanaya N, Kamada N, Kimura S and
Namiki A: Differential effects of propofol and sevoflurane on
ischemia-induced ventricular arrhythmias and phosphorylated
connexin 43 protein in rats. Anesthesiology. 110:50–57. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dhein S, Polontehouk L, Salameh A and
Haefliger JA: Pharmacological modulation and differential
regulation of the cardiac gap junction proteins connexin 43 and
connexin 40. Biol Cell. 94:409–422. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De Vuyst E, Decrock E, De Bock M, Yamasaki
H, Naus CC, Evans WH and Leybaert L: Connexin hemichannels and gap
junction channels are differentially influenced by
lipopolysaccharide and basic fibroblast growth factor. Mol Biol
Cell. 18:34–46. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nielsen MS, Axelsen LN, Sorgen PL, Verma
V, Delmar M and Holstein-Rathlou NH: Gap junctions. Compr Physiol.
2:1981–2035. 2012.PubMed/NCBI
|
|
39
|
Palatinus JA, Rhett JM and Gourdie RG: The
connexin 43 carboxyl terminus and cardiac gap junction
organization. Biochim Biophys Acta. 1818:1831–1843. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gendaszewska-Darmach E: Lysophosphatidic
acids, cyclic phosphatidic acids and autotaxin as promising targets
in therapies of cancer and other diseases. Acta Biochim Pol.
55:227–240. 2008.PubMed/NCBI
|
|
41
|
Chan LC, Peters W, Xu Y, Chun J, Farese RV
Jr and Cases S: LPA3 receptor mediates chemotaxis of immature
murine dendritic cells to unsaturated lysophosphatidic acid (LPA).
J Leukoc Biol. 82:1193–1200. 2007. View Article : Google Scholar : PubMed/NCBI
|