Introduction
Multiple trauma, which is commonly associated with
severe injuries and multiple organ failure, may lead to various
complications, including sepsis and septic shock, which are major
healthcare problems worldwide (1–3). There
are 400,000–500,000 cases of sepsis in the United States annually
(4). Antimicrobial therapy may be
applied for the management of sepsis; however, the mortality rate
associated with sepsis has increased, and was reported to be as
high as 40% in 2003 (5).
Tumor necrosis factor (TNF)-α, a cytokine that is
predominantly secreted by macrophages, has been shown to be
involved in the regulation of numerous biological processes,
including cell proliferation, differentiation, apoptosis, lipid
metabolism and coagulation (6–8).
However, the role of TNF-α in tumorigenesis remains unclear. This
cytokine has been reported to induce tumor necrosis and apoptosis,
as well as to promote tumor development (9). However, previous studies investigating
the role of TNF-α in clinical sepsis syndrome or septic shock have
reported conflicting results (10–12).
TNF-α has been established as an effective marker in the diagnosis
of neonatal sepsis (13); however,
the mechanisms underlying the regulatory role of TNF-α in the
development of sepsis syndrome remain undefined. Genetic variations
have previously been implicated in the progression of numerous
types of cancer (14,15). In addition, the clinical outcomes of
sepsis have been associated with genetic polymorphisms in the genes
encoding various inflammatory cytokines (16). Menges et al (17) demonstrated that common variants of
the TNF-α gene were associated with sepsis syndrome and mortality
following severe injury. Reportedly, the common TNF-α gene variant
carrying the TNF rs1800629 A allele is correlated with higher TNF-α
serum concentrations and alteration of genes strongly associated
with proinflammatory and apoptosis (17). Furthermore, TNF rs1800629 A is
closely associated with sepsis syndrome and mortality following
multiple trauma (18).
The present study re-analyzed the GSE5760 microarray
data deposited in the Gene Expression Omnibus (GEO) database by
Menges et al (17), in order
to detect genes that were differentially expressed between patients
with and without TNF-α genetic variations, and to identify their
potential functions and pathways. Furthermore, the regulatory
associations between differentially expressed genes (DEGs) and
microRNAs (miRNAs) were analyzed in order to elucidate the
regulatory mechanisms underlying sepsis in patients with TNF-α
genetic variations, at the transcriptional and post transcriptional
levels.
Materials and methods
Microarray data
The GSE5760 gene expression profile data was
downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). Based on the
description provided by Menges et al (17), the profile data consisted of 30
wild-type (WT) peripheral blood samples from 12 injured patients
without the TNF-α rs1800629 A variant and 28 mutation (MUT)
peripheral blood samples from 10 injured patients carrying the
TNF-α rs1800629 A variant. The technical replicate numbers for the
WT samples were three replicates for 6 patients and two replicates
for the other 6 patients. The technical replicate numbers for the
MUT samples were three replicates for 8 patients and two replicates
for 2 patients. Menges et al (17) had used the GPL4204 platform (GE
Healthcare/Amersham Biosciences CodeLink UniSet Human I Bioarray;
GE Healthcare, Little Chalfont, UK). The annotation information in
the platform was also downloaded.
Data preprocessing and identification
of DEGs
The gene expression value was calculated from raw
microarray data of the probe value. In the case that multiple
probes corresponded to one gene, the average value was calculated
as the expression level of this gene, whereas in the case that one
probe corresponded to multiple genes, the probe value was removed.
Following transformation of the data by log2 and
normalization using the median method (19), DEGs between the WT and MUT samples
were identified using the Limma package in R software (20) and Student's t-test. The
Benjamini-Hochberg procedure (21)
was applied, in order to control the false discovery rate (FDR).
The threshold criteria for the DEGs were FDR<0.05 and
|log2 fold change (FC)| of >0.05.
Selection of miRNAs targeted to
DEGs
Following identification of the DEGs, WebGestal
software (version 2.0; Vanderbilt University, Nashville, TN, USA;
http://bioinfo.vanderbilt.edu/webgestalt/) (22) was used in order to identify miRNAs
that were associated with the DEGs. A threshold of adjusted
P<0.05 was used.
Construction of an integrated
regulatory network between miRNAs and their target DEGs
The DEGs were mapped using the Search Tool for the
Retrieval of Interacting Genes/Proteins database (http://string-db.org/) (23), in order to identify potential
protein-protein interactions between the DEGs. Interaction pairs
were identified using the default parameter of a combined score of
≥0.6. The integrated regulatory miRNA-DEG network was constructed
and visualized using the Cytoscape software (24), based on the DEG interaction pairs and
the interactions between the 11 miRNAs and their target DEGs.
Function and pathway enrichment
analyses for the identified DEGs
The biological functions of the DEGs in the
established network were investigated by Gene Ontology (GO)
enrichment analysis, using the Database for Annotation,
Visualization and Integrated Discovery online software (https://david.ncifcrf.gov/) (25). P<0.05 was considered to indicate a
significantly enriched GO term. In addition, pathway enrichment
analyses were conducted using the KEGG Orthology Based Annotation
System (KOBAS, version 2; http://kobas.cbi.pku.edu.cn/home.do), in order to
identify the pathways in which the DEGs were involved. In addition,
the statistical method of cumulative hypergeometric distribution
was applied and P<0.05 was considered to indicate a
significantly enriched pathway.
Results
Identification of DEGs between the WT
and MUT samples, and the associated miRNAs
Based on the preset criteria of FDR<0.05 and
|log2FC|>0.05, a total of 390 genes were shown to be
differentially expressed between the WT and MUT samples, including
238 genes that were upregulated and 152 genes that were
downregulated. Based on an established threshold for miRNA
searching, 11 miRNAs, including miR-141, miR-374, miR-204, miR-23,
miR-182, miR-26, miR-15, miR-30, miR-34, miR-181 and miR-130, were
significantly associated with the identified DEGs, and were
selected for inclusion in the integrated miRNA-DEG regulatory
network (Table I).
 | Table I.miRNAs associated with DEGs. |
Table I.
miRNAs associated with DEGs.
| miRNA | ID | DEG counts | Raw P-value | Adjusted
P-value |
|---|
| Has_CAGTGTT,
miR-141 | DB_ID:690 | 15 |
7.29×10−8 |
1.39×10−6 |
| Has_TATTATA,
miR-374 | DB_ID:727 | 14 |
1.72×10−7 |
1.63×10−6 |
| Has_AAAGGGA,
miR-204 | DB_ID:682 | 12 |
4.49×10−7 |
2.84×10−6 |
| Has_AATGTGA,
miR-23 | DB_ID:683 | 16 |
6.71×10−7 |
3.19×10−6 |
| Has_TTGCCAA,
miR-182 | DB_ID:757 | 13 |
4.62×10−6 |
1.76×10−5 |
| Has_TACTTGA,
miR-26 | DB_ID:687 | 12 |
9.99×10−6 |
3.16×10−5 |
| Has_TGCTGCT,
miR-15 | DB_ID:666 | 17 |
1.48×10−5 |
4.02×10−5 |
| Has_TGTTTAC,
miR-30 | DB_ID:667 | 16 |
3.52×10−5 |
7.43×10−5 |
| Has_CACTGCC,
miR-34 | DB_ID:673 | 10 | 0.0001 | 0.0002 |
| Has_TGAATGT,
miR-181 | DB_ID:669 | 13 | 0.0003 | 0.0005 |
| Has_TTGCACT,
miR-130 | DB_ID:676 | 11 | 0.0006 | 0.0009 |
Construction of the integrated
miRNA-DEG regulatory network
A total of 36 DEG interaction pairs were identified,
according to their predicted protein-protein interactions. Combined
with the identified DEGs targeted by miRNAs, the integrated
miRNA-DEG regulatory network was constructed. This network
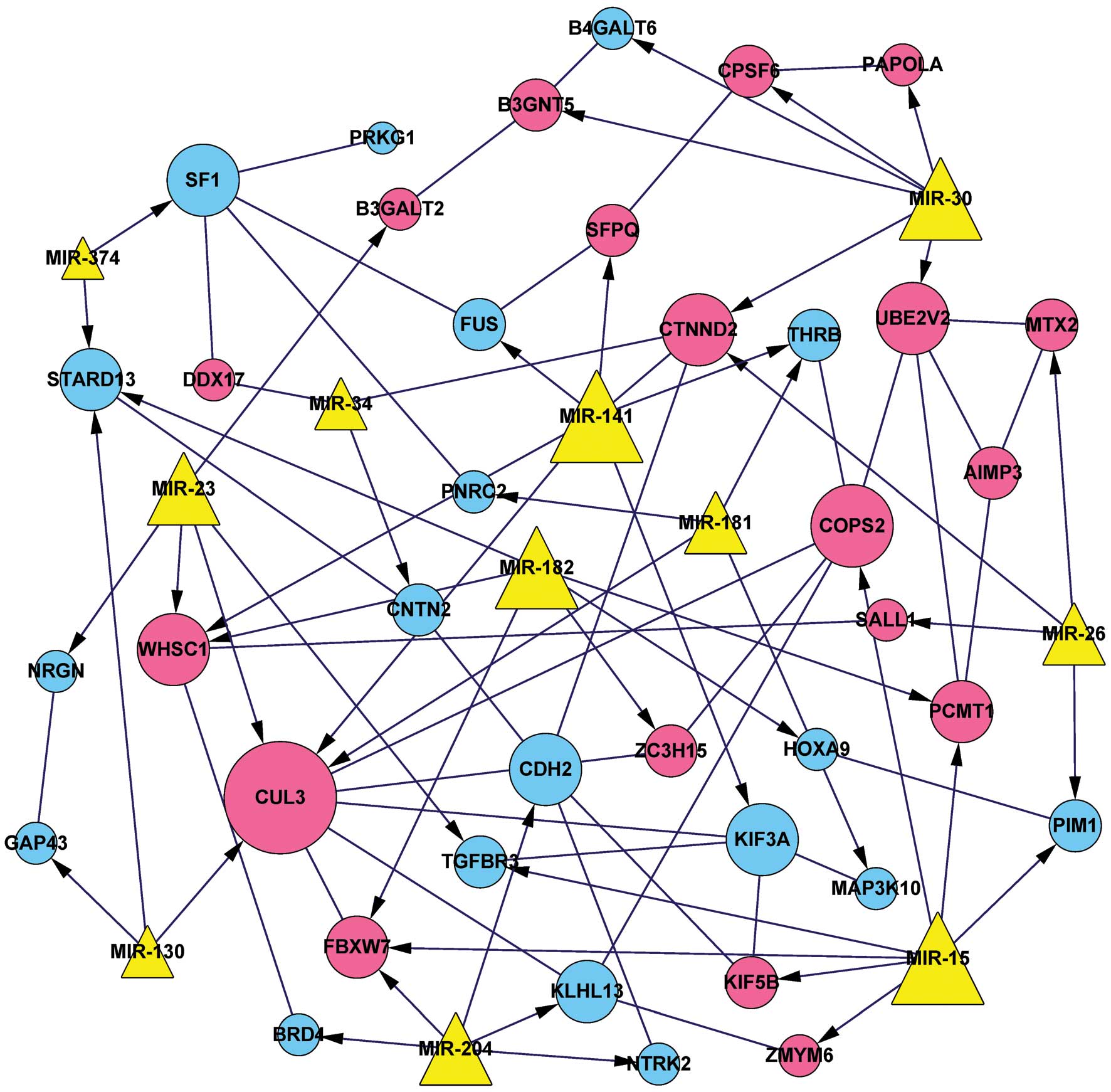
comprised 49 nodes and 88 edges, involving 11 miRNAs and 38 DEGs,
including 19 upregulated and 19 downregulated genes (Fig. 1).
Biological function and pathway
annotation of the DEGs
In order to investigate the functions of the DEGs
associated with TNF-α genetic variations, the identified DEGs were
subjected to GO analysis. As presented in Table II, the DEGs were predominantly
enriched in seven GO terms associated with transcriptional events,
phosphorylation and RNA functions. DEGs associated with
transcriptional regulation were as follows: COPS2,
THRB, SFPQ, SALL1, PNRC2,
CTNND2, PIM1, SF1, MAP3K10,
TGFBR3, HOXA9 and WHSC1. DEGs associated with
transcription were as follows: COPS2, PAPOLA,
THRB, SFPQ, SALL1, PNRC2,
CTNND2, SF1, HOXA9 and WHSC1. In
addition, DEGs associated with GO terms such as phosphorylation and
protein amino acid phosphorylation were NTRK2, PIM1,
MAP3K10, TGFBR3, BRD4 and PRKG1. The
six DEGs associated with RNA processing were FUS,
DDX17, PAPOLA, SFPQ, SF1 and
CPSF6. Those involved in mRNA metabolic processes, which was
the most significantly enriched GO term (P=0.001632), included
FUS, PAPOLA, SFPQ, PNRC2, SF1
and CPSF6. DEGs, including NTRK2, PIM1,
MAP3K10, TGFBR3, BRD4 and GAP43, were
significantly associated with positive regulation of molecular
function.
| Table II.GO enrichment analysis of the DEGs in
the integrated regulatory network. |
Table II.
GO enrichment analysis of the DEGs in
the integrated regulatory network.
| Term | DEGs | P-value |
|---|
| GO:0016071 - mRNA
metabolic process | FUS, PAPOLA,
SFPQ, PNRC2, SF1, CPSF6 | 0.001632 |
| GO:0006396 - RNA
processing | FUS, DDX17,
PAPOLA, SFPQ, SF1, CPSF6 | 0.008649 |
| GO:0044093 -
positive regulation of molecular function | NTRK2, PIM1,
MAP3K10, TGFBR3, BRD4, GAP43 | 0.011448 |
| GO:0006468 -
protein amino acid phosphorylation | NTRK2, PIM1,
MAP3K10, TGFBR3, BRD4, PRKG1 | 0.019140 |
| GO:0045449 -
regulation of transcription | COPS2, THRB,
SFPQ, SALL1, PNRC2, CTNND2, PIM1, SF1, MAP3K10, TGFBR3, HOXA9,
WHSC1 | 0.031480 |
| GO:0016310 -
phosphorylation | NTRK2, PIM1,
MAP3K10, TGFBR3, BRD4, PRKG1 | 0.038131 |
| GO:0006350 -
transcription | COPS2, PAPOLA,
THRB, SFPQ, SALL1, PNRC2, CTNND2, SF1, HOXA9, WHSC1 | 0.049845 |
KOBAS analysis was also conducted in order to
identify significantly enriched pathways. Two significantly
enriched pathways were identified, including the glycosphingolipid
biosynthesis (including B3GNT5 and B3GALT2) and
ubiquitin-mediated proteolysis, which was the most significantly
enriched pathway with three DEGs (including CUL3,
FBXW7 and KLHL13) (Table
III). Based on the information from the integrated regulatory
network (Fig. 1), both COPS2
and CUL3 were found to be targets of miR-15.
| Table III.Enriched pathways of the
differentially expressed genes in the integrated regulatory
network. |
Table III.
Enriched pathways of the
differentially expressed genes in the integrated regulatory
network.
| ID | Pathway | P-value | Genes |
|---|
| hsa04120 | Ubiquitin mediated
proteolysis | 0.02628 | CUL3, FBXW7,
KLHL13 |
| hsa00601 | Glycosphingolipid
biosynthesis | 0.04213 | B3GNT5,
B3GALT2 |
Discussion
The present study identified 390 genes that were
differentially expressed between the WT and MUT (carrying TNF-α
rs1800629 A variant) samples, and established a regulatory network
containing 38 DEGs and 11 miRNAs. This network suggested that
COPS2 was predominantly associated with transcriptional
functions, whereas FUS was primarily involved in mRNA
metabolic processes. Other DEGs, including FBXW7 and
CUL3, were enriched in the ubiquitin-mediated proteolysis
pathway. Furthermore, miR-15 was predicted to target both
COPS2 and CUL3.
COPS2, also known as COP9 signalosome subunit
2 or CSN2, is an essential component of the COP9 signalosome
complex, which is involved in diverse cellular processes and acts
as a critical regulator of the ubiquitin conjugation pathway
(26). In a previous study it was
demonstrated that TNF-α was able to promote the stabilization of
Snail (the most important transcriptional repressor of E-cadherin)
and β-catenin, by inhibiting GSK-3β-mediated phosphorylation of
these proteins via the nuclear factor-κB and Akt signalling
pathways, which in turn promoted tumor cell invasion. Notably,
COPS2 had a crucial role in this process since it was able
to inhibit the phosphorylation and ubiquitination of Snail by
preventing the binding of Snail to its receptors (27).
The protein encoded by FUS is a
multifunctional component of the heterogeneous nuclear
ribonucleoprotein complex, which is involved in pre-mRNA splicing
and the export of fully processed mRNA to the cytoplasm (28). FUS, which contains multiple
RNA binding domains and an N-terminal glutamine-rich domain, acts
as a mediator of RNA binding and as a potent transcriptional
activator (29). In addition,
FUS has previously been associated with familial amyotrophic
lateral sclerosis (30) and the
pathogenesis of myxoid liposarcoma (31); however, to the best of our knowledge,
no previous studies have investigated the association between
FUS and sepsis. In the present study, COPS2 was shown
to be upregulated in the MUT samples, and was strongly associated
with transcriptional functions, whereas FUS was
downregulated in the TNF-α rs1800629 A allele variant samples, and
was associated with mRNA metabolic processes. Furthermore,
COPS2 and FUS were shown to interact in the
integrated regulatory network. These results suggested that
COPS2 may play a vital role in the pathogenesis of sepsis in
patients with TNF-α rs1800629 A allele variation by regulating the
phosphorylation and ubiquitination of the protein encoded by
FUS at the transcriptional level.
FBXW7 and CUL3 are two critical DEGs,
which were found to be upregulated in the TNF-α rs1800629 A allele
variant samples. CUL3 (also known as cullin 3) encodes a
member of the cullin protein family, and is the core component and
scaffold protein of an E3 ubiquitin-protein ligase complex, which
mediates the ubiquitiation and subsequent proteasomal degradation
of target proteins (32). As major
subunits of the cullin-RING ligase (CRL) family, cullins are active
components of ~50% of human E3 ubiquitin ligases and are
responsible for one fifth of all cellular proteasome-dependent
protein degradation (33).
FBXW7 (also known as F-box and WD repeat domain-containing
7) is a substrate receptor for CRL1, and facilitates the
ubiquitination and degradation of numerous proteins (34). Cyclin E, a member of the cyclin
family, is a positive regulator of proliferation in mammalian
fibroblasts. CUL1 and CUL3 have previously been
implicated in the degradation of cyclin E via two distinct pathways
(35). In comparison with the
CUL1/FBXW7-based E3 ligase-dependent pathway, cyclin
E does not require phosphorylation at threonine 380, and may be
recognized as a substrate by CUL3 in the CUL3-based
E3 ligase pathway (36), which may
be essential for the maintenance of quiescence in mammalian cells
(35). Therefore, consistent with
the findings of the present study, previous reports have suggested
that FBXW7 and CUL3 may be involved in the
ubiquitin-mediated proteolysis pathway (37,38).
Based on a novel mechanism of autophagy, CaMKIV
(also known as calcium/calmodulin-dependent protein kinase IV) may
inhibit ubiquitin-mediated mTOR degradation through the inhibition
of FBXW7 recruitment. However, in sepsis, mTOR expression is
required for autophagy (39),
indicating that FBXW7 may regulate mTOR. Although Qiao et
al (40) did not identify
CUL3 as a DEG in sepsis, it is associated with DEGs in the
PPI network, suggesting their potential involvement in sepsis.
However, no study has previously reported a direct interaction
between FBXW7 and CUL3. The aforementioned findings
collectively suggest that FBXW7 and CUL3 may exert
proteolytic functions via ubiquitination in sepsis progression in
patients with TNF-α genetic variations. In addition, there may be
regulators between FBXW7 and CUL3 that facilitate the
functions of the two genes.
miRNAs have a critical role in the regulation of
gene expression at the post-transcriptional level (41). miR-15 has been demonstrated to be
associated with apoptosis in various types of cancer. For instance,
a previous study identified that miR-15 was closely associated with
apoptosis in chronic lymphocytic leukemia, and it was demonstrated
to induce apoptosis by negatively regulating the expression of
B-cell lymphoma 2 (42).
Furthermore, miR-15(a/b) was reported to have inhibited the
expression of cyclin E and was identified as a novel
transcriptional target of E2F1, which is a vital transcription
factor that induces proliferation and cell death (43). Thus, miR-15 and CUL3 may
co-regulate the expression of cyclin E. Up-regulation of miR-15 has
been found in the serum of neonatal sepsis patients, and this miRNA
is proposed as a potential biomarker for neonatal sepsis prognosis
(44). Notably, the present study
identified CUL3 as a potential target of miR-15, which is
consistent with this hypothesis. In addition, although previous
investigations have been unable to uncover a regulatory association
between miR-15 and COPS2, the present study predicted that
COPS2 was a target of miR-15, thus suggesting that
COPS2 may be a novel target gene for miR-15 in the
progression of sepsis.
In conclusion, in the present study, COPS2,
FUS, FBXW7 and CUL3 were found to be
associated with the progression of sepsis in patients with the
TNF-α rs1800629 A variant. Of these genes, FBXW7 and
CUL3 may exert regulatory roles in the ubiquitin-mediated
proteolysis pathway, whereas COPS2 may affect the
development of sepsis by regulating the phosphorylation and
ubiquitination of the FUS protein. In addition, COPS2 and
CUL3 may be novel targets of miR-15. The aforementioned
genes and miRNAs may be used as therapeutic biomarkers for sepsis
diagnosis in patients with the TNF-α rs1800629 A variant.
Acknowledgements
The present study was supported by the Shanghai
Medical Key Subject Construction Project (grant no. ZK2012A28), the
Shanghai Special Project for Advanced and Suitable Technology
(grant no. 2013SY039) and the National Clinical Key Specialty
Construction Project.
References
|
1.
|
Wafaisade A, Lefering R, Bouillon B, Sakka
SG, Thamm OC, Paffrath T, Neugebauer E and Maegele M: Trauma
Registry of the German Society for Trauma Surgery: Epidemiology and
risk factors of sepsis after multiple trauma: An analysis of 29,829
patients from the Trauma Registry of the German Society for Trauma
Surgery. Crit Care Med. 39:621–628. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Coimbra R: From the trauma surgeon's
viewpoint: Multiple injuries - which cavity to open first? J Trauma
Nurs. 12:7–9. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Dellinger RP, Levy MM, Carlet JM, Bion J,
Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale
R, et al: Surviving Sepsis Campaign: International guidelines for
management of severe sepsis and septic shock: 2008. Intensive Care
Med. 34:17–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Balk RA: Severe sepsis and septic shock:
Definitions, epidemiology, and clinical manifestations. Crit Care
Clin. 16:179–192. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Hanna NF: Sepsis and septic shock. Adv
Emerg Nurs J. 25:158–165. 2003.
|
|
6.
|
Widera D, Mikenberg I, Elvers M,
Kaltschmidt C and Kaltschmidt B: Tumor necrosis factor α triggers
proliferation of adult neural stem cells via IKK/NF-kappaB
signaling. BMC Neurosci. 7:642006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lemaître N, Sebbane F, Long D and
Hinnebusch BJ: Yersinia pestis YopJ suppresses tumor
necrosis factor alpha induction and contributes to apoptosis of
immune cells in the lymph node but is not required for virulence in
a rat model of bubonic plague. Infect Immun. 74:5126–5131. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Greenberg JD, Kremer JM, Curtis JR,
Hochberg MC, Reed G, Tsao P, Farkouh ME, Nasir A, Setoguchi S and
Solomon DH: CORRONA Investigators: Tumour necrosis factor
antagonist use and associated risk reduction of cardiovascular
events among patients with rheumatoid arthritis. Ann Rheum Dis.
70:576–582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Yu M, Zhou X, Niu L, Lin G, Huang J, Zhou
W, Gan H, Wang J, Jiang X, Yin B and Li Z: Targeting transmembrane
TNF-α suppresses breast cancer growth. Cancer Res. 73:4061–4074.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Liang Y, Li X, Zhang X, Li Z, Wang L, Sun
Y, Liu Z and Ma X: Elevated levels of plasma TNF-α are associated
with microvascular endothelial dysfunction in patients with sepsis
through activating the NF-κB and p38 mitogen-activated protein
kinase in endothelial cells. Shock. 41:275–281. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Rice TW and Bernard GR: Therapeutic
intervention and targets for sepsis. Annu Rev Med. 56:225–248.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Alexander JJ, Jacob A, Cunningham P,
Hensley L and Quigg RJ: TNF is a key mediator of septic
encephalopathy acting through its receptor, TNF receptor-1.
Neurochem Int. 52:447–456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kocabaş E, Sarikçioğlu A, Aksaray N,
Seydaoğlu G, Seyhun Y and Yaman A: Role of procalcitonin,
C-reactive protein, interleukin-6, interleukin-8 and tumor necrosis
factor-alpha in the diagnosis of neonatal sepsis. Turk J Pediatr.
49:7–20. 2007.PubMed/NCBI
|
|
14.
|
Shigematsu H, Lin L, Takahashi T, Nomura
M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, et
al: Clinical and biological features associated with epidermal
growth factor receptor gene mutations in lung cancers. J Natl
Cancer Inst. 97:339–346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Lièvre A, Bachet J-B, Le Corre D, Boige V,
Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al:
KRAS mutation status is predictive of response to cetuximab therapy
in colorectal cancer. Cancer Res. 66:3992–3995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Sutherland AM and Walley KR:
Bench-to-bedside review: Association of genetic variation with
sepsis. Crit Care. 13:2102009. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Menges T, König IR, Hossain H, Little S,
Tchatalbachev S, Thierer F, Hackstein H, Franjkovic I, Colaris T,
Martens F, et al: Sepsis syndrome and death in trauma patients are
associated with variation in the gene encoding tumor necrosis
factor. Crit Care Med. 36:1456–1462, e1–e6. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Chen G, Han N, Li G, Li X, Li G, Liu Y, Wu
W, Wang Y, Chen Y, Sun G, et al: Prediction of feature genes in
trauma patients with the TNF rs1800629 A allele using support
vector machine. Comput Biol Med. 64:24–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Fujita A, Sato JR, Rodrigues LO, Ferreira
CE and Sogayar MC: Evaluating different methods of microarray data
normalization. BMC Bioinformatics. 7:4692006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Smyth GK: Limma: Linear models for
microarray data. Bioinformatics and computational biology solutions
using R and Bioconductor. Gentleman R, Carey VJ, Huber W, Irizarry
RA and Dudoit S: (New York). Springer. 397–420. 2005. View Article : Google Scholar
|
|
21.
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J R Stat Soc Series B Stat Methodol. 57:289–300.
1995.
|
|
22.
|
Duncan D, Prodduturi N and Zhang B:
WebGestalt2: An updated and expanded version of the Web-based Gene
Set Analysis Toolkit. BMC Bioinformatics. 11(Suppl 4): 102010.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9.1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
41:D808–D815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Smoot ME, Ono K, Ruscheinski J, Wang P-L
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Wei N and Deng XW: The COP9 signalosome.
Annu Rev Cell Dev Biol. 19:261–286. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Wu Y and Zhou BP:
TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and
invasion. Br J Cancer. 102:639–644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Schwartz JC, Podell ER, Han SS, Berry JD,
Eggan KC and Cech TR: FUS is sequestered in nuclear aggregates in
ALS patient fibroblasts. Mol Biol Cell. 25:2571–2578. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Bentmann E, Neumann M, Tahirovic S, Rodde
R, Dormann D and Haass C: Requirements for stress granule
recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein
of 43 kDa (TDP-43). J Biol Chem. 287:23079–23094. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Maruyama H, Morino H, Ito H, Izumi Y, Kato
H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, et al: Mutations of
optineurin in amyotrophic lateral sclerosis. Nature. 465:223–226.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Riggi N, Cironi L, Provero P, Suvà ML,
Stehle JC, Baumer K, Guillou L and Stamenkovic I: Expression of the
FUS-CHOP fusion protein in primary mesenchymal progenitor cells
gives rise to a model of myxoid liposarcoma. Cancer Res.
66:7016–7023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Pintard L, Willis JH, Willems A, Johnson
JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B and
Peter M: The BTB protein MEL-26 is a substrate-specific adaptor of
the CUL-3 ubiquitin-ligase. Nature. 425:311–316. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Saito N, Sakakibara K, Sato T, Friedman
JM, Kufe DW, VonHoff DD and Kawabe T: CBS9106-induced CRM1
degradation is mediated by cullin ring ligase activity and the
neddylation pathway. Mol Cancer Ther. 13:3013–3023. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Tron AE, Arai T, Duda DM, Kuwabara H,
Olszewski JL, Fujiwara Y, Bahamon BN, Signoretti S, Schulman BA and
DeCaprio JA: The glomuvenous malformation protein Glomulin binds
Rbx1 and regulates cullin RING ligase-mediated turnover of Fbw7.
Mol Cell. 46:67–78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
McEvoy JD, Kossatz U, Malek N and Singer
JD: Constitutive turnover of cyclin E by Cul3 maintains quiescence.
Mol Cell Biol. 27:3651–3666. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Singer JD, Gurian-West M, Clurman B and
Roberts JM: Cullin-3 targets cyclin E for ubiquitination and
controls S phase in mammalian cells. Genes Dev. 13:2375–2387. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Eide PW, Cekaite L, Danielsen SA,
Eilertsen IA, Kjenseth A, Fykerud TA, Ågesen TH, Bruun J, Rivedal
E, Lothe RA and Leithe E: NEDD4 is overexpressed in colorectal
cancer and promotes colonic cell growth independently of the
PI3K/PTEN/AKT pathway. Cell Signal. 25:12–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Yu S, Yi H, Wang Z and Dong J: Screening
key genes associated with congenital heart defects in Down syndrome
based on differential expression network. Int J Clin Exp Pathol.
8:8385–8393. 2015.PubMed/NCBI
|
|
39.
|
Zhang X, Howell GM, Guo L, Collage RD,
Loughran PA, Zuckerbraun BS and Rosengart MR: CaMKIV-dependent
preservation of mTOR expression is required for autophagy during
lipopolysaccharide-induced inflammation and acute kidney injury. J
Immunol. 193:2405–2415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Qiao FS, Wei C, Yun J and Qian LX:
Insights into the molecular mechanisms in sepsis with microarray
technology. Eur Rev Med Pharmacol Sci. 18:2405–2412.
2014.PubMed/NCBI
|
|
41.
|
Geng J, Luo H, Pu Y, Zhou Z, Wu X, Xu W
and Yang Z: Methylation mediated silencing of miR-23b expression
and its role in glioma stem cells. Neurosci Lett. 528:185–189.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Cimmino A, Calin GA, Fabbri M, Iorio MV,
Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et
al: miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Ofir M, Hacohen D and Ginsberg D: MiR-15
and miR-16 are direct transcriptional targets of E2F1 that limit
E2F-induced proliferation by targeting cyclin E. Mol Cancer Res.
9:440–447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Wang X, Wang X, Liu X, Wang X, Xu J, Hou
S, Zhang X and Ding Y: miR-15a/16 are upregulated in the serum of
neonatal sepsis patients and inhibit the LPS-induced inflammatory
pathway. Int J Clin Exp Pathol. 8:5683–5690. 2015.
|