Introduction
The exact cause of Parkinson disease (PD) is poorly
understood, even though nearly 200 years has passed since James
Parkinson first described PD in 1817, and the treatment is limited
to maintaining the balance of dopamine and cholinergic transmitter
through the administration of L-dopa, inhibitors of L-dopa
decarboxylase, dopaminergic agonists and/or monoamine oxidase B
inhibitors (1,2). These measures are only
symptom-relieving options, and cannot prevent the neuronal
degenerative process. Therefore, the exploration of new therapeutic
targets is necessary.
Cyclin-dependent kinase 5 (Cdk5) is a unique cell
cycle kinase homolog that exhibits serine/threonine kinase activity
in the brain (3). Complexing with
its activator, p35, Cdk5 is essential for early neurodevelopment in
mammals (4,5). However, endogenous calpain-dependent
cleavage of p35 to p25 is associated with neuron death and
neurodegenerative disease as the product p25 aberrantly activates
Cdk5 and has a long half-life (6).
Studies have found that the Cdk5/p25 complex is involved in
dopaminergic neuron loss in the substantia nigra pars compacta
(SNpc), which is responsible for the symptoms of PD patients and
animal models (7–10). This occurs through the
hyper-phosphorylation of several substrates involved in PD, such as
the neuronal survival factor myocyte enhancer factor 2 (MEF2)
(11), Parkin (10), EndoB1 (9) and Raf kinase inhibitor protein (RKIP)
(10).
Treatments targeted at the modification of Cdk5
activity include Cdk inhibitors such as roscovitine, and calpain
inhibitors such as kenpaullone and induribins (12). However, these drugs lack specificity
and also inhibit normal Cdk5/p35 activity and other Cdks essential
for normal development and function. This may lead to serious side
effects and thereby reduce the clinical value. Addressing this
problem, the use of truncated peptides from p35 that can
specifically inhibit Cdk5/p25 formation and aberrant activity is a
promising approach. A peptide (p10′) encoding the N-terminal domain
of p35 has been shown to prevent the death of neurons exposed to
the neurotoxic 1-methyl-4-phenylpyridinium ion (MPP+),
which induces the conversion of endogenous p35 to p25 (13). Another short peptide [amino acid
residues 154–279; Cdk5 inhibitory peptide (CIP)] derived from p35,
also inhibits Cdk5/p25 activity in neurons without affecting
Cdk5/p35 activity (14,15). To address the problem that p10′ and
CIP are too large for successful therapeutic regimens, a smaller
peptide, p5, spanning CIP residues Lys245-Ala277, was developed and
is able to reduce amyloid β-induced tau hyperphosphorylation and
the apoptosis of cortical neurons without effecting endogenous
Cdk5/p35 activity (16). To
facilitate passage through the blood-brain barrier, p5 peptide has
been modified by conjugating an 11-amino acid peptide derived from
transactivator of transcription (Tat) protein at the C terminus and
attaching fluorescein isothiocyanate (FITC; a green fluorescent
tag) with a linker (GGG) at the N terminus (17). This modified peptide is known as
TFP5, and intraperitoneal injections of TFP5 into mutant mice have
been shown to rescue behavioral and motor deficits in an
Alzheimer's disease (AD) transgenic mouse model (17). Furthermore, TFP5 treatment reduces
Cdk5/p25 hyperactivity significantly and, in turn decreases tau
hyperphosphorylation, phospho-neurofilament proteins, plaques and
gliosis in the brain (18).
Encouraged by the aforementioned successful results
obtained with TFP5, and since Cdk5/p25 plays a critical role in the
nigrostriatal degeneration of PD patients and animal models, the
present study aimed to test the protective function of TFP5 on
MPP+-induced neurotoxicity in primary mouse cortical
neurons and explore the possible therapeutic effect of TFP5 on
PD.
Materials and methods
Reagents
MPP+ was purchased from Sigma-Aldrich
(Merck Millipore, Darmstadt, Germany). TFP5
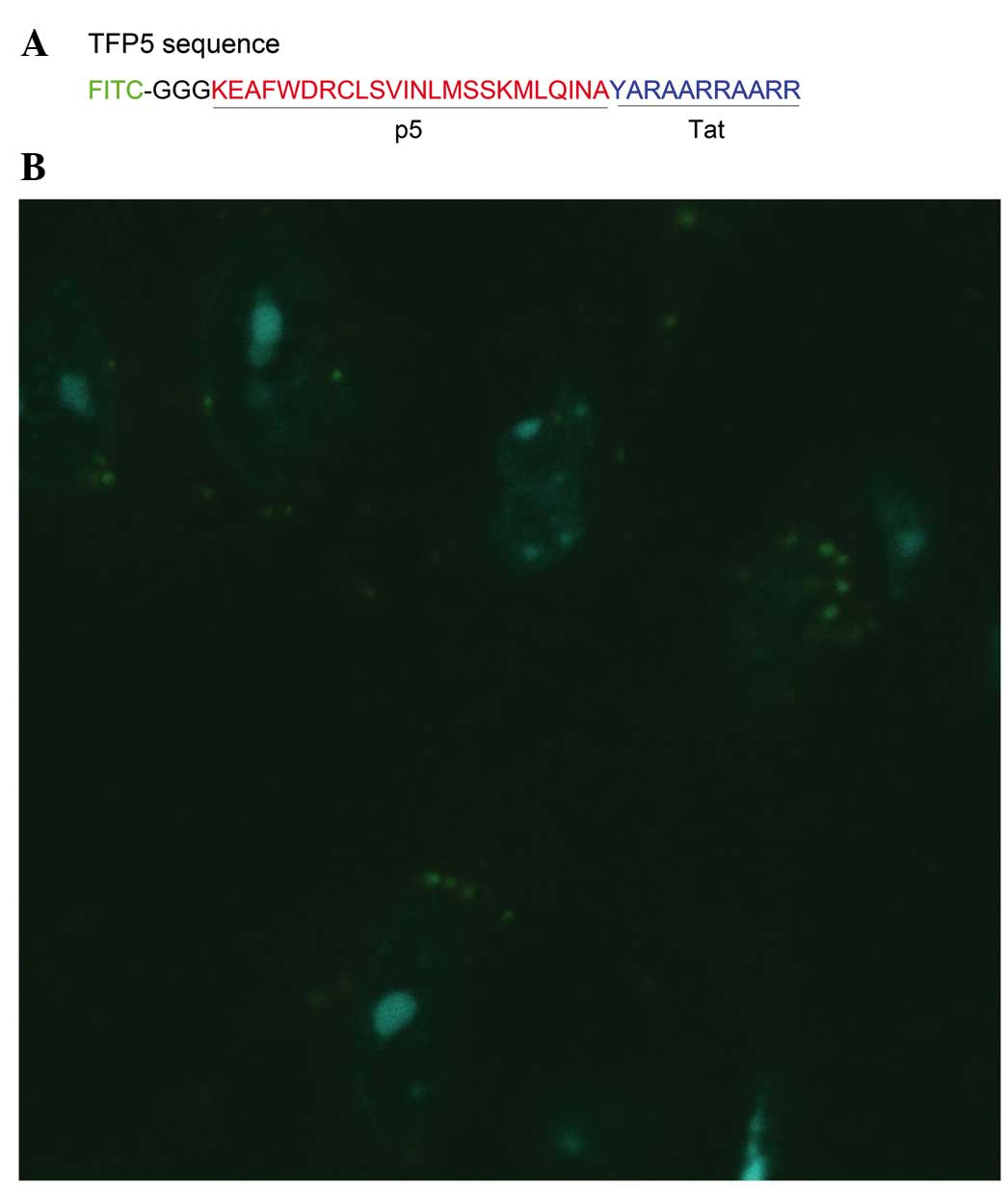
(FITC-GGGKEAFWDRCLSVINLMSSKMLQINAYARAARRAARR) and scrambled (Scb)
TFP5 peptide (Scb peptide;
FITC-GGGGGGFWDRCLSGKGKMSSKGGGINAYARAARRAARR) were synthesized by
Qiangyao Biotechnology Co., Ltd. (Shanghai, China). All drugs were
dissolved in phosphate-buffered saline (PBS) and applied to primary
mice neuron culture.
In vitro PD model establishment and
treatment
Primary cortical neuron cultures were prepared from
C57BL/6 mouse embryos at embryonic day 18 and maintained as
previously described (19). Mice
were purchased from the Laboratory Animal Center of Southern
Medical University (Guangzhou, China) and all animal procedures
were approved by the University Committee on Animal Care of
Southern Medical University [certificate No. SCXK (Yue) 2006–0015]
and conducted in accordance with the Guide for the Care and Use of
Laboratory Animals (8th edition, 2011).
Briefly, forebrains were isolated, the hippocampus
and the midbrain were removed, cortical tissue was obtained and the
meninges and blood vessels on it were stripped. All above
procedures were processed in medium comprising 1X HBSS (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), 10 ml HEPES (pH
7.3) and 10 ml 100X penicillin/streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) placed on ice. The cortical tissue was then cut
into small pieces and incubated with papain at 37°C for 30 min.
Following this, it was digested for 20–30 sec with 200 µl DNA
enzyme I, neutralized with 20 ml 10% glial minimum essential media
(MEM; Hyclone; GE Healthcare Life Sciences, Logan, USA), then
dispersed by repeated aspiration with a Pasteur pipette. Finally,
tissue was homogenized in 10% modified glial MEM (425 ml MEM, 15 ml
20% glucose, 5 ml 100X penicillin/streptomycin and 50 ml horse
serum; Gibco; Thermo Fisher Scientific, Grand Island, NY, USA).
Cells were counted and plated in 10% glial MEM in polylysine-coated
6- and 96-well cell culture plates respectively (5×106
cells per well in 6-well culture plates) for 3 h. The medium was
then replaced and cultures were maintained in neurobasal media
supplemented with B-27, N2 nutritional supplements, 0.5 mM
glutamine and 0.05 mg/ml penicillin/streptomycin (all Invitrogen;
Thermo Fisher Scientific, Inc.). After 48 h, cultures were
subjected to treatment with different concentrations of
MPP+ (0, 50, 100, 200, 400, 800 and 1,600 µM). Cell
viability was evaluated at 24 and 48 h after MPP+
treatment using a Cell Counting kit-8 (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan). From the results, 800 µM
MPP+ was selected for use in subsequent experiments.
In the later experiments, neuron cultures were
divided into four groups: Blank control, model (MPP+),
Scb (Scb + MPP+) and TFP5 (TFP5 + MPP+)
groups. For pretreatment, PBS, PBS, 0.2 mM Scb and 0.2 mM TFP5 was
applied, respectively. At 3 h after the start of pretreatment, 800
µM MPP+ was added to the neurons, with the exception of
those in the blank control group. Neuronal culture was continued
for an additional 24 h. Finally, the viability of the neurons was
determined by CCK8 assay, as described above. All cell toxicity
assays were performed in triplicate more than twice to ascertain
reproducibility.
Cell membrane penetration
detection
In order to observe the cell membrane penetration of
TFP5, 3 h after the application of 0.2 mM TFP5, neurons cultures
(400–500 neurons/mm2) were washed with 1X PBS at room
temperature for 3 min. 1X PBS was removed and 40 ml 4%
paraformaldehyde was added, then the neurons were fixed with
paraformaldehyde at room temperature for 10 min. Following this,
neurons were washed with 1X PBS for 3 min, the nuclei were stained
with 4′,6-diamidino-2-phenylindole and observed with a fluorescence
microscope at 3 h after the application of TFP5.
Western blot analysis
For preparation of whole well extract, neurons were
sonicated in sample buffer (30 mm Tris-HCl, pH 6.8, 5% glycerol,
1.6% sodium dodecyl sulfate and 2.5% β-mercaptoethanol), and
concentrated by centrifugation at 12,000 × g for 20 min at 4°C. The
supernatant was obtained and the concentration of protein was
determined by BCA protein assay (Pierce Biotechnology, Inc.,
Rockford, IL, USA). Samples were then boiled at 95°C for 5 min and
separated by electrophoresis on 12% acrylamide gels, with proteins
subsequently blotted to polyvinylidene fluoride membrane (Immobilon
TM-P; Millipore Corporation, Bedford, MA, USA). The membrane was
then incubated with primary antibodies in Tris-buffered saline and
Tween 20 (TBS-T) buffer containing 3% bovine serum albumin
(Sigma-Aldrich) at 4°C overnight. The following antibodies were
used: Phospho-MEF2D (pSer444) antibody (1:1,000; SAB4503938;
Sigma-Aldrich), p35/25 (C64B10) antibody (1:1,000; 2680S; Cell
Signaling Technology, Inc., Beverly, MA, USA) and β-actin antibody
(1:5,000; SC-130301; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA). The blots were then washed in TBS-T buffer and incubated for
1 h with horseradish peroxidase-labeled goat anti-mouse IgG or goat
anti-rabbit IgG (1:2,000; BA1054; Wuhan Boster Biological
Technology, Ltd., Wuhan, China). Immunoreactive protein was
visualized using an enhanced chemiluminescence method (Amersham ECL
Plus; GE Healthcare Life Sciences, Chalfont, UK). Chemiluminescence
was detected by exposing the membrane in a Kodak in-vivo
Imaging System FX Pro suite from 30 sec to 15 min (Kodak,
Rochester, NY, USA). Protein load was periodically monitored via
the immunodetection of β-actin. Semi-quantitative values were
obtained using Quantity One® software (version 4.62;
Bio-Rad Laboratories, Inc., Hercules, CA, USA). Results are
expressed as arbitrary units of optical density.
Statistical analysis
Data are expressed as means ± standard deviation.
Statistical analysis was performed using SPSS 16.0 statistical
software (SPSS, Inc., Chicago, IL, USA). One-way analysis of
variance (ANOVA) and the Scheffé post-hoc comparison method were
applied to compare the means of groups and P<0.05 was considered
to indicate a statistically significant difference among the
compared groups.
Results
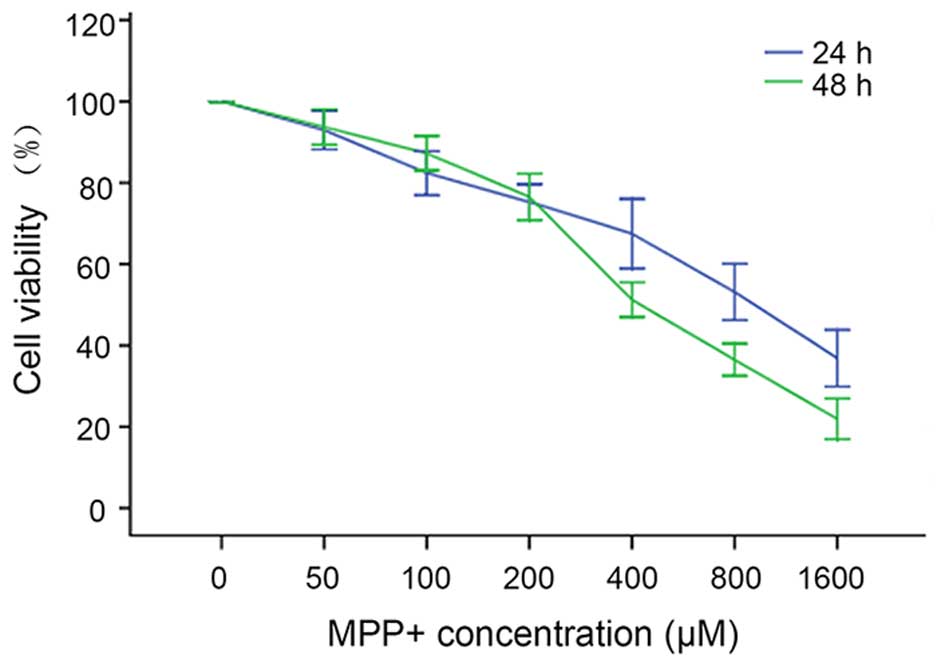
MPP+ induces neurotoxicity
in mouse cortical neurons in a dose- and time-dependent manner
To evaluate the neurotoxicity of different
concentrations of MPP+ in mouse cortical neurons,
various concentrations of MPP+ were applied to primary
mouse cortical neurons cultured in 96-well plates, and after 24 and
48 h a CCK8 assay was employed to evaluate the viability of
neurons. The results show that as the MPP+ concentration
increased from 0 to 1,600 µM and the exposure time was prolonged,
neuronal variability decreased in a dose- and time-dependent manner
(Fig. 1). The viabilities of neurons
treated with 0, 50, 100, 200, 400, 800 and 1,600 µM MPP+
for 24 h were as follows: 99±2, 93±4, 82±5, 75±4, 68±7, 53±6 and
36±6%, respectively. When the treatment time was prolonged to 48 h,
the neuronal viabilities at these concentrations of MPP+
were: 100±0, 93±4, 87±4, 77±5, 51±3, 36±3 and 22±4%, respectively.
One-way ANOVA indicated that the neuronal viability was
significantly different among the groups (P<0.01), and the
median lethal dose (LD50) of MPP+ to mouse
cortical neurons after 24 h exposure was ~800 µM. This
concentration was chosen for establishing the in vitro PD
model.
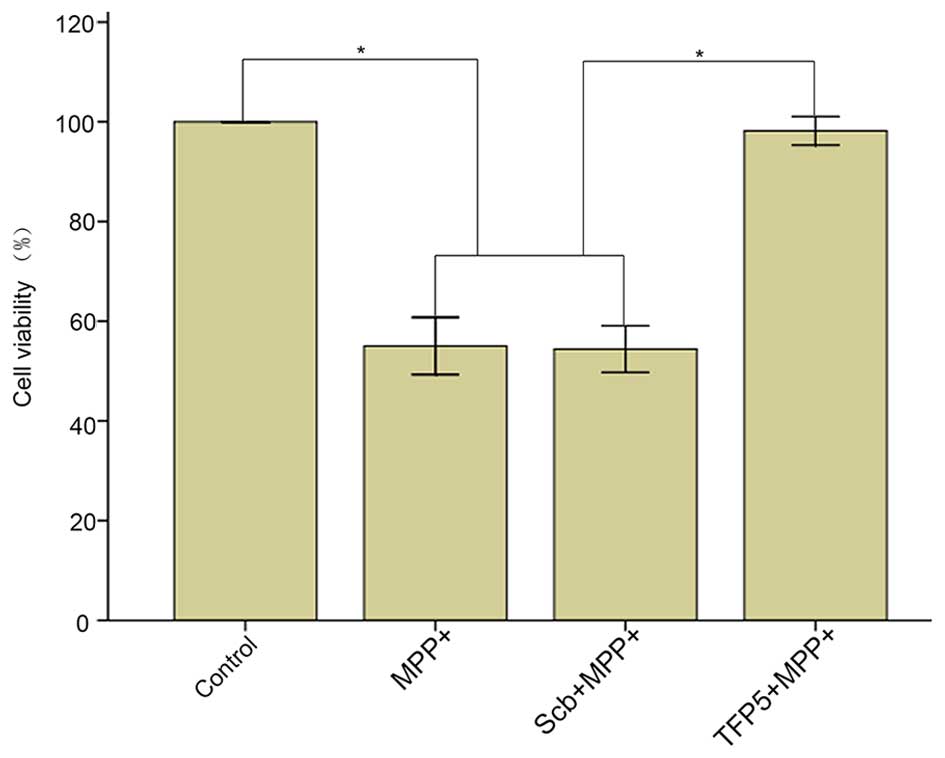
TFP5 suppresses
MPP+-induced neurotoxicity in primary mouse cortical
neurons
The protective function of TFP5 in
MPP+-treated primary mouse cortical neurons was tested.
The results demonstrated that the viability of neurons in the model
group was significantly decreased compared with that in the blank
control group (55±5 vs. 100±0%; P<0.01), and TFP5 (0.2 mm)
significantly inhibited MPP+-induced toxicity compared
with Scb peptide (98±2 vs. 54±4%; P<0.01) and compared with the
model group (98±2 vs. 55±5%; P<0.01; Fig. 2).
TFP5 successfully penetrates through
the cell membrane of primary mouse cortical neurons
Whether TFP5 is able to penetrate through cell
membranes following application to cultured primary neurons was
investigated. The neurons were observed with a fluorescence
microscope 3 h after the application of TFP5. As indicated in
Fig. 3, green fluorescence was
visible in the cytoplasm of neurons. Thus, TFP5, with FITC as a
fused fluorescence label, successfully penetrated through the cell
membranes of primary mouse cortical neurons.
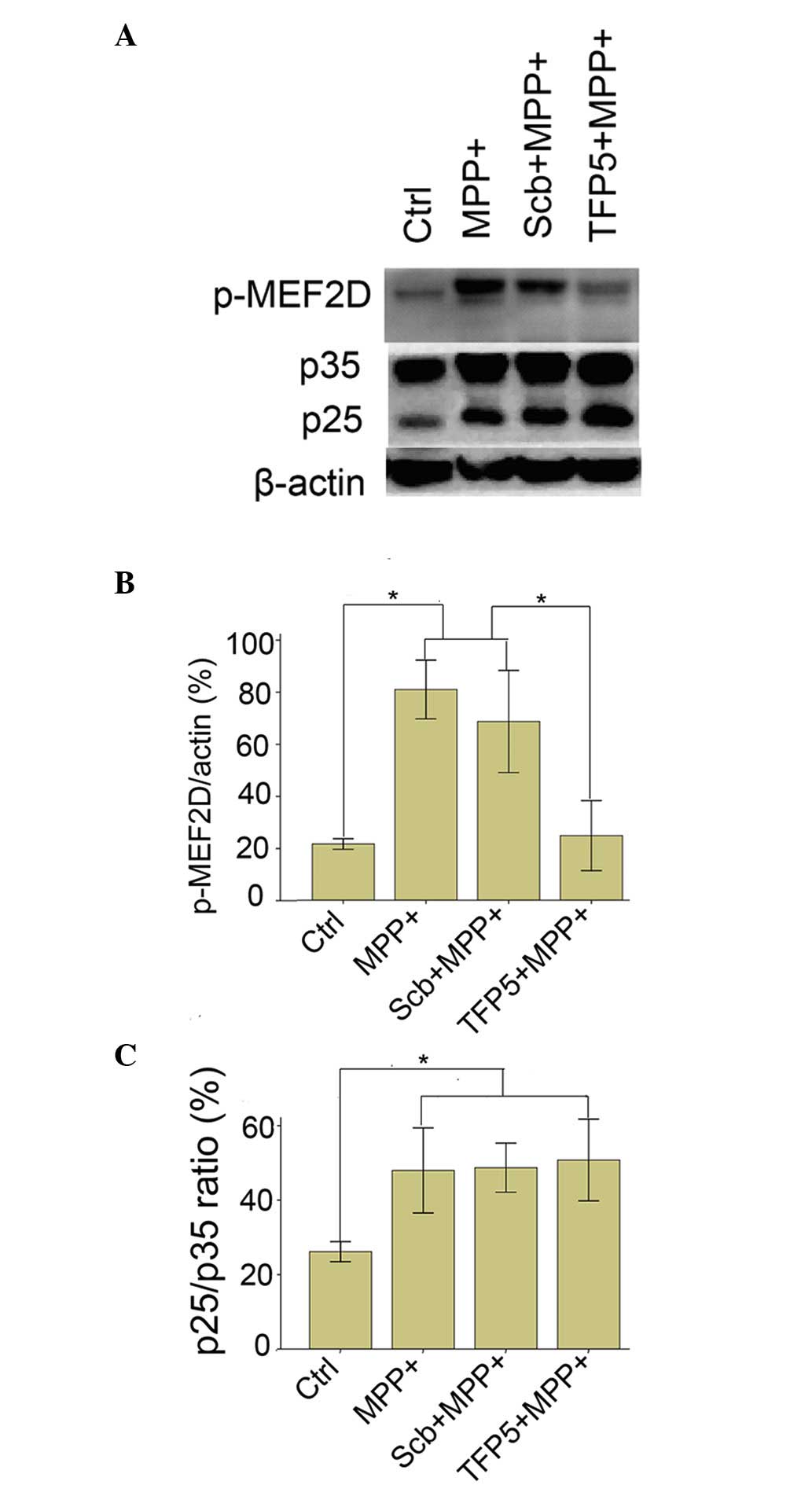
TFP5 inhibits MPP+-induced
hyperphosphorylation of MEF2D in mouse cortical neurons
Cdk5/p25 formation and the subsequent
hyperphosphorylation of MEF2D are involved in the pathology of PD
disease and PD model mice (11). To
investigate the mechanism underlying the protective effect of TFP5
on dopaminergic neuron death in PD, p25 expression and the
phosphorylation status of MEF2D were analyzed in
MPP+-treated mouse cortical neurons. As indicated in
Fig. 4, MPP+ application
induced p35 cleavage in mouse cortical neurons and induced the
phosphorylation of MEF2D (81±10 vs. 22±2%; P<0.01). TFP5 did not
alter MPP+-induced p25 expression, but significantly
inhibited the phosphorylation of MEF2D compared with the model
group (25±12 vs. 81±10%; P<0.01). Neither PBS nor Scb peptide
had any effect on p25 expression or the phosphorylation level of
MEF2D.
Discussion
Cdk5/p25 hyperactivity is involved in the
development of neurodegenerative diseases such as AD and PD
(20,21). The specific Cdk5/p25-inhibiting
peptide p5 has been demonstrated to be protective in AD (22). It has been demonstrated that
MPP+ induces oxidative damage by selectively inhibiting
the activity of mitochondrial complex I in dopaminergic cells, or
cortical neurons when used at a relatively high dose (23); therefore, such treated cells serve as
a well-established PD model (24).
In the present study, the neuroprotective role of a derivative of
the peptide p5, namely TPF5, was further investigated in this
MPP+-induced cellular PD model. The mechanism by which
TFP5 decreases Cdk5 activity is hypothesized to proceed via the
competitive inhibition of formation of the Cdk5/p25 complex, which
in turn inhibits the hyperphosphorylation of MEF2D.
The Cdk5/p25 complex is involved in the loss of
dopaminergic neurons from the SNpc, which is responsible for the
symptoms of PD patients and animal models (7). P5 has been demonstrated to inhibit
Ckd5/p25 interaction and decrease Cdk5 activity in AD (22). Therefore, it may be hypothesized that
p5 could play the same role in a PD model. To facilitate passage
through the blood brain barrier and/or cell membranes, the p5
peptide was used in the modified form TFP5. The results showed that
TFP5 was able to penetrate the cell membrane of neurons and was
distributed around the nucleus. Further in vivo experiments
using an MPTP-induced PD model are now ongoing in the present
authors' laboratory.
Hyperphosphorylation of MEF2D is important in the
pathological pathway of PD (8,13). As a
downstream target of Cdk5/p25, how the phosphorylation status of
MEF2D was affected by the inhibition of Cdk5/p25 activity was
investigated. The results of the present study indicate that TFP5
reduced the hyperphosphorylation of MEF2D and thus may have
prevented neuronal apoptosis.
A limitation of this study is the lack of activity
measurements of Cdk5/p25 due to experimental limitations. However,
the specific inhibitory effect of TFP5 on Cdk5/p25 has been well
documented in the literature (20,25).
Inflammatory factors also play an important role in the pathology
of PD. The ability of TFP5 to interfere with inflammation in PD
requires investigation in further studies. Another focus of future
studies is the protective role of TFP5 in a MPTP-induced mouse
model of PD.
In conclusion, the findings of the present study
provide new information that increases our understanding of the
neuroprotective role of TFP5 and its potential for use in clinical
trials.
Acknowledgements
This study was supported by grants from the Nature
Science Foundation of Guangdong Province (grant no.
S2012010008993), the National Nature Science Foundation of China
(NSFC81271430) and the Scientific Research Foundation of the First
People's Hospital of Chenzhou (CZYY20130009).
References
|
1
|
Rascol O: Drugs and drug delivery in PD:
Optimizing control of symptoms with pramipexole prolonged-release.
Eur J Neurol. 18 Suppl 1:S3–S10. 2011. View Article : Google Scholar
|
|
2
|
Salat D and Tolosa E: Levodopa in the
treatment of Parkinson's disease: Current status and new
developments. J Parkinsons Dis. 3:255–269. 2013.PubMed/NCBI
|
|
3
|
Wood-Kaczmar A, Gandhi S and Wood NW:
Understanding the molecular causes of Parkinson's disease. Trends
Mol Med. 12:521–528. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Su SC and Tsai LH: Cyclin-dependent
kinases in brain development and disease. Annu Rev Cell Dev Biol.
27:465–491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee MS, Kwon YT, Li M, Peng J, Friedlander
RM and Tsai LH: Neurotoxicity induces cleavage of p35 to p25 by
calpain. Nature. 405:360–364. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alvira D, Ferrer I, Gutierrez-Cuesta J,
Garcia-Castro B, Pallàs M and Camins A: Activation of the
calpain/cdk5/p25 pathway in the girus cinguli in Parkinson's
disease. Parkinsonism Relat Disord. 14:309–313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hattori N and Mizuno Y: Pathogenetic
mechanisms of parkin in Parkinson's disease. Lancet. 364:722–724.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith PD, Crocker SJ, Jackson-Lewis V,
Jordan-Sciutto KL, Hayley S, Mount MP, O'Hare MJ, Callaghan S,
Slack RS, Przedborski S, et al: Cyclin-dependent kinase 5 is a
mediator of dopaminergic neuron loss in a mouse model of
Parkinson's disease. Proc Natl Acad Sci USA. 100:13650–16655. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wong AS, Lee RH, Cheung AY, Yeung PK,
Chung SK, Cheung ZH and Ip NY: Cdk5-mediated phosphorylation of
endophilin B1 is required for induced autophagy in models of
Parkinson's disease. Nat Cell Biol. 13:568–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Avraham E, Rott R, Liani E, Szargel R and
Engelender S: Phosphorylation of Parkin by the cyclin-dependent
kinase 5 at the linker region modulates its ubiquitin-ligase
activity and aggregation. J Biol Chem. 282:12842–12850. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Smith PD, Mount MP, Shree R, Callaghan S,
Slack RS, Anisman H, Vincent I, Wang X, Mao Z and Park DS:
Calpain-regulated p35/cdk5 plays a central role in dopaminergic
neuron death through modulation of the transcription factor myocyte
enhancer factor 2. J Neurosci. 26:440–447. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Camins A, Verdaguer E, Folch J, Canudas AM
and Pallàs M: The role of CDK5/P25 formation/inhibition in
neurodegeneration. Drug News Perspect. 19:453–460. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang L, Liu W, Szumlinski KK and Lew J:
p10, the N-terminal domain of p35, protects against
CDK5/p25-induced neurotoxicity. Proc Natl Acad Sci USA.
109:20041–20046. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosales JL and Lee KY: Extraneuronal roles
of cyclin-dependent kinase 5. Bioessays. 28:1023–1034. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lopes JP and Agostinho P: Cdk5:
Multitasking between physiological and pathological conditions.
Prog Neurobiol. 94:49–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Piedrahita D, Hernández I, López-Tobón A,
Fedorov D, Obara B, Manjunath BS, Boudreau RL, Davidson B, Laferla
F, Gallego-Gómez JC, et al: Silencing of CDK5 reduces
neurofibrillary tangles in transgenic alzheimer's mice. J Neurosci.
30:13966–13976. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shukla V, Zheng YL, Mishra SK, Amin ND,
Steiner J, Grant P, Kesavapany S and Pant HC: A truncated peptide
from p35, a Cdk5 activator, prevents Alzheimer's disease phenotypes
in model mice. FASEB J. 27:174–186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gong CX and Iqbal K: Hyperphosphorylation
of microtubule-associated protein tau: A promising therapeutic
target for Alzheimer disease. Curr Med Chem. 15:2321–2328. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu SY, Wu YM, Ji Z, Gao XY and Pan SY: A
modified technique for culturing primary fetal rat cortical
neurons. J Biomed Biotechnol. 2012:8039302012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shukla V, Skuntz S and Pant HC:
Deregulated Cdk5 activity is involved in inducing Alzheimer's
disease. Arch Med Res. 43:655–662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wen Z, Shu Y, Gao C, Wang X, Qi G, Zhang
P, Li M, Shi J and Tian B: CDK5-mediated phosphorylation and
autophagy of RKIP regulate neuronal death in Parkinson's disease.
Neurobiol Aging. 35:2870–2880. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng YL, Amin ND, Hu YF, Rudrabhatla P,
Shukla V, Kanungo J, Kesavapany S, Grant P, Albers W and Pant HC: A
24-residue peptide (p5), derived from p35, the Cdk5 neuronal
activator, specifically inhibits Cdk5-p25 hyperactivity and tau
hyperphosphorylation. J Biol Chem. 285:34202–34212. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Namura I, Douillet P, Sun CJ, Pert A,
Cohen RM and Chiueh CC: MPP+ (1-methyl-4-phenylpyridine)
is a neurotoxin to dopamine-, norepinephrine- and
serotonin-containing neurons. Eur J Pharmacol. 136:31–37. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie HR, Hu LS and Li GY: SH-SY5Y human
neuroblastoma cell line: In vitro cell model of dopaminergic
neurons in Parkinson's disease. Chin Med J (Engl). 123:1086–1092.
2010.PubMed/NCBI
|
|
25
|
Binukumar BK, Zheng YL, Shukla V, Amin ND,
Grant P and Pant HC: TFP5, a peptide derived from p35, a Cdk5
neuronal activator, rescues cortical neurons from glucose toxicity.
J Alzheimers Dis. 39:899–909. 2014.PubMed/NCBI
|