Introduction
Tubulointerstitial fibrosis, which is considered the
final common pathway of progressive kidney diseases and eventually
leads to end-stage renal disease, is a result of the tubular
epithelial-to-mesenchymal transition (EMT) and excessive
accumulation of extracellular matrix (ECM) that characterize the
majority of chronic kidney diseases, including diabetic nephropathy
(DN) (1). Previous studies have
demonstrated that high glucose conditions induce the EMT in renal
proximal tubular cells in vitro and in vivo (2–5). In
addition, it has been reported that myofibroblast formation is a
critical step in the pathogenesis of tubulointerstitial fibrosis,
and it has been shown to be a key step in DN progression (5,6).
Myofibroblasts, which are considered to be one of the principle
effective cells derived from the renal tubular EMT, are responsible
for the ECM and have a central role in progressive renal fibrosis
(7,8). In the process of the EMT, renal
proximal tubular cells have been shown to contribute to renal
interstitial fibrosis; the cells lose their epithelial phenotype
and acquire a myofibroblastic phenotype, which is characterized by
an increased motility, extracellular protein synthesis and
invasiveness (9). Irrespective of
the initial causes, interstitial fibrosis is a remarkable process
that is characterized by de novo activation of the
mesenchymal markers, α-smooth muscle actin (α-SMA) and vimentin,
and the excessive deposition of ECM components in the
tubulointerstitium under pathological conditions by the
myofibroblasts (8,10,11).
Therefore, it is important to investigate the molecular mechanisms
underlying tubulointerstitial fibrosis in order to identify novel
targets for the effective treatment, prevention and delay of
DN.
The EMT is regulated by numerous growth factors and
hormones (12). Transforming growth
factor (TGF)-β1 has been shown to be a potent growth factor that
has a pivotal role in renal fibrogenesis and induces various
biological effects via numerous signal transduction pathways
(8,13,14). At
present, TGF-β1 is recognized as the major cytokine responsible for
the ECM pathology that accompanies DN (15).
The mitogen-activated protein kinase (MAPK)
signaling pathway is one of the most important signal transduction
pathways and is found widely in cells (16). Activation of extracellular
signal-regulated kinase 1/2 (ERK1/2), a downstream signaling
molecule of TGF-β1 and the first member of the MAPK family to be
identified, also has an important role in the progression of
tubular EMT and renal fibrosis (17). Previous studies have demonstrated
that TGF-β1 induces the EMT primarily via the activation of MAPK
and ERK in proximal tubular epithelial cells (18). Notably, the chemical inhibition of
ERK1/2 was able to restrain the EMT process by inhibiting TGF-β1
(18). These findings suggested that
phosphorylated (p)-ERK1/2 blocking therapies may attenuate renal
interstitial fibrosis.
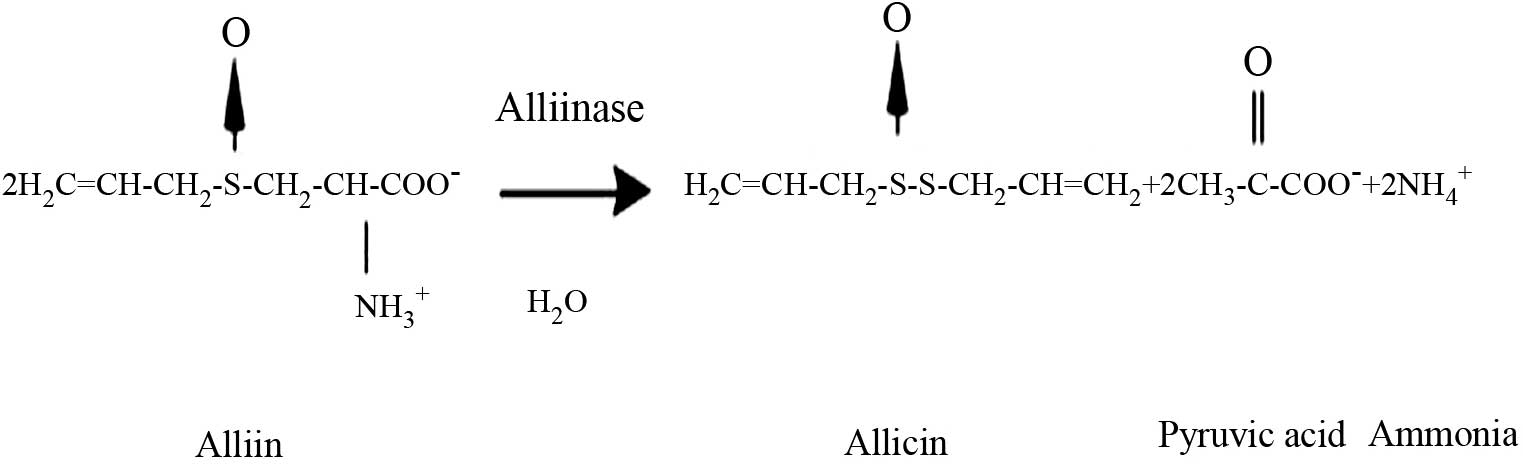
Allicin is one of the most biologically active
compounds of garlic (Allium sativum) (19), and its chemical structure is shown in
Fig. 1. Allicin has a broad spectrum
of physiological activities, including antimicrobial (20,21),
antifungal (22), antioxidant
(23), antihypertensive (24), cardioprotective (25–27),
antiinflammatory (28), anticancer
(29–32) and immunomodulatory (28) properties. Previous studies (33–36) have
demonstrated that allicin may have a role in the prevention of
tissue fibrosis, particularly in the liver, lungs and heart, by
inhibiting fibroblast proliferation, fibrogenic cytokine secretion
and ECM synthesis (37). The present
study aimed to investigate the effects of allicin on high
glucose-induced EMT in human tubule epithelial cells (HK-2) and the
potential underlying mechanisms.
Materials and methods
Reagents and antibodies
Allicin (purity, >88.4%), which is also known as
diallyl thiosulfinate, was purchased from the China National
Institute for Food and Drug Control (Beijing, China). Allicin was
dissolved in serum-free culture medium (Jinuo Bio-Pharmaceutical
Tech. Co. Ltd., Hangzhou, China) and further diluted to the
recommended concentration (2.5, 5, 10 or 20 µg/ml) with culture
medium. The HK-2 normal human renal tubular epithelial cell line
was purchased from the American Type Culture Collection (Manassas,
VA, USA). Human recombinant anti-α-SMA (cat. no. BM0002; dilution
1:200), anti-collagen I (cat. no. PB0980; dilution 1:100) and
anti-ERK1/2 (cat. no. BA1246; dilution 1:500) antibodies were
purchased from Boster Biotechnology Inc. (Wuhan, China). Human
recombinant anti-vimentin (cat. no. SC6260; dilution 1:200),
anti-TGF-β1 (cat. no. SC146; dilution 1:500) and anti-p-ERK1/2
(cat. no. SC16982; dilution 1:500) antibodies were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Human recombinant
anti-E-cadherin antibody was obtained from Epitomics (Burlingame,
CA, USA; cat. no. 1702–1; dilution 1:200). PD98059, a selective
inhibitor of the MAPK/ERK kinase, was purchased from Promega
Corporation (Madison, WI, USA). The mouse anti-β-actin monoclonal
antibody (cat. no. A5441; dilution 1:5,000) and other antibodies
for the western blot analysis were purchased from Sigma-Aldrich
(St. Louis, MO, USA). Fluorescein isothiocyanate (FITC)-conjugated
anti-mouse (cat. no. 70-GAM001; dilution, 1:200), anti-rabbit (cat.
no. 70-GAR001; dilution, 1:200) and anti-goat (cat. no. 70-RAG001;
dilution, 1:200) secondary antibodies were obtained from Liankebio
Biomart, Inc., (Hangzhou, China). Horseradish peroxidase
(HRP)-conjugated anti-mouse (cat. no. ZB-5305; dilution, 1:10,000),
anti-rabbit (cat. no. ZB-5301; dilution, 1:10,000) and anti-goat
(cat. no. ZB-5306; dilution, 1:10,000) secondary antibodies were
obtained from Zhongshan Belling Biotechnology Co., Ltd. (Beijing,
China). DAPI, used for nuclear staining, and RNA Extraction reagent
were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). The PCR primers, PrimeScript™ RT Reagent kit and SYBR Premix
Ex Taq kit were purchased from Takara Bio, Inc. (Otsu, Japan).
Cell culture
HK-2 cells from passages 3 to 5 were used throughout
the studies. Cells were cultured at 37°C under 5% CO2 in
Dulbecco's modified Eagle's medium: Nutrient Mixture F-12
(DMEM/F12; Thermo Fisher Scientific, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Biological Industries
USA, Cromwell, CT, USA), 5.5 mmol/l D-glucose, glutamine and
antibiotics (penicillin and streptomycin). Cells were grown on
6-well plates, on glass coverslips or on 10-cm dishes (Corning Life
Sciences, Tokyo, Japan) to either 100% confluence or subconfluence,
then subjected to various treatments. Briefly, i) in the normal
glucose group, cells were cultured in DMEM supplemented with 5.5
mmol/l D-glucose (normal glucose); ii) in the high glucose group,
cells were cultured in high glucose medium supplemented with 25
mmol/l D-glucose; iii) allicin (at concentrations of 2.5, 5, 10 or
20 µg/ml) was added when the cell culture medium was changed from
normal glucose to high glucose (25 mmol/l) medium; iv) PD98059 (at
concentrations of 20 µmol/l) was added when the cell culture medium
was changed from normal glucose to high glucose (25 mmol/l) medium.
HK-2 cells were passaged when 80% confluent. For experiments,
subconfluent cells were incubated with serum-free medium for 24 h
and divided into four groups, as follows: i) Normal glucose (5.5
mmol/l; control group); ii) high glucose group (25 mmol/l); iii)
high glucose (25 mmol/l) plus allicin (2.5, 5, 10 or 20 µg/ml)
group; and iv) high glucose (25 mmol/l) plus PD98059 (20 µmol/l)
group. Following treatment, the cells were incubated for 48 h prior
to harvesting and further experiments. Each experiment was repeated
at least three times.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the cultured HK-2 cells
using RNA Extraction reagent and RT-qPCR was performed according to
a previous study (38). Briefly,
total RNA (500 ng) was reverse transcribed into cDNA using the
ThermoScript™ RT-PCR System (Thermo Fisher Scientific, Inc.), after
which qPCR was performed using the SYBR Premix Ex Taq kit and PCR
primers on an ABI Prism 7500 thermal cycler (Applied Biosystems;
Thermo Fisher Scientific, Inc.), according to manufacturer's
protocol. The PCR cycling conditions were as follows: 95°C for 5
min, followed by 40 cycles at 95°C for 15 sec, 60°C for 20 sec and
72°C for 20 sec, and a final extension at 72°C for 10 min. The
primer sequences were: TGF-β1 (362 bp) forward,
5′-ACTACGCCAAAGAAGTCACCC-3′ and reverse,
5′-AAGCCCTGTATTCCGTCTCC-3′; and β-actin (317 bp) forward,
5′-CGTACCACTGGCATTGTGAT-3′ and reverse, 5′-TTGCCGATAGTGATGACCTG-3′.
Reaction specificity was confirmed by agarose gel electrophoresis
analysis of PCR products. Ratios for TGF-β1/β-actin mRNA were
calculated for each sample and are expressed as the mean ± standard
error of the mean. Each sample was run in triplicate. The
expression of each gene was normalized against that of β-actin. The
relative quantity of mRNA was calculated using the
2−ΔΔCq method (39).
Western blotting
HK-2 cells were plated in 10-cm culture plates with
or without stimuli and various treatments: HK-2 cells were passaged
until 80% confluent. Subconfluent cells were incubated with
serum-free medium for 24 h and divided into four groups, as
follows: i) Normal glucose group (5.5 mmol/l; control group); ii)
high glucose group (25 mmol/l); iii) high glucose (25 mmol/l) and
allicin (2.5, 5, 10 or 20 µg/ml) group; and iv) high glucose (25
mmol/l) and PD98059 (20 µmol/l) group. Following treatment, the
cells were incubated for 48 h. The cells were then analyzed by
western blotting, as described previously (40). Cells were collected and lysed using
lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1%
Triton, 1% NP-40, 2.5 mM sodium pyrophosphate, 1 mM
β-glycerophosphate, 1 mM leupeptin, 1 mM phenylmethylsulfonyl
fluoride], and samples were centrifuged at 12,000 × g for 30 min at
4°C. The concentration of protein in each cell lysate was
determined using a BCA Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Equal quantities of cell protein lysates (20 µg)
were mixed with 2X sodium dodecyl sulfate (SDS) loading buffer
containing dithiothreitol and heated at 100°C for 10 min, prior to
separation by 10% SDS-PAGE. Subsequently, the proteins were
transferred to a polyvinylidene difluoride membrane and
non-specific binding was blocked with 5% non-fat dry milk in
phosphate-buffered saline containing 0.02% v/v Tween-20. The
membrane was incubated overnight at 4°C with one of the following
primary antibodies: Rabbit anti-p-ERK1/2 (1:500), anti-ERK1/2
(1:500) and anti-TGF-β1 (1:500) polyclonal antibodies, and mouse
anti-β-actin monoclonal antibody (1:5,000). After three washes with
Tris-buffered saline with Tween 20, the membranes were incubated
for 2 h at room temperature with HRP-conjugated anti-rabbit or
anti-mouse IgG (1:10,000). After further washing, the membrane was
detected with ECL chemiluminescence, and band intensities were
quantified by densitometry using Image Lab software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). β-actin was used as a
loading control.
Immunocytochemistry
HK-2 cells in the various groups were analyzed for
tubular EMT using microwave-based two-color immunostaining.
Briefly, cells were fixed in 4% paraformaldehyde and pre-incubated
with 10% FBS and 10% normal goat serum (Bejing Zhongshan Golden
Bridge Biotechnology Co., Ltd., Beijing, China) to block
non-specific binding. Subsequently, the cells were incubated with
rabbit anti-α-SMA, rabbit anti-vimentin, mouse anti-E-cadherin and
mouse anti-collagen I monoclonal antibodies or an isotype control
IgG at 4°C overnight. Following inactivation of endogenous
peroxidase activity, the cells were incubated with HRP-conjugated
goat anti-rabbit or goat anti-mouse IgG, then by rabbit or mouse
anti-peroxidase complexes. Slides were then developed with
3,3′-diaminobenzidine to produce a brown product. Finally, all
sections were counterstained with hematoxylin and mounted on
cover-slips using aqueous mounting medium. All procedures were
performed at room temperature. Brown-yellow granules, as assessed
by light microscopy, were regarded as positive cells. Images were
analyzed using Image-Pro Plus 6.0 image analysis software (Media
Cybernetics, Inc., Rockville, MD, USA). The stained field sections
were then assessed for morphological changes using a light
microscope at ×400 magnification. For all groups, sections were
taken from the same region. An average gray-scale value represented
the measurement value. The gray-scale value of positive protein
expression was determined and statistically analyzed.
Fluorescence immunocytochemistry
HK-2 cells were cultured in DMEM containing 5.5
mmol/l D-glucose. Upon reaching 80% confluence, cells were
synchronized with FBS-free medium (5.5 mmol/l D-glucose) for 24 h,
then cultured with or without various treatments for 48 h.
Subconfluent cells were incubated with serum-free medium for 24 h
and divided into four groups, as follows: i) Normal glucose (5.5
mmol/l; control group); ii) high glucose group (25 mmol/l); iii)
high glucose (25 mmol/l) plus allicin (2.5, 5, 10 or 20 µg/ml)
group; and iv) high glucose (25 mmol/l) plus PD98059 (20 µmol/l)
group. Following treatment, the cells were incubated for 48 h.
Cells were then fixed in 4% paraformaldehyde for 30 min,
permeabilized with 0.1% Triton X-100 for 15 min and incubated with
10% normal goat serum blocking buffer for 1 h at 37°C.
Subsequently, the cells were incubated overnight at 4°C with rabbit
anti-α-SMA (1:200), rabbit anti-vimentin (1:200), mouse
anti-E-cadherin (1:200) and mouse anti-collagen I (1:100)
monoclonal antibodies. Cells were then incubated with
FITC-conjugated secondary antibody (1:200) for 1 h at 37°C in the
dark, then stained with propidium iodide for 1 h. The negative
control consisted of cells incubated with IgG instead of primary
antibody. Cells were visualized and photographed using a laser
scanning confocal microscope (Olympus Corp., Tokyo, Japan). Olympus
FluoView (Olympus Corp.) and Velocity 4.1 (Improvision; Velocity
Software Inc, Mountain View, CA, USA) software were used for image
processing, deconvolution, and quantitative imaging analyses
wherever appropriate. Confocal images acquired under the identical
exposure time and instrument settings among different groups were
used for colocalization and quantitative fluorescence intensity
analyses.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Statistical significance was determined using one-way
analysis of variance followed by Fisher's least significant
difference test. Statistical analyses were performed using SPSS
16.0 software for Windows (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
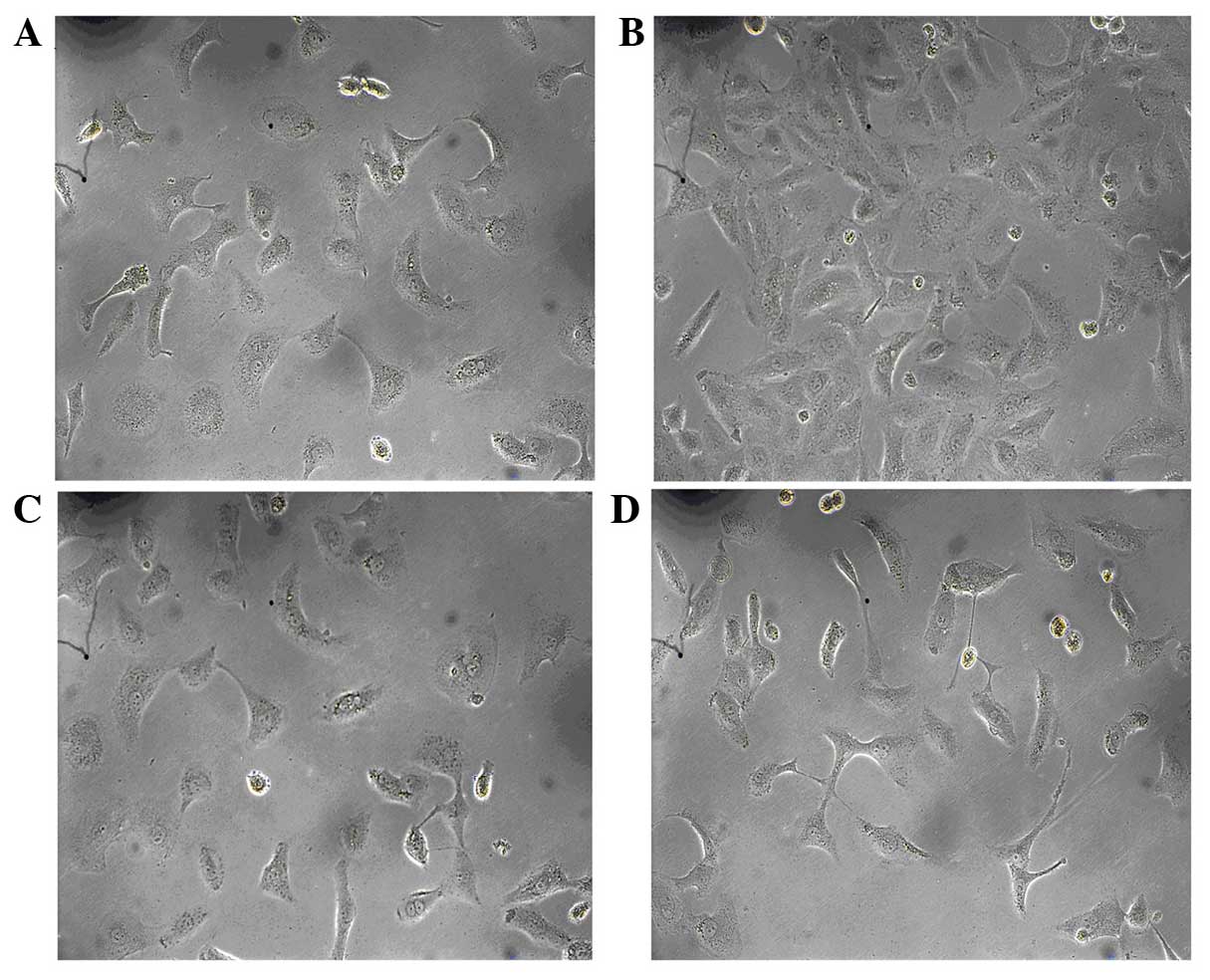
Effect of allicin on high
glucose-induced morphological changes in HK-2 cells
To assess the effect of allicin on cell morphology,
HK-2 cells were serum deprived for 24 h and exposed to high glucose
conditions for 48 h, after which the cells were observed by
inverted phase-contrast microscopy. The normal group had the
typical epithelial cuboidal shape, with a cobblestone morphology
(Fig. 2A). Conversely, cells in the
high glucose group exhibited an elongated, fibroblast-like
phenotype (Fig. 2B). Simultaneous
incubation with allicin (20 µg/ml) or PD98059 (20 µg/ml) prevented
the high glucose-induced morphological changes in the majority of
cells, with cells retaining epithelial polarity and a cobblestone
growth pattern, in the absence of hypertrophy and an elongated
morphology (Fig. 2C and D).
Effect of allicin on the expression
levels of E-cadherin, α-SMA, vimentin and collagen I in HK-2 cells
cultured under high glucose conditions
To confirm the transformation of HK-2 cells into a
fibroblast-like phenotype, the expression levels of the epithelial
marker, E-cadherin, and the mesenchymal markers, α-SMA and
vimentin, were determined by immunohistochemistry and fluorescence
immunocytochemistry. In addition, the expression levels of collagen
I, an important component of the ECM, were also evaluated. The
expression levels of α-SMA, vimentin and collagen I were
significantly increased and peaked at 48 h in the high glucose
group, as compared with the control group (P<0.01; Figs. 3 and 4). Conversely, the expression levels of
E-cadherin were markedly decreased in the high glucose group, as
compared with the control group (P<0.01; Figs. 3 and 4). Allicin reversed the high
glucose-induced changes at 48 h in a dose-dependent manner, with
the difference being significant at 20 µg/ml allicin (P<0.01 vs.
the high glucose group). Upon incubation with PD98059 for 48 h, the
expression levels of α-SMA, vimentin and collagen I were markedly
decreased and those of E-cadherin were markedly increased, as
compared with those of the high glucose cells (P<0.01), although
they were not significantly different from the control cells
(P>0.05).
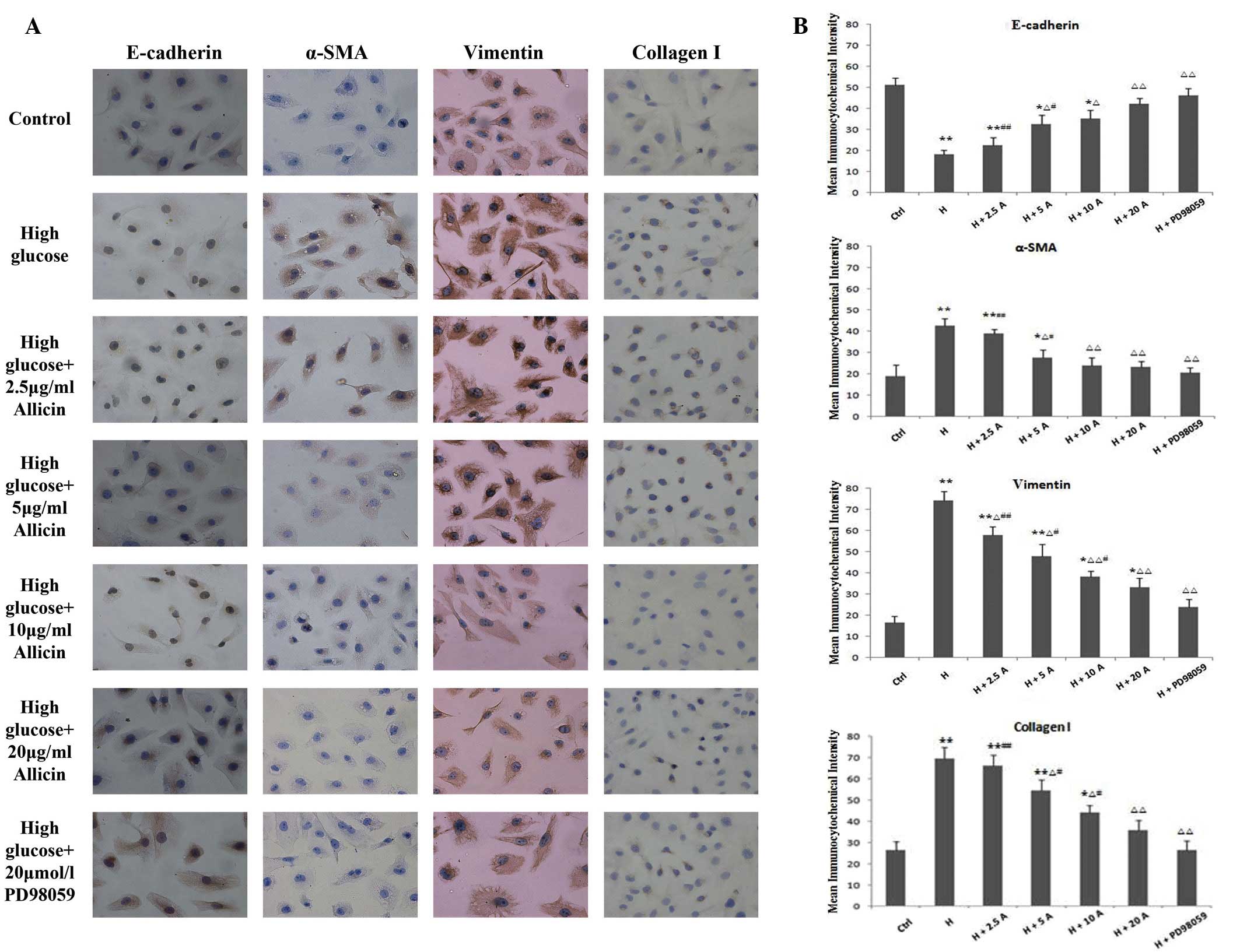 | Figure 3.(A) Immunocytochemical staining of
E-cadherin, α-SMA, vimentin and collagen I in HK-2 cells cultured
for 48 h (magnification, ×200). (B) HK-2 cells were exposed to
normal glucose (ctrl), high glucose, high glucose plus 2.5, 5, 10
and 20 µg/ml allicin or high glucose plus 20 µmol/l PD98059.
*P<0.05 and **P<0.01 vs. Ctrl; ∆P<0.05 and
∆∆P<0.01 vs. H; #P<0.05 and
##P<0.01 vs. H + PD98059. α-SMA, α-smooth muscle
actin; Ctrl, control; H, high glucose; A, allicin. |
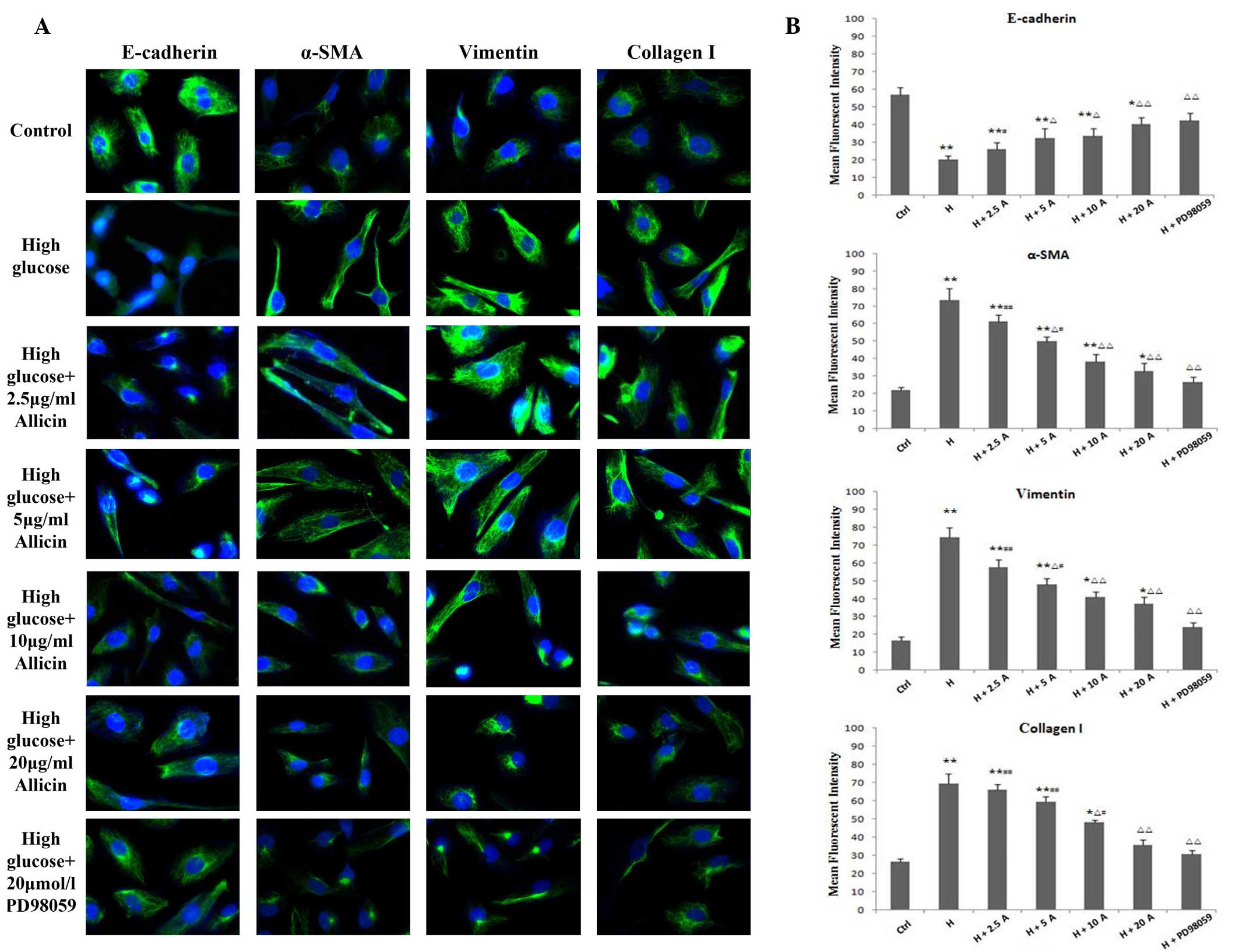 | Figure 4.Expression of E-cadherin, α-SMA,
vimentin and collagen I in HK-2 cells. (A) Fluorescein
isothiocyanate-labeled proteins are shown in green; DAPI-labeled
nuclei are shown in blue. Cells were exposed to normal glucose
(ctrl), high glucose, high glucose plus 5 or 2.5, 5, 10 and 20
µg/ml allicin or high glucose plus 20 µmol/l PD98059 for 48 h, as
detected by fluorescence immunohistochemistry (magnification,
×400). (B) Statistical analysis is also shown. *P<0.05 and
**P<0.01 vs. Ctrl; ∆P<0.05 and
∆∆P<0.01 vs. H; #P<0.05 and
##P<0.01 vs. H + PD98059. α-SMA, α-smooth muscle
actin; Ctrl, control; H, high glucose; A, allicin. |
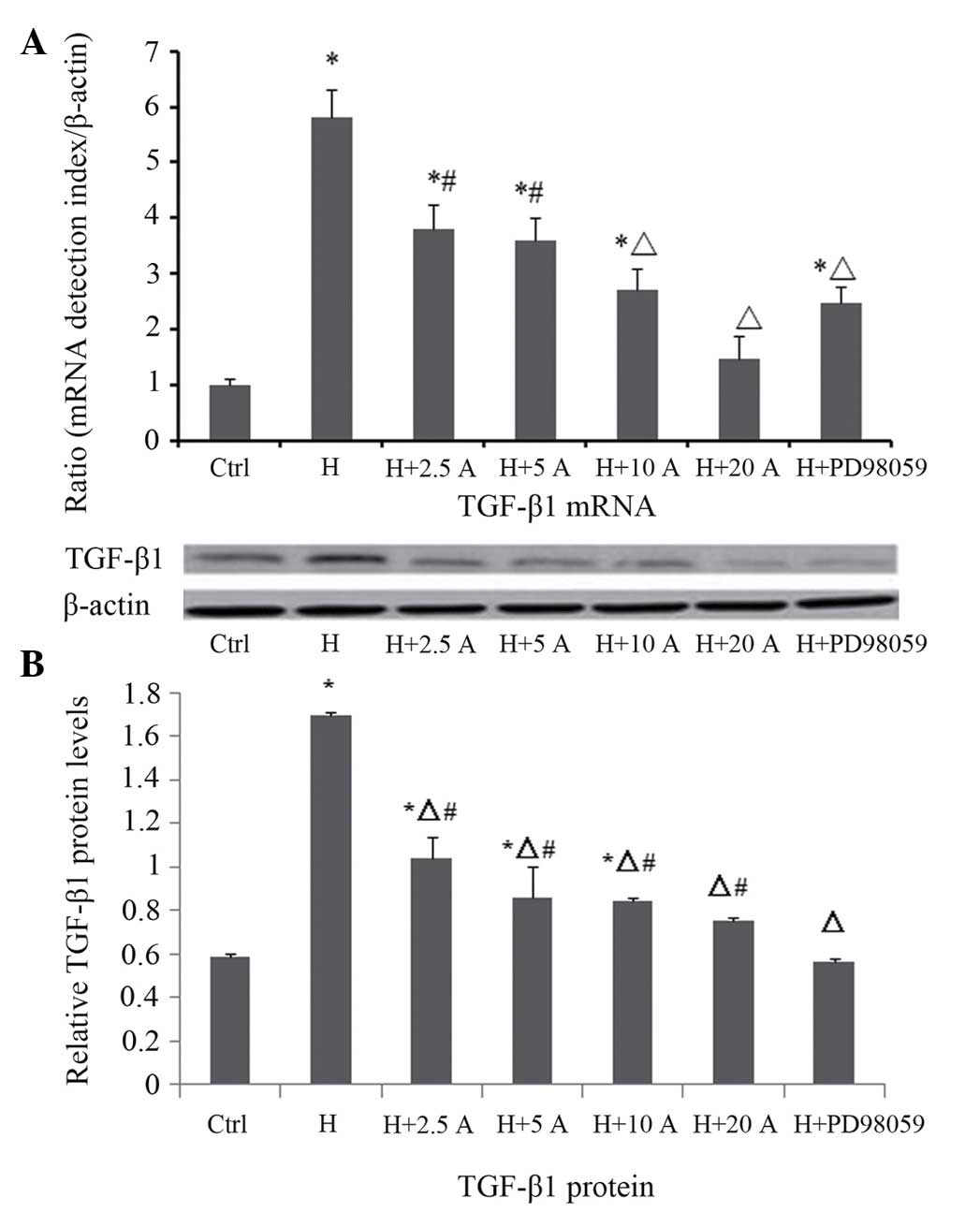
Effect of allicin on the expression
level of TGF-β1 in HK-2 cells cultured under high glucose
conditions
The mRNA and protein expression levels of TGF-β1
were measured by RT-qPCR and western blotting, respectively
(Fig. 5). RT-qPCR demonstrated that
the mRNA expression levels of TGF-β1 were significantly increased
at 48 h in the high glucose group, as compared with the control
group (P<0.05). Allicin treatment resulted in a dose-dependent
decrease in the mRNA expression levels of TGF-β1 at 48 h; in
particular the differences were significant at 10 and 20 µg/ml
allicin (P<0.05 vs. the high glucose group). Upon intervention
with PD98059, the mRNA expression levels of TGF-β1 were
significantly reduced, as compared with the high glucose group
(P<0.05), although they were increased, as compared with the
normal control cells (P<0.05). These results were consistent
with the results of the western blot analysis. The protein
expression levels of TGF-β1 were significantly increased and peaked
at 48 h in the high glucose group, as compared with the control
group (P<0.05). Allicin decreased the protein expression levels
of TGF-β1 at 48 h in a dose-dependent manner, in particular at 20
µg/ml where the inhibition rate was 55.7%, as compared with the
high glucose group (P<0.05). Upon intervention with PD98059, the
protein expression levels of TGF-β1 were also significantly
reduced, as compared with the high glucose group (P<0.05), and
were not significantly different, as compared with the control
group (P>0.05).
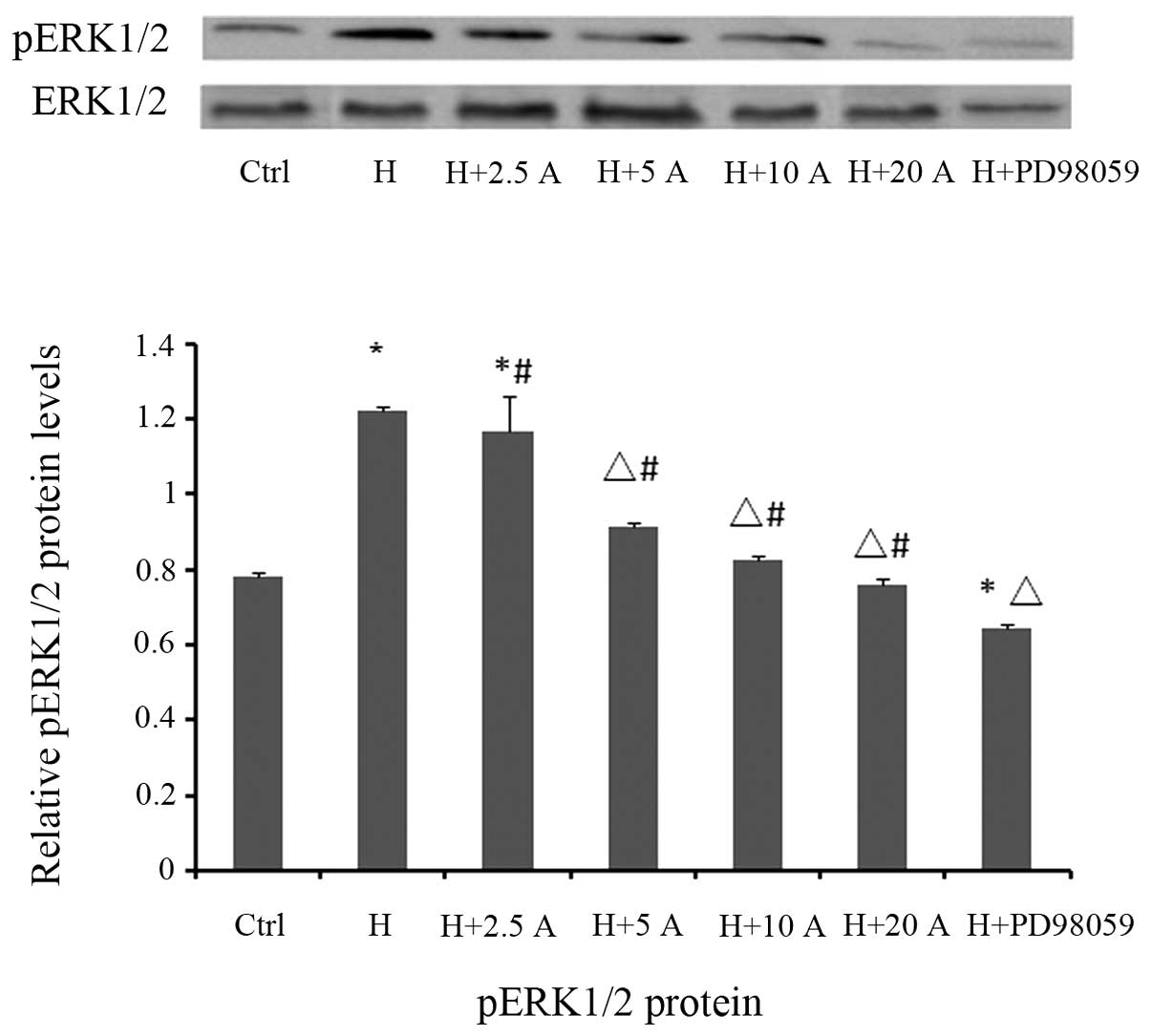
Effect of allicin on p-ERK1/2
expression in HK-2 cells cultured under high glucose
conditions
To further elucidate the molecular mechanisms
underlying the allicin-mediated inhibition of the EMT process in
HK-2 cells cultured under high glucose conditions, the potential
involvement of the ERK1/2 signaling pathway was investigated by
western blotting. The levels of p-ERK1/2 were significantly
increased in the high glucose group at 48 h, as compared with the
control group (P<0.05; Fig. 6).
However, high glucose-induced ERK1/2 phosphorylation was
significantly attenuated by pre-treatment with PD98059 (P<0.05;
Fig. 6), a specific inhibitor of the
MAPK/ERK kinase, which is the upstream activator of ERK1/2.
Similarly, treatment with allicin significantly decreased the
protein expression levels of p-ERK1/2 in a dose-dependent manner,
in particular at 20 µg/ml, with inhibition rates of 37.7%, as
compared with the high glucose group (P<0.05). However, the
levels were significantly increased, as compared with the control
group (P<0.05).
Discussion
The EMT is a key process in tissue development,
carcinogenesis and organ fibrosis (41). In addition, it has emerged as a
central mechanism underlying tubulointerstitial fibrosis, a
progressive pathology common to numerous chronic kidney diseases,
including DN (8,42). Interstitial myofibroblasts have a
critical role in the development of tubulointerstitial fibrosis in
diabetic and non-diabetic kidney diseases (43). A large proportion of interstitial
myofibroblasts originate from transformed tubular epithelial cells
experiencing pathological conditions during renal fibrogenesis
(44). Interstitial fibrosis is
characterized by de novo activation of α-SMA and
vimentin-positive myofibroblasts (45). Collagen I is a key component of the
ECM. In the process of EMT, tubular epithelial cells acquire the
myofibroblast marker α-SMA and vimentin, display a myofibroblastic
morphology and secrete interstitial matrix components such as
collagen I and fibronectin (8).
Previous studies have demonstrated that selective blockade of
tubular EMT may protect the kidneys from developing fibrotic
lesions following obstructive injury (46), and that tubular EMT has an important
role in renal tubulointerstitial fibrosis (8,10,47).
Therefore, a potentially effective therapeutic strategy for
progressive renal fibrosis may involve the prevention of tubular
EMT in the diseased kidney.
TGF-β1 is a key mediator responsible for
transdifferentiation in vivo and in vitro (7,46,48,49).
It has previously been shown that TGF-β1 has an important role in
altering the phenotype of renal epithelial cells, and that this
significantly contributes to the profibrotic effects (50). Previous studies have demonstrated
that advanced glycation end products, which accumulate in the
diabetic kidney, are powerful mediators of EMT (51), and act via TGF-β1-dependent pathways
involving various intracellular signaling molecules, including Smad
and MAPK, in response to high glucose conditions (51,52). Our
previous study reported overexpression of TGF-β1 during the EMT of
renal tubular epithelial cells in a diabetic rat model (53). In the present study, it was shown
that high glucose concentrations induced the EMT of HK-2 cells and
significantly increased the expression levels of TGF-β1 and
collagen I. These results suggested that the tubular EMT and
increased ECM synthesis induced by hyperglycemia may at least
partly depend on TGF-β1, while increased TGF-β1 secretion following
transdifferentiation may form a positive feedback loop. Therefore,
TGF-β1 may represent an additional key component of the pathway
leading to EMT.
Activation of the Smad and/or MAPK signaling
pathways is required for TGF-β1-induced EMT (54). In addition, phosphorylation of
ERK1/2, a downstream signaling molecule of TGF-β1, is required for
an optimal response to TGF-β1 (55).
Rhyu et al (18) reported
that PD98059, a specific inhibitor of the MAPK/ERK kinase signaling
pathway, was able to effectively inhibit the TGF-β1-induced EMT
process in NRK52E cells. In the present study, high glucose
conditions activated the ERK1/2/MAPK and TGF-β signaling pathways
in the process of EMT. Previous studies have demonstrated that high
glucose induces ERK1/2 phosphorylation in vitro and in
vivo (56,57). Furthermore, high glucose-mediated
activation of p-ERK1/2 and high-glucose induced EMT were shown to
be blocked by PD98059 (58). These
results suggested that blockade of high glucose-mediated activation
of the ERK1/2/MAPK signaling pathway was able to inhibit the EMT, a
critical process in renal tubulointerstitial fibrosis (59). Further studies are required in order
to validate that high glucose mediates EMT via the ERK/MAPK and
TGF-β1 signaling pathways, which are involved in numerous
intracellular processes.
Allicin, which is a major active component isolated
from garlic, has been used as a popular folk medicine for thousands
of years (60). Allicin has
previously been shown to inhibit fibroblast proliferation and
collagen synthesis by regulating the TGF-β signaling pathway, and
inhibited myocardial fibrosis caused by abdominal aortic
coarctation via its inhibition of myocardial collagen (33,34). In
a rat model of liver fibrosis, allicin was able to significantly
inhibit the transdifferentiation of stellate cells to
myofibroblasts via the downregulation of TGF-β1 expression
(35). Zhang et al (36) demonstrated that allicin significantly
attenuated the development of myocardial fibrosis and exerted
significant anti-proliferative effects in rabbit arterial smooth
muscle cells induced by angiotensin II in a dose- and
time-dependent manner. These findings suggested that allicin may
have a role in the prevention of tissue fibrosis. However, whether
allicin has a role in preventing renal fibrosis remains
unknown.
The present study demonstrated that allicin was able
to block the EMT and decrease the expression levels of collagen I
in HK-2 cells cultured under high glucose conditions. Notably, 25
mM glucose was able to induce the transdifferentiation of tubular
cells into myofibroblasts that showed fibroblast-like morphologies,
a loss of E-cadherin epithelial marker expression and α-SMA and
vimentin positivity. In addition, collagen I expression was shown
to be increased in high glucose-induced HK-2 cells, which indicated
that the transformed cells had begun to produce components of the
ECM. Importantly, allicin treatment increased the expression of
E-cadherin, prevented the de novo expression of α-SMA and
vimentin, and reduced collagen I expression in a dose-dependent
manner. Furthermore, the present study demonstrated that
simultaneous incubation of HK-2 cells with allicin markedly
decreased the expression of p-ERK1/2 at 48 h in a dose-dependent
manner, in particular at 20 µg/ml. In addition, allicin reduced the
expression of TGF-β1, potentially by inhibiting the high
glucose-mediated activation of the ERK1/2 signaling pathway,
thereby inhibiting HK-2 cell morphological changes, the EMT and ECM
synthesis, and resulting in the attenuation of tubular
fibrosis.
In conclusion, the present study demonstrated that
high glucose concentrations induced the EMT of renal tubule
epithelial cells, and this was associated with upregulation of
TGF-β1 and collagen I. In addition, it was shown that the MAPK
inhibitor PD98059 was able to reverse high glucose-induced
transdifferentiation of HK-2 cells by inhibiting the expression of
p-ERK1/2 and TGF-β1. These results suggested that TGF-β1 is an
important regulator of the EMT and that ERK1/2 signaling pathway
may be involved in renal interstitial fibrosis associated with DN.
Furthermore, allicin treatment restrained the EMT and prevented
subsequent interstitial matrix accumulation in vitro.
However, further studies are required in order to clarify the
effects of allicin on renal fibrosis.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81270924) and the
Major Science Technology Program of Zhejiang Province (grant no.
2009C03010-4).
References
|
1
|
Liu Y: Renal fibrosis: New insights into
the pathogenesis and therapeutics. Kidney Int. 69:213–217. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee JH, Kim JH, Kim JS, Chang JW, Kim SB,
Park JS and Lee SK: AMP-activated protein kinase inhibits TGF-β-,
angiotensin II-, aldosterone-, high glucose-, and albumin-induced
epithelial-mesenchymal transition. Am J Physiol Renal Physiol.
304:F686–F697. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang WC, Liu SF, Chang WT, Shiue YL, Hsieh
PF, Hung TJ, Hung CY, Hung YJ, Chen MF and Yang YL: The effects of
diosgenin in the regulation of renal proximal tubular fibrosis. Exp
Cell Res. 323:255–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei J, Shi Y, Hou Y, Ren Y, Du C, Zhang L,
Li Y and Duan H: Knockdown of thioredoxin-interacting protein
ameliorates high glucose-induced epithelial to mesenchymal
transition in renal tubular epithelial cells. Cell Signal.
25:2788–2796. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ban CR and Twigg SM: Fibrosis in diabetes
complications: Pathogenic mechanisms and circulating and urinary
markers. Vasc Health Risk Manag. 4:575–596. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Simonson MS: Phenotypic transitions and
fibrosis in diabetic nephropathy. Kidney Int. 71:846–854. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Burns WC and Thomas MC: The molecular
mediators of type 2 epithelial to mesenchymal transition (EMT) and
their role in renal pathophysiology. Expert Rev Mol Med.
12:e172010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Y: Epithelial to mesenchymal
transition in renal fibrogenesis: Pathologic significance,
molecular mechanism, and therapeutic intervention. J Am Soc
Nephrol. 15:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rastaldi MP, Ferrario F, Giardino L,
Dell'Antonio G, Grillo C, Grillo P, Strutz F, Müller GA, Colasanti
G and D'Amico G: Epithelial-mesenchymal transition of tubular
epithelial cells in human renal biopsies. Kidney Int. 62:137–146.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Y and Yang J: Hepatocyte growth
factor: New arsenal in the fights against renal fibrosis? Kidney
Int. 70:238–240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Y, Zhang J, Fang L, Luo P, Peng J and
Du X: Lefty A attenuates the TGF-beta1-induced epithelial to
mesenchymal transition of human renal proximal epithelial tubular
cells. Mol Cell Biochem. 339:263–270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W, Koka V and Lan HY: Transforming
growth factor-beta and Smad signalling in kidney diseases.
Nephrology (Carlton). 10:48–56. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
El Mesallamy HO, Ahmed HH, Bassyouni AA
and Ahmed AS: Clinical significance of inflammatory and fibrogenic
cytokines in diabetic nephropathy. Clin Biochem. 45:646–650. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang W and Liu H: MAPK signal pathways in
the regulation of cell proliferation in mammalian cells. Cell
Research. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jang HS, Han SJ, Kim JI, Lee S, Lipschutz
JH and Park KM: Activation of ERK accelerates repair of renal
tubular epithelial cells, whereas it inhibits progression of
fibrosis following ischemia/reperfusion injury. Biochim Biophys
Acta. 1832:1998–2008. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh
ST and Lee HB: Role of reactive oxygen species in TGF-beta1-induced
mitogen-activated protein kinase activation and
epithelial-mesenchymal transition in renal tubular epithelial
cells. J Am Soc Nephrol. 16:667–675. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ali M, Thomson M and Afzal M: Garlic and
onions: Their effect on eicosanoid metabolism and its clinical
relevance. Prostaglandins Leukot Essent Fat Acids. 62:55–73. 2000.
View Article : Google Scholar
|
|
20
|
Cutler RR and Wilson P: Antibacterial
activity of a new, stable, aqueous extract of allicin against
methicillin-resistant staphylococcus aureus. Br J Biomed Sci.
61:71–74. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goncagul G and Ayaz E: Antimicrobial
effect of garlic (Allium sativum). Recent Pat Antiinfect Drug
Discov. 5:91–93. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Davis SR: An overview of the antifungal
properties of allicin and its breakdown products-the possibility of
a safe and effective antifungal prophylactic. Mycoses. 48:95–100.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ramoutar RR and Brumaghim JL: Antioxidant
and anticancer properties and mechanisms of inorganic selenium,
oxo-sulfur and oxo-selenium compounds. Cell Biochem Biophys.
58:1–23. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Elkayam A, Mirelman D, Peleg E, Wilchek M,
Miron T, Rabinkov A, Sadetzki S and Rosenthal T: The effects of
allicin and enalapril in fructose-induced hyperinsulinemic
hyperlipidemic hypertensive rats. Am J Hypertens. 14:377–381. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eilat S, Oestraicher Y, Rabinkov A, Ohad
D, Mirelman D, Battler A, Eldar M and Vered Z: Alteration of lipid
profile in hyperlipidemic rabbits by allicin, an active constituent
of garlic. Coron Artery Dis. 6:985–990. 1995.PubMed/NCBI
|
|
26
|
Abramovitz D, Gavri S, Harats D, Levkovitz
H, Mirelman D, Miron T, Eilat-Adar S, Rabinkov A, Wilchek M, Eldar
M and Vered Z: Allicin-induced decrease in formation of fatty
streaks (atherosclerosis) in mice fed a cholesterol-rich diet.
Coron Artery Dis. 10:515–519. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gonen A, Harats D, Rabinkov A, Miron T,
Mirelman D, Wilchek M, Weiner L, Ulman E, Levkovitz H, Ben-Shushan
D and Shaish A: The antiatherogenic effect of allicin: Possible
mode of action. Pathobiology. 72:325–334. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lang A, Lahav M, Sakhnini E, Barshack I,
Fidder HH, Avidan B, Bardan E, Hershkoviz R, Bar-Meir S and Chowers
Y: Allicin inhibits spontaneous and TNF-alpha induced secretion of
proinflammatory cytokines and chemokines from intestinal epithelial
cells. Clin Nutr. 23:1199–1208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Antony ML and Singh SV: Molecular
mechanisms and targets of cancer chemoprevention by garlic-derived
bioactive compound diallyl trisulfide. Indian J Exp Biol.
49:805–816. 2011.PubMed/NCBI
|
|
30
|
Nagini S: Cancer chemoprevention by garlic
and its organosulfur compounds-panacea or promise? Anticancer
Agents Med Chem. 8:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park BJ, Cho SJ, Kwon HC, Lee KR, Rhee DK
and Pyo S: Caspase independent cell death by allicin in human
epithelial carcinoma cells: Involvement of PKA. Cancer Lett.
224:123–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oommen S, Anto RJ, Srinivas G and
Karunagaran D: Allicin (from garlic) induces caspase-mediated
apoptosis in cancer cells. Eur J Pharmacol. 485:97–103. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang HX, Shi ZX and Jia HZ: Effect of
allicin on NIH3T3 cells on the proliferation and collagen
synthesis. Zhong Guo Zhong Xi Yi Jie He Za Zhi. 27:431–434.
2007.(In Chinese).
|
|
34
|
Zhang HX, Jia HZ and Li G: Effect of
allicin on myocardial fibrosis in rats with pressure overload.
Zhongguo Zhong Yi Ji Chu Yi Xue. 14:149–151. 2008.(In Chinese).
|
|
35
|
Zhu LX, Cheng WC and Liu SZ: Effect of
allicin on experimental hepatic fibrosis in rats. Chin J Dig Dis.
7:441–443. 2003.
|
|
36
|
Zhang DX, Ren YS and Liu B: Effect of
allicin on angiotensin II-induced vascular smooth muscle cell
proliferation. Zhong Guo Xiandai Yi Xue Za Zhi. 15:2136–2138.
2005.(In Chinese).
|
|
37
|
Zhu LX, Chen WC, Liu SZ and Gu ZL: Effect
of allicin on experimental liver fibrosis in rats. Chin J Digest.
23:441–443. 2003.(In Chinese).
|
|
38
|
Xu C, Ding W, Zhang M and Gu Y: Protective
effects of angiotensin-(1–7) administered with an
angiotensin-receptor blocker in a rat model of chronic kidney
disease. Nephrology (Carlton). 18:761–769. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu J, Ma KL, Zhang Y, Wu Y, Hu ZB, Lv LL,
Tang RN, Liu H, Ruan XZ and Liu BC: Activation of mTORC1 disrupted
LDL receptor pathway: A potential new mechanism for the progression
of non-alcoholic fatty liver disease. Int J Biochem Cell Biol.
61:8–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development and disease. J Cell Biol. 172:973–981. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Strutz F, Okada H, Lo CW, Danoff T, Carone
RL, Tomaszewski JE and Neilson EG: Identification and
characterization of a fibroblast marker: FSP1. J Cell Biol.
130:393–405. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Barnes JL and Glass WF: Renal Interstitial
Fibrosis: A Critical Evaluation of the Origin of Myofibroblasts.
Contrib Nephrol. 169:73–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Iwano M, Plieth D, Danoff TM, Xue C, Okada
H and Neilson EG: Evidence that fibroblasts derive from epithelium
during tissue fibrosis. J Clin Invest. 110:341–350. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li MX and Liu BC: Epithelial to
mesenchymal transition in the progression of tubulointerstitial
fibrosis. Chin Med J (Engl). 120:1925–1930. 2007.PubMed/NCBI
|
|
46
|
Yang J and Liu Y: Blockage of tubular
epithelial to myofibroblast transition by hepatocyte growth factor
prevents renal interstitial fibrosis. J Am Soc Nephrol. 13:96–107.
2002.PubMed/NCBI
|
|
47
|
Burns WC, Kantharidis P and Thomas MC: The
role of tubular epithelial-mesenchymal transition in progressive
kidney disease. Cells Tissues Organs. 185:222–231. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fan JM, Huang XR, Ng YY, Nikolic-Paterson
DJ, Mu W, Atkins RC and Lan HY: Interleukin-1 induces tubular
epithelial- myofibroblast transdifferentiation through a
transforming growth factor-beta1-dependent mechanism in vitro. Am J
Kidney Dis. 37:820–831. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jinde K, Nikolic-Paterson DJ, Huang XR,
Sakai H, Kurokawa K, Atkins RC and Lan HY: Tubular phenotypic
change in progressive tubulointerstitial fibrosis in human
glomerulonephritis. Am J Kidney Dis. 38:761–769. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lan HY: Tubular epithelial-myofibroblast
transdifferentiation mechanisms in proximal tubule cells. Curr Opin
Nephrol Hypertens. 12:25–29. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Oldfield MD, Bach LA, Forbes JM,
Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T,
Jerums G and Cooper ME: Advanced glycation end products cause
epithelial-myofibroblast transdifferentiation via the receptor for
advanced glycation end products (RAGE). J Clin Invest.
108:1853–1863. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li JH, Wang W, Huang XR, Oldfield M,
Schmidt AM, Cooper ME and Lan HY: Advanced glycation end products
induce tubular epithelial-myofibroblast transition through the
RAGE-ERK1/2 MAP kinase signaling pathway. Am J Pathol.
164:1389–1397. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ye X, Li H and Zhang JH: Phenotypic
conversion of renal cortex cells in streptozotocin-induced diabetic
rats. Zhong Guo Bing Li Sheng Li Za Zhi. 23:1645–1647. 2007.(In
Chinese).
|
|
54
|
Liu RY, Zeng YY, Lei Z, Wang LQ, Yang HP,
Liu ZY, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 2:1643–1651. 2014.
|
|
55
|
Nakerakanti S and Trojanowska M: The role
of TGF-β receptors in fibrosis. Open Rheumat J. 6:156–162. 2012.
View Article : Google Scholar
|
|
56
|
Zhou L, Xue H, Yuan P, Ni J, Yu C, Huang Y
and Lu LM: Angiotensin AT1 receptor activation mediates high
glucose-induced epithelial-mesenchymal transition in renal proximal
tubular cells. Clin Exp Pharmacol Physiol. 37:e152–e157. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cheng X, Gao W, Dang Y, Liu X, Li Y, Peng
X and Ye X: Both ERK/MAPK and TGF-Beta/Smad signaling pathways play
a role in the kidney fibrosis of diabetic mice accelerated by blood
glucose fluctuation. J Diabetes Res. 2013:4637402013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dai B, Cui M, Zhu M, Su WL, Qiu MC and
Zhang H: STAT1/3 and ERK1/2 synergistically regulate cardiac
fibrosis induced by high glucose. Cell Physiol Biochem. 32:960–971.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nakasatomi M, Maeshima A, Mishima K,
Ikeuchi H, Sakairi T, Kaneko Y, Hiromura K and Nojima Y: Novel
approach for the detection of tubular cell migration into the
interstitium during renal fibrosis in rats. Fibrogenesis Tissue
Repair. 8:122015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Block E: The chemistry of garlic and
onions. Sci Am. 252:114–119. 1985. View Article : Google Scholar : PubMed/NCBI
|