Introduction
Ischemia/reperfusion (I/R) injury has previously
been demonstrated to be the cause of debilitating complications and
mortality associated with stroke, myocardial infarction and
traumatic brain injury (1). I/R
injury in cerebral vessels is characterized as an early secondary
injury leading to inflammation and edema (1). Cerebral hypoxia and ischemia can lead
to vascular cell damage, which manifests as impairments in
autoregulation and vascular reactivity and endothelial cell
apoptosis (2). Morbidity from
reperfusion and post-ischemia damage remains common, despite
improvements in imaging, interventional techniques and
pharmacological agents. Cerebral I/R injury activates various
distinct, yet overlapping, cell signaling pathways, which lead to
vascular dysfunction characterized by endothelial cell injury or
apoptosis (2).
A large proportion (>80%) of the human genome is
transcribed; however, only <2% is subsequently translated into
proteins, suggesting that the vast majority of DNA sequences are
transcribed as non-protein coding RNAs (3). A novel class of noncoding RNAs, which
are >200 nucleotides and termed long noncoding RNAs (lncRNAs),
have been characterized (4,5). lncRNAs are localized in the nucleus or
cytoplasm and are involved in the activation and inhibition of gene
expression through epigenetic mechanisms, including chromatin
remodeling, regulation of splicing, and by acting as sponges for
microRNAs (6,7). A recent study showed significant
changes in the expression profiles of lncRNA after oxygen-glucose
deprivation (OGD), suggesting a potential pathological role of
lncRNAs in mediating endothelial responses to ischemic stimuli
(8).
Metastasis-associated lung adenocarcinoma transcript
1 (MALAT1), which is a 6.5-Kb nuclear residing lncRNA, was
initially demonstrated to control tumor metastasis and cancer cell
survival (9). Recent studies have
shown that MALAT1 is enriched in endothelial cells and is closely
associated with endothelial cell functions and dysfunctions,
including hyperglycaemia-induced inflammatory processes (9), proliferation (10) and angiogenesis (11).
In the present study, the role of MALAT1 in
I/R-induced cerebral vascular endothelial cell apoptosis was
investigated, using OGD and reoxygenation (OGD-R) as an in
vitro I/R injury model (1).
Materials and methods
Establishment of an in vitro model of
OGD-R
Primary human brain microvascular endothelial cells
were purchased from Cell Systems Corp., (cat. no. ACBRI 376;
Kirkland, WA, USA) and cultured in CSC Complete Medium, which
includes 10% fetal bovine serum (cat. no. 4Z0-500; Cell Systems
Corp.), in a humidified atmosphere (relative humidity >95%)
containing 5% CO2 at 37°C. To avoid undue depletion of
glucose in the culture medium, experiments were performed 24 h
after the final medium renewal. To simulate ischemia-like
conditions in vitro, cells were subjected to OGD as
previously described (12). Cells
were exposed to OGD for 4 h by replacing the culture medium with a
glucose- and serum-free medium that had been equilibrated in an
anaerobic atmosphere (<0.1% O2, 5% CO2 and
95% N2) at 37°C inside a hypoxic workstation (H35; Don
Whitley Scientific Ltd., Shipley, UK). The pH of the medium
remained constant throughout the experiments. As reoxygenation
consisted of a return to the initial culture conditions after OGD,
cells were withdrawn from the hypoxic workstation and the OGD
medium was replaced with glucose- and serum-containing culture
medium. Cells grown under normal culture conditions were used as a
control.
Lentiviral transduction
Human MALAT1 lentiviral vector (cat. no. LV212703)
and blank control lentivector (cat. no. LV587) were purchased from
Applied Biological Materials Inc., (Richmond, BC, Canada). MALAT1
and blank control lentivectors were transfected with the packaging
vectors (psPAX2 and pMD2.G) into 293T cells by calcium chloride to
produce the lentivirus. Primary human brain microvascular
endothelial cells were subsequently transduced with the blank
control or MALAT1-expressing lentivirus. Human MALAT1 siRNA
lentivirus (cat. no. iV012699) and scrambled siRNA lentivirus (cat.
no. LVP015-G) were purchased from Applied Biological Materials
Inc., and were directly used to transduce primary human brain
microvascular endothelial cells. Six hours after lentiviral
transduction, cells were subjected to OGD-R.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from cultured cells was isolated using
miRNeasy kits (Qiagen China Co., Ltd., Shanghai, China) according
to the manufacturer's protocol, followed by purification with TURBO
DNA-free System (Ambion; Thermo Fisher Scientific, Inc., Waltham,
MA, USA). To measure lncRNAs, 1,000 ng total RNA was reverse
transcribed using MulV reverse transcriptase and random hexamer
primers (both Thermo Fisher Scientific, Inc.) in a 20-µl reaction.
cDNA was used as a template for RT-qPCR using Fast SYBR Green and
an Applied Biosystems StepOnePlus machine (both Applied Biosystems;
Thermo Fisher scientific, Inc.). Human ribosomal P0 (RPLP0) mRNA
was sequenced and used as a reference gene for normalization.
Primer sequences were as follows: MALAT1, forward
5′-GTGATGCGAGTTGTTCTCCG-3′ and reverse 5′-CTGGCTGCCTCAATGCCTAC-3′;
and RPLP0, forward 5′-TCGACAATGGCAGCATCTAC-3′ and reverse
5′-ATCCGTCTCCACAGACAAGG-3′. The PCR reaction mixture contined 12.5
µl of SYBR Green Master Mix, 400 ng of template DNA, forward and
reverse primers (0.25 µM each), and 12 µl of nuclease-free water.
PCR amplification conditions were as follows: 20 sec at 94°C,
followed by 40 cycles of 3 sec at 95°C and 30 sec at 62°C, and a
final extension at 62°C for 10 mins. Analysis of relative gene
expression levels was performed using the following formula:
2−ΔΔCq with ΔΔCT=Cq(target
gene)-Cq(control) (13).
Cell apoptosis assay
Primary human brain microvascular endothelial cells
were cultured at 9×104 cells/well in 96-well tissue
culture plates and subjected to OGD-R. Cell apoptosis was measured
at 6, 9, 12, 24 and 36 h post-reoxygenation with a microplate
reader-based TiterTACS in situ apoptosis detection kit (cat.
no. 4822-96-K; R&D Systems, Inc., Minneapolis, MN, USA)
according to the manufacturer's protocol. Each experiment was
performed in duplicate and repeated thrice.
Terminal deoxynucleotidyl transferase
dUTP nick-end labeling (TUNEL) assay
Cells were subjected to a TUNEL assay using an
ApopTag InSitu apoptosis detection kit (cat. no. S7111; EMD
Millipore, Billerica, MA, USA) according to the manufacturer's
protocol. TUNEL-positive/apoptosis rate was presented as the
percentage of total cells. Images were captured at ×40
magnification with a fluorescence/phase contrast microscope
(FSX100; Olympus Corp., Tokyo, Japan). Three independent
experiments were performed.
Caspase 3 activity assay
Activity of caspase 3 was determined using a
colorimetric caspase 3 assay kit (cat. no. ab39401) from Abcam
(Cambridge, MA, USA). The assays were performed in 96-well plates
by incubating 20 µl cell lysate protein/sample in 70 µl reaction
buffer [1% NP-40, 20 mM Tris-HCl (pH 7.5), 137 mM Nad and 10%
glycerol] containing 10 µl caspase-3 substrate (2 mM). Lysates were
then incubated at 37°C for 6 h, after which the samples were
assayed using a spectrophotometer (cat. no. 1702525; Bio-Rad
Laboratories, Hercules, CA, USA) at 405 nm.
Reactive oxygen species (ROS)
detection
ROS were measured using a dichlorofluorescin
diacetate (DCFDA) Cellular Reactive Oxygen Species Detection Assay
kit (cat. no. ab113851; Abcam, Cambridge, UK) according to the
manufacturer's protocol. Cells were plated at 9×104
cells/well in black 96-well plates and incubated with 25 µM DCFDA
for 45 min. Fluorescence was detected using a Victor3 1420
Multilabel Counter (PerkinElmer Instruments, Shanghai, China).
Phosphatidylinositol 3-kinase (PI3K)
activity assay
PI3K activity was determined with a PI3K activity
ELISA kit (cat. no. K-1000s; Echelon Biosciences Inc., Salt Lake
City, UT, USA) according to the manufacturer's protocol (12). To functionally assess PI3K activity,
PI3K was isolated by immunoprecipitation using a rabbit polyclonal
anti-human PI3K antibody (cat. no. 06-195; EMD Millipore) to the
p85 adapter subunit. The ability with which the co-precipitated
catalytic p110 catalytic subunit was able to convert a standard
PIP2 to PIP3 in a kinase reaction was subsequently assessed by
measuring the PIP3, generated using an ELISA kit. Each experiment
was repeated performed in duplicate, and was repeated three
times.
Western blot analysis
Cells were lysed in a solution containing hypotonic
buffer containing 0.5% Nonidet-P40 and a protease inhibitor
cocktail (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) by
sonication three times for 3 sec on ice. Following centrifugation
at 2,000 × g for 15 min at 4°C, the resulting supernatant
was used for protein concentration determination by the Coomassie
blue method (Thermo Fisher Scientific, Inc.) and for subsequent
steps. From each sample, 5 µg protein for each sample were
separated by 10% SDS-polyacrylamide gel electrophoresis and were
subsequently blotted onto a polyvinylidene difluoride microporous
membrane (EMD Millipore). Membranes were blocked with 5% skim milk
powder in Tris-buffered saline with tween 20 (TBS-T) for 2 h and
incubated for 1 h at room temperature with rabbit polyclonal
anti-human phosphorylated Akt (Ser 473) antibody (cat. no.
sc-101629) or mouse monoclonal anti-human Akt antibody (cat. no.
sc-81434) at a dilution of 1:1,000, and were subsequently washed
three times in TBS-T (cat. no. SRE0031; Sigma-Aldrich; Merck
Millipore) for 5 min and revealed using bovine anti-rabbit
(sc-2370) or anti-mouse (sc-2371; all Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) secondary antibody (both 1:5,000) for 1 h at
room temperature. Peroxidase was revealed with an enhanced
chemiluminescence kit (GE Healthcare Life Sciences, Shanghai,
China). Three independent experiments were performed.
Statistical analysis
Statistical analyses were performed using SPSS for
Windows 19.0 (IBM SPSS, Armonk, NY, USA). All values were expressed
as the mean ± standard deviation. Comparisons of the means among
multiple groups were performed with one-way analysis of variance
followed by post-hoc pairwise comparisons using Tukey's tests. A
two-tailed P<0.05 was considered to indicate a statistically
significant difference.
Results
MALAT1 inhibits OGD-R-induced
apoptosis and caspase 3 activity, but not ROS production, in human
brain vascular endothelial cells
In the present study, primary human brain
microvascular endothelial cells exposed to OGD-R were employed as
an in vitro model of cerebral vascular endothelial cell I/R
injury, as previously described (1).
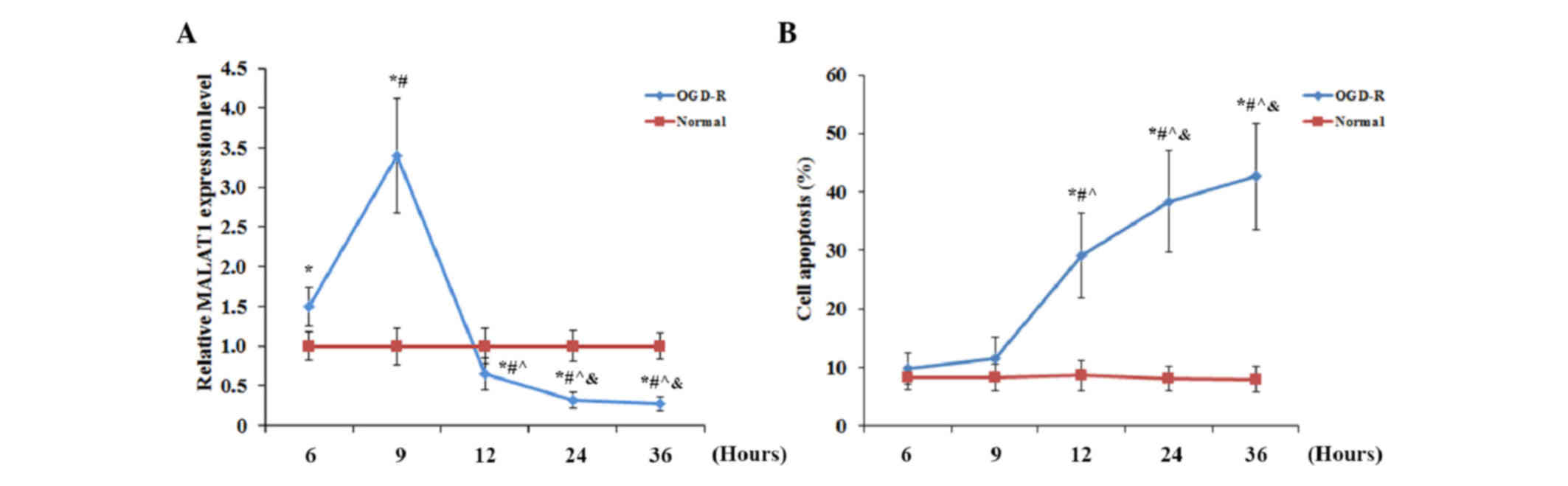
As shown in Fig. 1, human brain
microvascular endothelial cells were cultured under OGD-R, and the
expression level of MALAT1 and apoptosis in the cells were measured
at 6, 9, 12, 24 and 36 h post-reoxygenation. As shown in Fig. 1A, using cells under normal culture
conditions for normalization, the expression level of MALAT1 in
cells under OGD-R significantly increased (P<0.05) after
reoxygenation, peaking at 1.5-fold that of cells under normal
culture conditions at 9 h post-reoxygenation, and significantly
decreased (P<0.05) below the normal level, reaching 0.27-fold
that of cells under normal culture conditions at 36 h
post-reoxygenation. The apoptosis rate of cells under normal
culture conditions did not exhibit any significant change over time
(Fig. 1B). Furthermore, no
significant change was identified in the apoptotic rate of cells
exposed to OGD-R at 6 h (9.8±2.7%) and 9 h (11.5±3.7%)
post-reoxygenation, compared with the cells under normal culture
conditions. However, the apoptotic rate of cells exposed to OGD-R
markedly increased to 29.1±7.2% at 12 h, reaching 42.6±9.1% at 36 h
post-oxygenation (Fig. 1B). The
contrasting trends of the expression levels of MALAT1 and cell
apoptosis in human brain microvascular endothelial cells following
OGD-R suggested that MALAT1 may inhibit OGD-R-induced apoptosis in
brain vascular endothelial cells.
To explore the effect of MALAT1 on OGD-R-induced
apoptosis of brain vascular endothelial cells, human brain
microvascular endothelial cells were transduced with human MALAT1
lentivirus or human MALAT1 siRNA lentivirus to overexpress or
knockdown MALAT1, respectively. Cells transduced with the blank
control lentivirus (VC) or the scrambled siRNA lentivirus (SC) were
used as controls. To investigate the potential role of PI3K/Akt
survival signaling in the effects induced by MALAT1, cells
overexpressing MALAT1 were treated with a PI3K inhibitor,
Wortmannin. As shown in Fig. 2, the
VC and SC controls exposed to OGD-R exhibited similar changes in
the expression level of MALAT1 as nontransduced cells in Fig. 1A, indicating that transduction of the
VC or SC did not significantly affect the expression level of
MALAT1. In the presence or absence of OGD-R, lentiviral
transduction of MALAT1 and MALAT1-siRNA resulted in a significant
increase and decrease (both P<0.05) in the expression levels of
MALAT1, when compared with the controls, respectively (Fig. 2). No significant change was detected
in the expression levels of MALAT1 over time in either of the
experimental groups in the absence of OGD-R (Fig. 2A). In the presence of OGD-R, all
experimental groups exhibited a significant increase in MALAT1
expression levels by 9 h post-reoxygenation, followed by a
continuous decrease (P<0.05) (Fig.
2B). Inhibition of PI3K signaling by Wortmannin showed no
significant effect on the expression level of MALAT1 in the
presence or absence of OGD-R (Fig.
2).
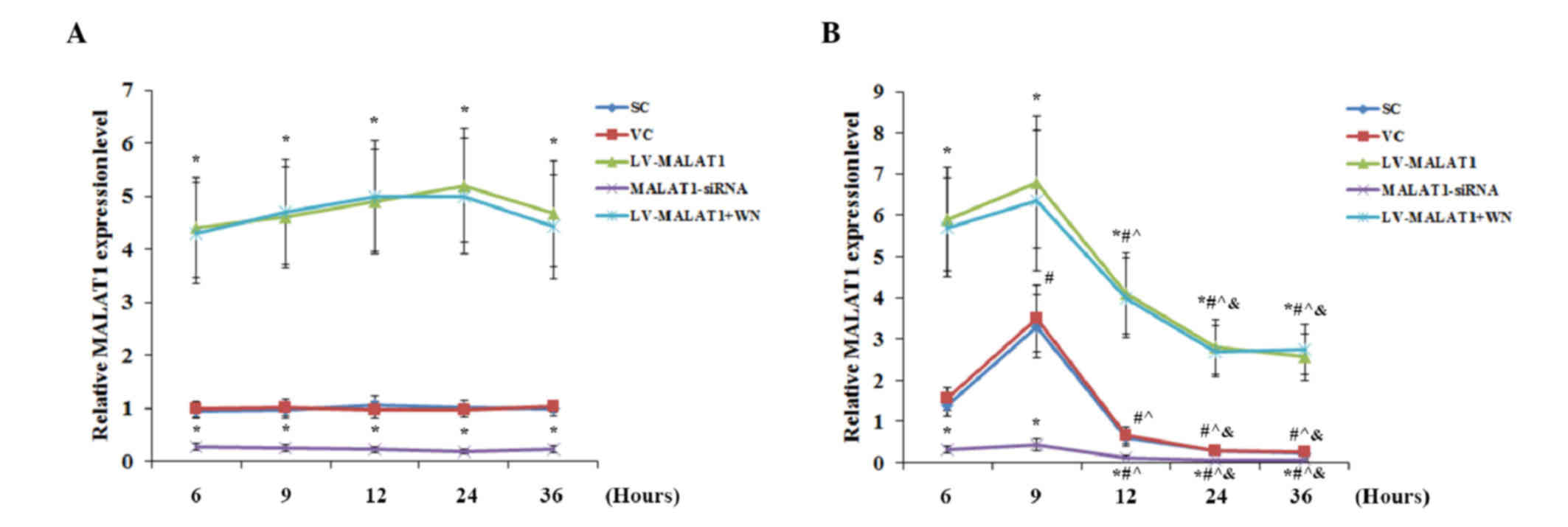 | Figure 2.Overexpression and knockdown of MALAT1
in brain vascular endothelial cells in the presence or absence of
OGD-R. Human brain microvascular endothelial cells were transduced
with human LV-MALAT1 or human MALAT1-siRNA to overexpress or
knockdown MALAT1, respectively. Cells transduced with VC or SC were
used as controls. Cells transduced with human LV-MALAT1 were also
treated with a phosphatidylinositol 3-kinase inhibitor, Wortmannin
(50 µM), during reoxygenation. The expression level of MALAT1 (A)
in the absence or (B) presence of OGD-R (specifically, at 6, 9, 12,
24 and 36 h post-reoxygenation) was measured via reverse
transcription-quantitative polymerase chain reaction and expressed
as fold changes to that of the control cells under normal culture
conditions (designated as 1); each group was normalized against
RPLP0 mRNA expression levels as a reference gene. Each experiment
was performed in duplicate and repeated three times. *P<0.05 vs.
SC and VC; #P<0.05 vs. 6 h; ^P<0.05 vs.
9 h; &P<0.05 vs. 12 h. MALAT1,
metastasis-associated lung adenoma transcript 1; OGD-R,
oxygen-glucose deprivation and reoxygenation; LV-MALAT1, MALAT1
lentivirus; MALAT1-siRNA, MALAT1 siRNA lentivirus; VC, blank
control lentivirus; SC, scrambled siRNA lentivirus; LV-MALAT1+WN,
MALAT1 lentivirus-Wortmannin. |
As shown in Fig. 3A,
in the absence of OGD-R, there was no significant change detected
in the cell apoptosis rate over time in each experimental group,
compared with the controls. Similarly, the overexpression and
knockdown of MALAT1 and inhibition of PI3K signaling by Wortmannin
also exhibited no significant effect on cell apoptosis. These
results suggested that MALAT1 had no significant effect on human
brain vascular endothelial cell apoptosis under normal conditions.
In the presence of OGD-R, however, overexpression of MALAT1
significantly decreased cell apoptosis compared with the controls
(P<0.05); this effect was abolished by Wortmannin (Fig. 3B). However, knockdown of MALAT1
significantly increased cell apoptosis compared with cells
transfected with the blank control lentivirus (P<0.05), up to
~90% at 36 h post-reoxygenation (Fig.
3B). TUNEL staining at 36 h post-reoxygenation corroborated
these findings (Fig. 4).
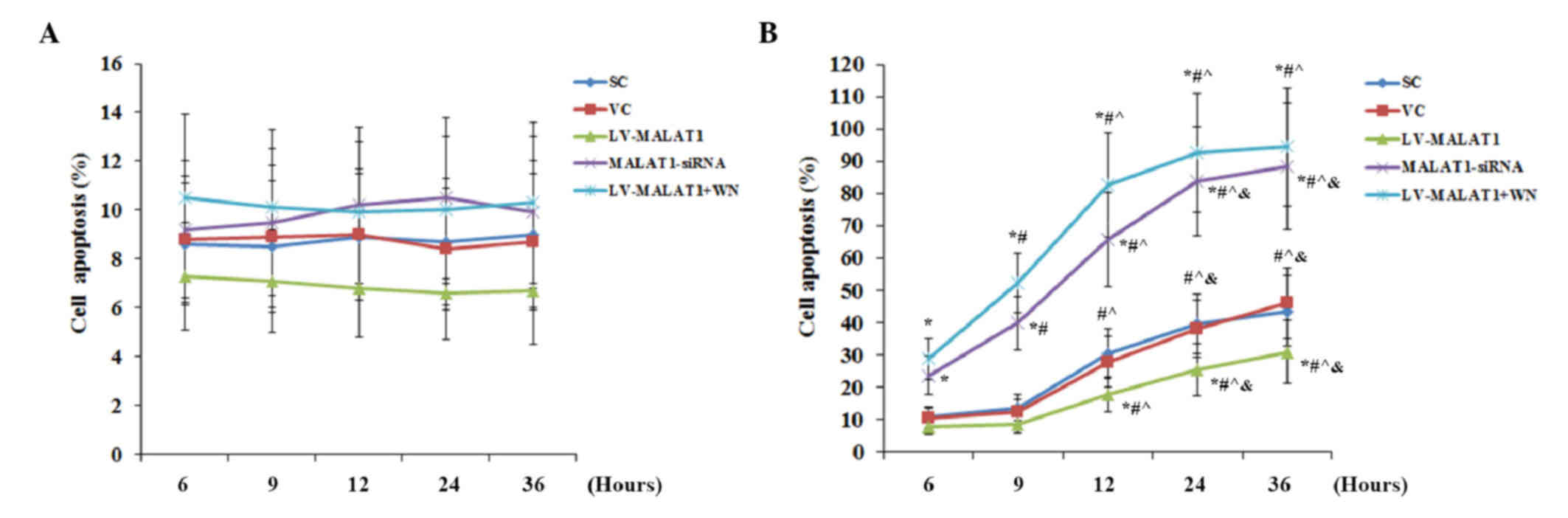 | Figure 3.Apoptosis in brain vascular
endothelial cells with overexpression or knockdown of MALAT1 in the
presence or absence of OGD-R. Human brain microvascular endothelial
cells were transduced with human LV-MALAT1 or MALAT1-siRNA to
overexpress or knockdown MALAT1, respectively. Cells transduced
with VC or SC were used as controls. Cells transduced with human
MALAT1 lentivirus were also treated with phosphatidylinositol
3-kinase inhibitor, Wortmannin (50 µM), during reoxygenation. The
apoptotic rate of cells (A) in the absence or (B) the presence of
OGD-R (specifically, at 6, 9, 12, 24 and 36 h post-reoxygenation)
was measured with a microplate reader-based apoptosis detection
kit. Each experiment was performed in duplicate and repeated three
times. *P<0.05 vs. SC and VC; #P<0.05 vs. 6 h;
^P<0.05 vs. 9 h; &P<0.05 vs. 12 h.
MALAT1, metastasis-associated lung adenoma transcript 1; OGD-R,
oxygen-glucose deprivation and reoxygenation; LV-MALAT1, MALAT1
lentivirus; MALAT1-siRNA, MALAT1 siRNA lentivirus; VC, blank
control lentivirus; SC, scrambled siRNA lentivirus; LV-MALAT1+WN,
MALAT1 lentivirus-Wortmannin. |
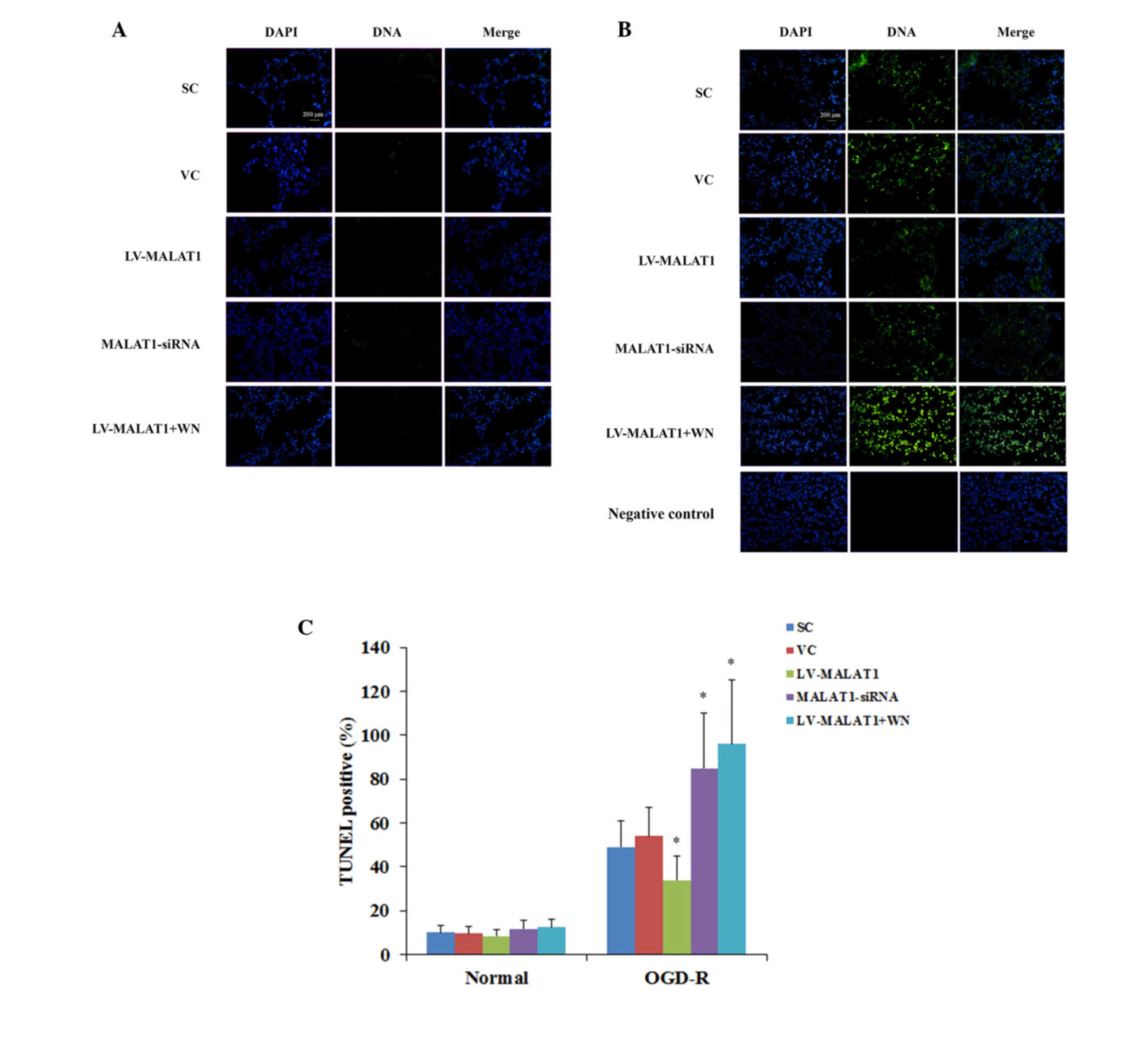 | Figure 4.TUNEL staining in brain vascular
endothelial cells with overexpression or knockdown of MALAT1 in the
presence or absence of OGD-R. Human brain microvascular endothelial
cells were transduced with human LV-MALAT1 or MALAT1-siRNA to
overexpress or knockdown MALAT1, respectively. Cells transduced
with VC or SC lentivirus were used as controls. Cells transduced
with human LV-MALAT1 were also treated with phosphatidylinositol
3-kinase inhibitor, Wortmannin (50 µM), during reoxygenation. The
apoptotic rate of cells (A) in the absence or (B) presence of OGD-R
at 36 h post-reoxygenation was measured with a TUNEL staining kit.
The images present cell nuclei stained by DAPI (blue) and DNA
fragmentation (green). (C) The TUNEL-positive/apoptosis rate is
shown as the percentage of total cells in histograms. Each
experiment was repeated three times.*P<0.05 vs. SC and VC.
TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end
labeling; MALAT1, metastasis-associated lung adenoma transcript 1;
OGD-R, oxygen-glucose deprivation and reoxygenation; LV-MALAT1,
MALAT1 lentivirus; MALAT1-siRNA, MALAT1 siRNA lentivirus; VC, blank
control lentivirus; SC, scrambled siRNA lentivirus; LV-MALAT1+WN,
MALAT1 lentivirus-Wortmannin. |
Consistent with the cell apoptosis data, in the
absence of OGD-R, there was no significant change identified
between the groups or over time in the activity of caspase 3
(Fig. 5A), a critical caspase in the
apoptotic pathway (14). In the
presence of OGD-R, however, overexpression of MALAT1 significantly
decreased caspase 3 activity compared with the controls
(P<0.05). This effect was abolished by Wortmannin; however,
knockdown of MALAT1 markedly increased the caspase 3 activity
compared with the controls (Fig.
5B).
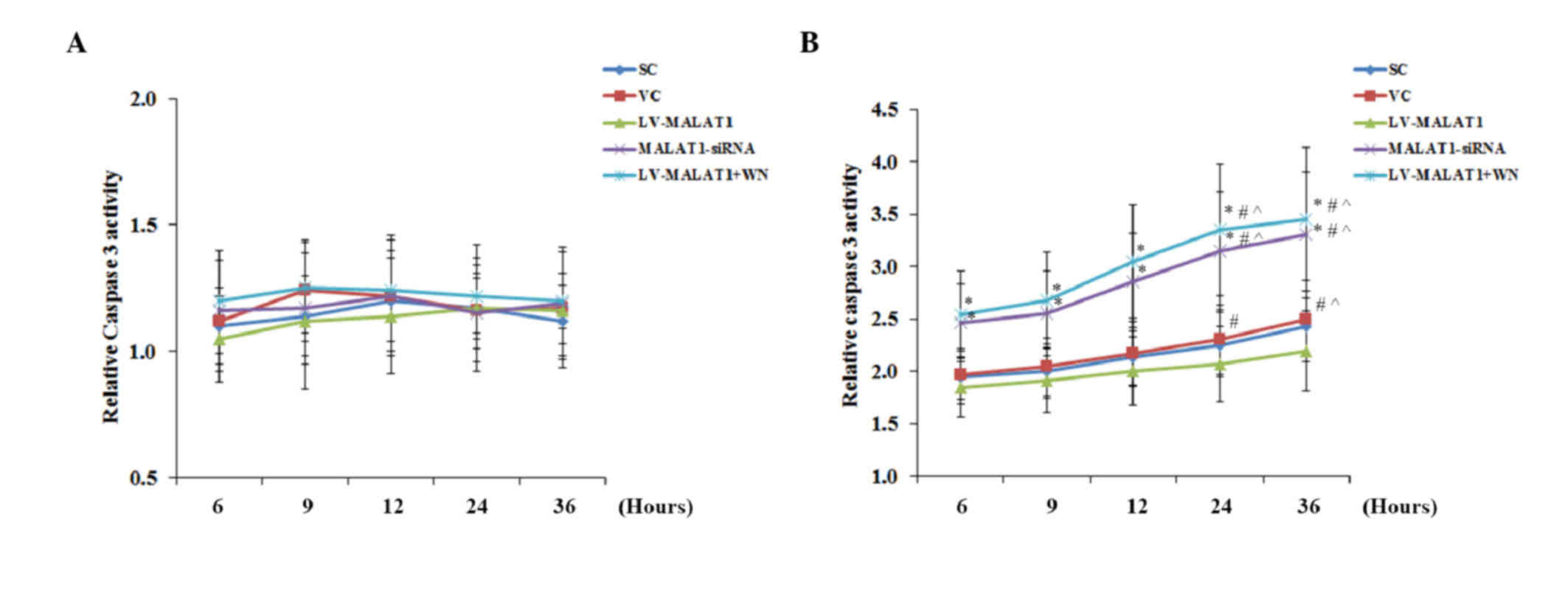 | Figure 5.Caspase 3 activity in brain vascular
endothelial cells with overexpression or knockdown of MALAT1 in the
presence or absence of OGD-R. Human brain microvascular endothelial
cells were transduced with human LV-MALAT1 MALAT1 or MALAT1-siRNA
to overexpress or knockdown MALAT1, respectively. Cells transduced
with VC or SC lentivirus were used as controls. Cells transduced
with human LV-MALAT1 were also treated with phosphatidylinositol
3-kinase inhibitor, Wortmannin (50 µM), during reoxygenation. The
caspase 3 activity in cells (A) in the absence or (B) presence of
OGD-R (specifically, at 6, 9, 12, 24 and 36 h post-reoxygenation)
was measured with a colorimetric caspase 3 assay kit and and
expressed as fold changes to that of cells under normal culture
conditions (designated as 1). Each experiment was performed in
duplicate and repeated three times. *P<0.05 vs. SC and VC;
#P<0.05 vs. 6 h; ^P<0.05 vs. 9 h;
&P<0.05 vs. 12 h. MALAT1, metastasis-associated
lung adenoma transcript 1; OGD-R, oxygen-glucose deprivation and
reoxygenation; LV-MALAT1, MALAT1 lentivirus; MALAT1-siRNA, MALAT1
siRNA lentivirus; VC, blank control lentivirus; SC, scrambled siRNA
lentivirus; LV-MALAT1+WN, MALAT1 lentivirus-Wortmannin. |
OGD-R has been shown to induce cellular
overproduction of ROS, which leads to cell apoptosis (15–18).
Therefore, the effect of MALAT1 on ROS production in human brain
microvascular endothelial cells in the presence and absence of
OGD-R was investigated. As shown in Fig.
6, in the absence of OGD-R, the ROS content in all experimental
groups remained similar to the levels exhibited by cells under
normal culture conditions (designated as 1) over time. However, in
the presence of OGD-R, the ROS content in all experimental groups
increases ~2.5-fold, compared with the cells under normal culture
conditions, with no significant differences identified between the
groups or over time.
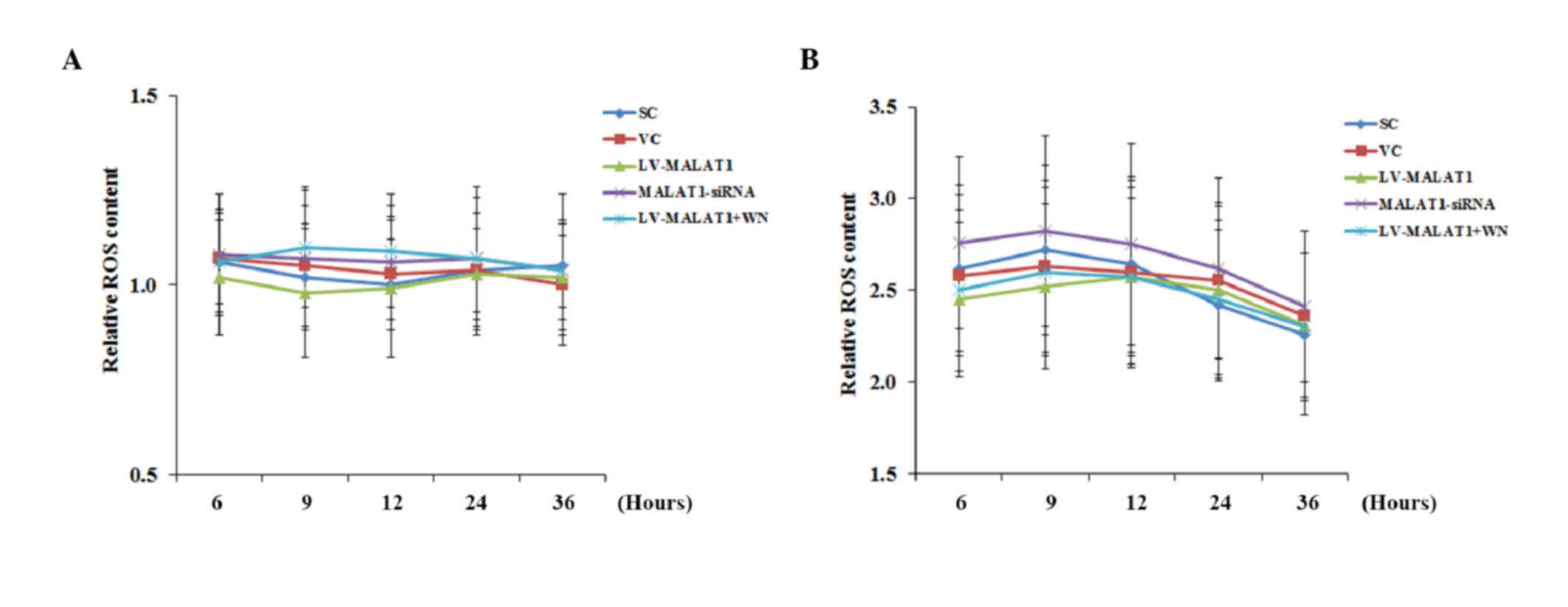 | Figure 6.ROS production in brain vascular
endothelial cells with overexpression or knockdown of MALAT1 in the
presence or absence of OGD-R. Human brain microvascular endothelial
cells were transduced with human LV-MALAT1 or MALAT1-siRNA to
overexpress or knockdown MALAT1, respectively. Cells transduced
with VC or SC lentivirus were used as controls. Cells transduced
with human LV-MALAT1 were also treated with phosphatidylinositol
3-kinase inhibitor, Wortmannin (50 µM), during reoxygenation. ROS
production in cells (A) in the absence or (B) presence of OGD-R
(specifically, at 6, 9, 12, 24 and 36 h post-reoxygenation) was
measured with a fluorescence cellular ROS detection assay kit and
expressed as fold changes to that of the control cells under normal
culture conditions (designated as 1). Each experiment was performed
in duplicate and repeated three times. *P<0.05 vs. SC and VC;
#P<0.05 vs. 6 h; ^P<0.05 vs. 9 h;
&P<0.05 vs. 12 h. ROS, reactive oxygen species;
MALAT1, metastasis-associated lung adenoma transcript 1; OGD-R,
oxygen-glucose deprivation and reoxygenation; LV-MALAT1, MALAT1
lentivirus; MALAT1-siRNA, MALAT1 siRNA lentivirus; VC, blank
control lentivirus; SC, scrambled siRNA lentivirus; LV-MALAT1+WN,
MALAT1 lentivirus-Wortmannin. |
These findings suggested that MALAT1 may protect
human brain vascular endothelial cells from OGD-R-induced
apoptosis, not by reducing OGD-R-induced ROS overproduction, but by
a PI3K-dependent survival mechanism. Therefore, the effect of
MALAT1 on PI3K/Akt survival signaling, which reportedly protects
cerebral endothelial cells from hypoxia/reoxygenation-induced
apoptosis (17), was subsequently
examined in human brain microvascular endothelial cells in the
presence and absence of OGD-R.
Effect of MALAT1 on PI3K/Akt survival
signaling in human brain microvascular endothelial cells
Overexpression and knockdown of MALAT1 had no
significant effect on PI3K activity in the absence of OGD-R
compared with the controls, except when Wortmannin completely
abolished PI3K activity (Fig. 7A).
There was no significant change detected in PI3K activity over time
in each experimental group. However, in the presence of OGD-R,
overexpression of MALAT1 markedly increased PI3K activity in human
brain microvascular endothelial cells compared with the controls;
this effect was completely abolished by Wortmannin (Fig. 7B). Knockdown of MALAT1 significantly
decreased (P<0.05) PI3K activity compared with the controls
(Fig. 7B). Similar data trends were
observed with the activation of the phosphorylation of Akt at
serine 473 (17) in human brain
microvascular endothelial cells in the presence and absence of
OGD-R, respectively (Fig. 8). These
findings indicated that MALAT1 may protect human brain vascular
endothelial cells from OGD-R-induced apoptosis via the PI3K/Akt
survival pathway.
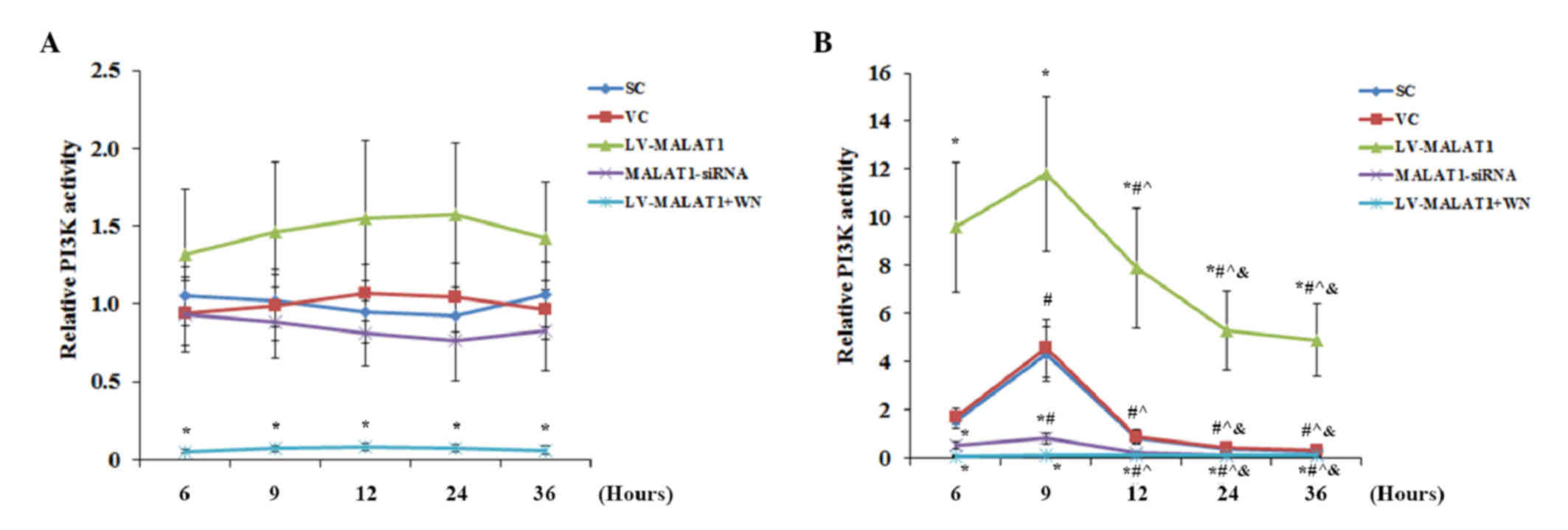 | Figure 7.PI3K activity in brain vascular
endothelial cells with overexpression or knockdown of MALAT1 in the
presence or absence of OGD-R. Human brain microvascular endothelial
cells were transduced with human LV-MALAT1) or MALAT1-siRNA to
overexpress or knockdown MALAT1, respectively. Cells transduced
with VC or SC were used as controls. Cells transduced with human
LV-MALAT1 were also treated with PI3K inhibitor, Wortmannin (50
µM), during reoxygenation. The PI3K activity in cells (A) in the
absence or (B) the presence of OGD-R (specifically, at 6, 9, 12, 24
and 36 h post-reoxygenation) was measured with a PI3K activity
ELISA kit and expressed as fold changes to that of the control
cells under normal culture conditions (designated as 1). Each
experiment was performed in duplicate and repeated three times.
*P<0.05 vs. SC and VC; #P<0.05 vs. 6 h;
^P<0.05 vs. 9 h; &P<0.05 vs. 12 h.
PI3K, phosphatidylinositol 3-kinase; MALAT1, metastasis-associated
lung adenoma transcript 1; OGD-R, oxygen-glucose deprivation and
reoxygenation; LV-MALAT1, MALAT1 lentivirus; MALAT1-siRNA, MALAT1
siRNA lentivirus; VC, blank control lentivirus; SC, scrambled siRNA
lentivirus; LV-MALAT1+WN, MALAT1 lentivirus-Wortmannin. |
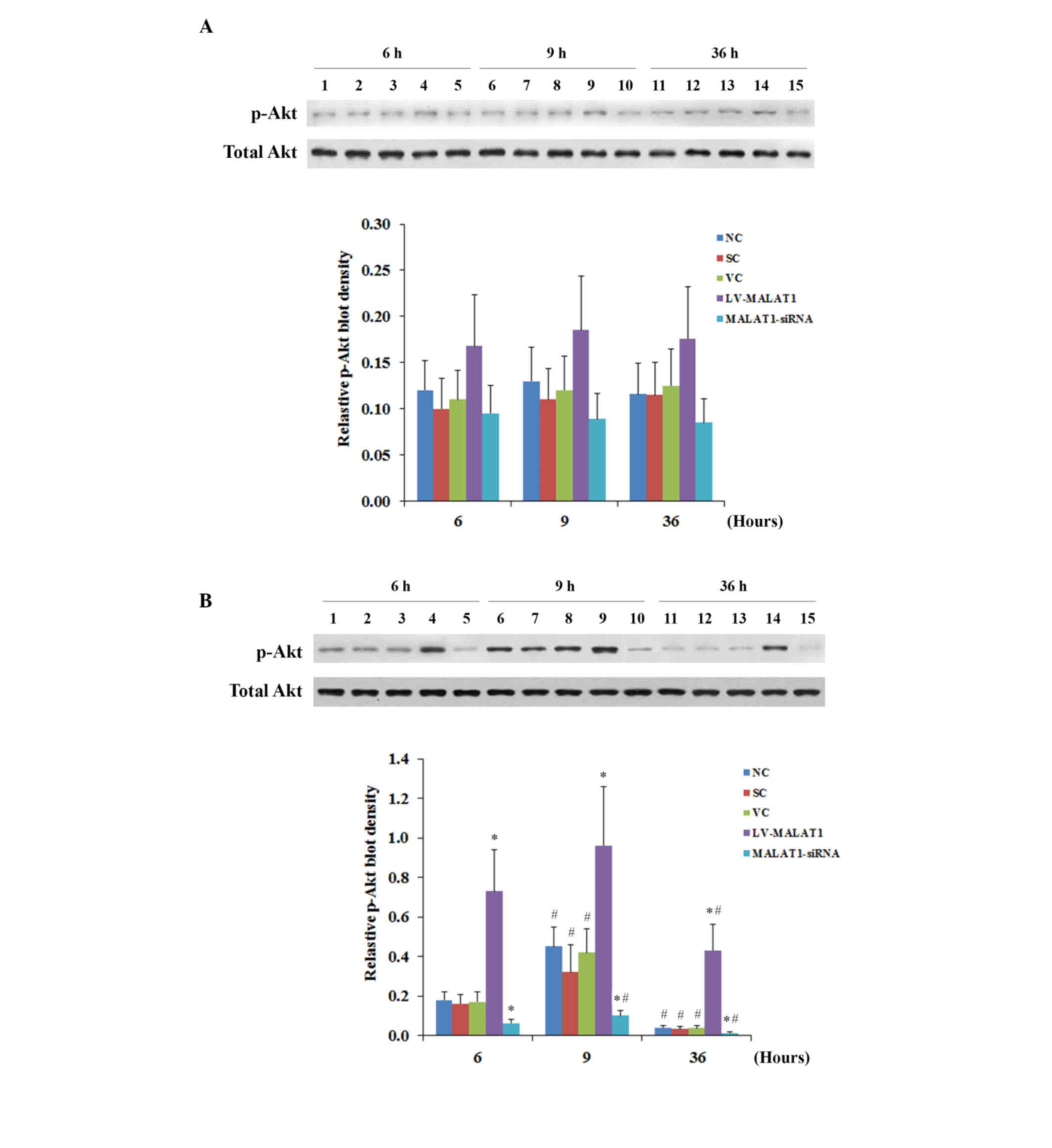 | Figure 8.p-Akt levels in brain vascular
endothelial cells with overexpression or knockdown of MALAT1 in the
presence or absence of OGD-R. Human brain microvascular endothelial
cells were transduced with human MALAT1 LV-MALAT1 or MALAT1-siRNA
to overexpress or knockdown MALAT1, respectively. Cells transduced
with VC or SC were used as controls. Cells transduced with human
LV-MALAT1 were also treated with phosphatidylinositol 3-kinase
inhibitor, Wortmannin (50 µM), during reoxygenation. Levels of
total Akt and p-Akt at ser473 were determined by western blot
analyses (A) in the absence or (B) the presence of OGD-R
(specifically, at 6, 9, 12, 24 and 36 h post-reoxygenation).
Lanes 1, 6 and 11: NC; lanes 2, 7 and 12: VC;
lanes 3, 8 and 13: SC; lanes 4, 9 and 14: LV-MALAT1;
lanes 5, 10 and 15: MALAT1-siRNA. Lanes 1–5: 6 h
post-reoxygenation; lanes 6–10: 9 h post-reoxygenation;
lanes 11–15: 36 h post-reoxygenation. The total Akt level
was not significantly altered by OGD-R. Density of the p-Akt
(ser473) blot was normalized against that of total Akt to obtain a
relative p-Akt blot density. Each experiment was repeated three
times. *P<0.05 vs. NC, SC and VC; #P<0.05 vs. 6 h.
p-Akt, phosphorylated Akt; MALAT1, metastasis-associated lung
adenoma transcript 1; OGD-R, oxygen-glucose deprivation and
reoxygenation; LV-MALAT1, MALAT1 lentivirus; MALAT1-siRNA, MALAT1
siRNA lentivirus; NC, normal control cells; VC, blank control
lentivirus; SC, scrambled siRNA lentivirus; LV-MALAT1+WN, MALAT1
lentivirus-Wortmannin; ser473, serine 473. |
Discussion
Cerebral I/R injury is a common cause of
debilitating complications and mortality associated with stroke and
traumatic brain injury (1), often
leading to brain vascular dysfunction characterized by endothelial
cell injury or apoptosis (2). A
recent study suggested that lncRNAs may function as a class of
novel master regulators in cerebrovascular endothelial pathologies
after ischemic stroke (8). MALAT1, a
lncRNA which was initially shown to control tumor metastasis and
cancer cell survival (9), has been
demonstrated to have a role in endothelial cell function and
dysfunction (9–11). In the present study, through the use
of primary human brain microvascular endothelial cells under an
in vitro I/R injury setting (1), it was observed that MALAT1 inhibits
OGD-R-induced human brain vascular endothelial cell apoptosis.
OGD-R has been used as an in vitro model of
cerebral vascular endothelial cell I/R injury in previous studies
and has been shown to cause significant (25–40%) endothelial cell
apoptosis (17,18). In the present study it was observed
that OGD-R induced ~40% endothelial cell apoptosis. The expression
level of MALAT1 and the apoptosis rate in human brain microvascular
endothelial cells under OGD-R exhibited contrasting trends after
reoxygenation. Notably, OGD-R induced a marked increase in the
expression levels of MALAT1 by 9 h post-reoxygenation, followed by
a continuous decrease. Conversely, the cell apoptosis rate remained
similar to that of cells under normal culture conditions by 9 h
post-reoxygenation, followed by a continuous increase.
Subsequently, overexpression and knockdown experiments revealed
that, in cells exposed to OGD-R, overexpression of MALAT1
significantly decreased cell apoptosis, and knockdown of MALAT1
markedly increased cell apoptosis by up to ~90%. The increase of
MALAT1 by 9 h post-oxygenation, which corresponded to inhibited
cell apoptosis at that time point, may be a cellular physiological
response to protect brain vascular endothelial cells against
OGD-R-induced apoptosis. These findings suggest that MALAT1 may be
a potent natural protector of brain vascular endothelium against
OGD-R insult in vitro, and thereby I/R injury in
vivo. Therefore, MALAT1 may be a potential novel therapeutic
target for cerebral I/R injury in stroke and traumatic brain
injury, although future in vivo studies are required to
verify its application potential. In addition, it is unclear why
the expression level of MALAT1 markedly decreased after peaking at
9 h post-reoxygenation. This trend remained following the
overexpression of MALAT1, which significantly elevated the overall
expression level of MALAT1 above those of the controls. This may be
a compensatory cellular response to accelerate the degradation of
MALAT1 after an acute elevation of its expression. Further study is
required to elucidate the underlying mechanisms and physiological
significance.
The activity of caspase 3, which is a highly active
caspase that is present in apoptotic cells (13), corroborated the findings from the
present study that indicated that MALAT1 inhibits OGD-R-induced
human brain microvascular endothelial cell apoptosis. It has been
reported that OGD-R induces cellular overproduction of ROS, which
leads to cell apoptosis (15–19). In
the present study, it was revealed that OGD-R significantly
increased cellular ROS content. However, unlike caspase 3 activity,
ROS content was not significantly altered by the expression level
of MALAT1, thereby excluding the possibility that MALAT1 protected
human brain vascular endothelial cells from OGD-R-induced apoptosis
by regulating OGD-R-induced ROS overproduction. Conversely, in
cells exposed to OGD-R, the Wortmannin PI3K inhibitor did not alter
the expression level of MALAT1; however, the apoptosis protective
effect of MALAT1 was abolished, suggesting that MALAT1 may protect
human brain vascular endothelial cells from OGD-R-induced apoptosis
via a PI3K-dependent mechanism. This was subsequently corroborated
by PI3K activity assay results and the phosphorylated levels of
Akt, indicating that the protective effect of MALAT1 against
OGD-R-induced apoptosis in human brain vascular endothelial cells
may be mediated through the PI3K/Akt survival signaling pathway.
These findings are in agreement with previous studies that have
reported that MALAT1 activates the PI3K/Akt pathway (20,21),
which reportedly protects human cerebral endothelial cells from
hypoxia/reoxygenation-induced oxidative stress and apoptosis
(18). In addition, it has been
shown that PI3K/Akt activation is able to suppress cell apoptosis
by inhibiting caspase activation (22), which supports the findings from the
present study that PI3K activity and caspase 3 activity in human
brain microvascular endothelial cells exposed to OGD-R exhibited
contrasting trends after reoxygenation. These findings suggest that
activating the PI3K/Akt survival pathway in response to apoptotic
stress may be a major mechanism underlying the protective effects
of MALAT1 on human brain vascular endothelial cells under
OGD-R.
Notably, MALAT1 did not significantly alter PI3K
signaling activity in cells under normal culture conditions, only
in cells exposed to OGD-R, which suggests that the presence of
in vitro OGD-R insult or in vivo I/R injury is
required to activate MALAT1/PI3K/Akt signaling in human brain
vascular endothelial cells. How MALAT1 activates the PI3K/Akt
pathway in human brain vascular endothelial cells exposed to OGD-R
remains unclear. Previous studies have shown that MALAT1 is able to
interact with polycomb 2 and thereby regulate histone modifications
to control cellular proliferation (23,24). In
addition, it has been reported that MALAT1 interacts with
serine-/arginine-rich proteins to regulate the subcellular
localization of proteins that regulate splicing (24). A recent study revealed that MALAT1 is
critical for maintaining the blood-brain/blood-tumor barrier, which
is characterized by the presence of tight junctions between brain
capillary endothelial cells that restrict paracellular diffusion
(25). This suggests that MALAT1 may
regulate cerebral vascular endothelial barrier functions. It is not
yet clear whether these mechanisms are associated with the
activation of MALAT1/PI3K/Akt signaling in human brain vascular
endothelial cells under OGD-R in vitro or I/R in
vivo.
In conclusion, to the best of our knowledge, the
present study was the first to indicate that lncRNA MALAT1 protects
human brain vascular endothelial cells from OGD-R-induced apoptosis
via a PI3K-dependent mechanism, suggesting that MALAT1 may be a
potential novel therapeutic target for cerebral I/R injury. Further
study is required in order to elucidate the underlying
mechanisms.
Acknowledgements
This work was supported by the Hunan Provincial
Bureau of Science and Technology, Changsha, China (grant no.
20151022).
References
|
1
|
Alluri H, Shaji C Anasooya, Davis ML and
Tharakan B: Oxygen-glucose deprivation and reoxygenation as an in
vitro ischemia-reperfusion injury model for studying blood-brain
barrier dysfunction. J Vis Exp. e526992015.PubMed/NCBI
|
|
2
|
Zhang Y, Zhang X, Park TS and Gidday JM:
Cerebral endothelial cell apoptosis after ischemia-reperfusion:
Role of PARP activation and AIF translocation. J Cereb Blood Flow
Metab. 25:868–877. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vausort M, Wagner DR and Devaux Y: Long
noncoding RNAs in patients with acute myocardial infarction. Circ
Res. 115:668–677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carninci P, Kasukawa T, Katayama S, Gough
J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al:
The transcriptional landscape of the mammalian genome. Science.
309:1559–1563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kapranov P, Cheng J, Dike S, Nix DA,
Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J,
Hofacker IL, et al: RNA maps reveal new RNA classes and a possible
function for pervasive transcription. Science. 316:1484–1488. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mercer TR and Mattick JS: Structure and
function of long noncoding RNAs in epigenetic regulation. Nat
Struct Mol Biol. 20:300–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu W, Alvarez-Dominguez JR and Lodish HF:
Regulation of mammalian cell differentiation by long non-coding
RNAs. EMBO Rep. 13:971–983. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Yuan L, Zhang X, Hamblin MH, Zhu
T, Meng F, Li Y, Chen YE and Yin KJ: Altered long non-coding RNA
transcriptomic profiles in brain microvascular endothelium after
cerebral ischemia. Exp Neurol. 277:162–170. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Puthanveetil P, Chen S, Feng B, Gautam A
and Chakrabarti S: Long non-coding RNA MALAT1 regulates
hyperglycaemia induced inflammatory process in the endothelial
cells. J Cell Mol Med. 19:1418–1425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Michalik KM, You X, Manavski Y,
Doddaballapur A, Zörnig M, Braun T, John D, Ponomareva Y, Chen W,
Uchida S, et al: Long noncoding RNA MALAT1 regulates endothelial
cell function and vessel growth. Circ Res. 114:1389–1397. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thum T and Fiedler J: LINCing MALAT1 and
angiogenesis. Circ Res. 114:1366–1368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mysiorek C, Culot M, Dehouck L, Derudas B,
Staels B, Bordet R, Cecchelli R, Fenart L and Berezowski V:
Peroxisome-proliferator-activated receptor-alpha activation
protects brain capillary endothelial cells from oxygen-glucose
deprivation-induced hyperpermeability in the blood-brain barrier.
Curr Neurovasc Res. 6:181–193. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−DeltaDelta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Woo M, Hakem R, Soengas MS, Duncan GS,
Shahinian A, Kägi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, et
al: Essential contribution of caspase 3/CPP32 to apoptosis and its
associated nuclear changes. Genes Dev. 12:806–819. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao XY, Huang JO, Hu YF, Gu Y, Zhu SZ,
Huang KB, Chen JY and Pan SY: Combination of mild hypothermia with
neuroprotectants has greater neuroprotective effects during
oxygen-glucose deprivation and reoxygenation-mediated neuronal
injury. Sci Rep. 4:70912014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abramov AY, Scorziello A and Duchen MR:
Three distinct mechanisms generate oxygen free radicals in neurons
and contribute to cell death during anoxia and deoxygenation. J
Neurosci. 27:1129–1138. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Park TS and Gidday JM: Hypoxic
preconditioning protects human brain endothelium from ischemic
apoptosis by Akt-dependent survivin activation. Am J Physiol Heart
Circ Physiol. 292:H2573–H2581. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J and Chen Y, Yang Y, Xiao X, Chen S,
Zhang C, Jacobs B, Zhao B, Bihl J and Chen Y: Endothelial
progenitor cells and neural progenitor cells synergistically
protect cerebral endothelial cells from
Hypoxia/reoxygenation-induced injury via activating the PI3K/Akt
pathway. Mol Brain. 9:122016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Zhang Y, Liu X, Ma J, Liu P, Hu C
and Zhang G: Annexin A5 inhibits diffuse large B-cell lymphoma cell
invasion and chemoresistance through phosphatidylinositol 3-kinase
signaling. Oncol Rep. 32:2557–2563. 2014.PubMed/NCBI
|
|
20
|
Xu S, Sui S, Zhang J, Bai N, Shi Q, Zhang
G, Gao S, You Z, Zhan C, Liu F and Pang D: Downregulation of long
noncoding RNA MALAT1 induces epithelial-to-mesenchymal transition
via the PI3K-AKT pathway in breast cancer. Int J Clin Exp Pathol.
8:4881–4891. 2015.PubMed/NCBI
|
|
21
|
Dong Y, Liang G, Yuan B, Yang C, Gao R and
Zhou X: MALAT1 promotes the proliferation and metastasis of
osteosarcoma cells by activating the PI3K/Akt pathway. Tumour Biol.
36:1477–1486. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen L, Wang J, Wang B, Yang J, Gong Z,
Zhao X, Zhang C and Du K: MiR-126 inhibits vascular endothelial
cell apoptosis through targeting PI3K/Akt signaling. Ann Hematol.
95:365–374. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang L, Lin C, Liu W, Zhang J, Ohgi KA,
Grinstein JD, Dorrestein PC and Rosenfeld MG: ncRNA- and Pc2
methylation-dependent gene relocation between nuclear structures
mediates gene activation programs. Cell. 147:773–788. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tripathi V, Ellis JD, Shen Z, Song DY, Pan
Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al: The
nuclear-retained noncoding RNA MALAT1 regulates alternative
splicing by modulating SR splicing factor phosphorylation. Mol
Cell. 39:925–938. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma J, Wang P, Yao Y, Liu Y, Li Z, Liu X,
Li Z, Zhao X, Xi Z, Teng H, et al: Knockdown of long non-coding RNA
MALAT1 increases the blood-tumor barrier permeability by
up-regulating miR-140. Biochim Biophys Acta. 1859:324–338. 2016.
View Article : Google Scholar : PubMed/NCBI
|