Introduction
Chronic obstructive pulmonary disease (COPD) is a
major chronic disease with increasing morbidity and mortality
worldwide and is characterized by reversible airflow limitation
(1). Pathologically, persistent
airway inflammation and airway remodeling are two key factors of
airway obstruction in COPD (1,2). In
airway remodeling, airway smooth muscle cells (ASMCs) act as the
main effector cells and their proliferation represents a major
characteristic of airway remodeling in COPD (3,4). Studies
in animal models and on human patients have shown that cigarette
smoke is one of the most important risk factors for the development
of COPD. In addition, studies have demonstrated that cigarette
smoke extract (CSE) can stimulate the proliferation of ASMCs
(5,6). The ASM layer was markedly thickened in
COPD patients and in a rat model of cigarette smoking (7,8).
CCAAT/enhancer-binding protein alpha (C/EBP-α), a
member of the C/EBP family, was recently implicated in the
pathogenesis of COPD. It belongs to the basic leucine zipper class
of transcription factors and has essential roles in the regulation
of cell cycle progression, differentiation and pro-inflammatory
gene expression. C/EBP-α-deficient mice displayed histopathological
and inflammatory characteristics of COPD (9). In asthma patients, the expression
levels of C/EBP-α are markedly decreased in lung ASMCs (10). In addition, a previous study has
revealed that C/EBP-α inhibited cell proliferation by directly
suppressing cyclin-dependent kinase (Cdk) 2 and Cdk4 (11).
A previous study reported that calreticulin, a
Ca2+-binding chaperone in the endoplasmic reticulum (ER)
of eukaryotic cells, transcriptionally regulated C/EBP-α through a
cis-regulatory CNG-rich loop in the mRNA of C/EBP-α (12). In the ER lumen, calreticulin acts as
a chaperone, which controls newly synthesized proteins and
glycoproteins and regulates intracellular Ca2+
homeostasis to affect a variety of cell processes such as adipocyte
differentiation, cardiogenesis and cell stress responses (13). Besides, calreticulin is also involved
in the regulation of wound healing, tumorigenesis and immunity.
Studies have also assessed the function of calreticulin in cell
proliferation, revealing that it is highly expressed in several
cancer types such as hepatoma, colon cancer and oral squamous cell
carcinoma with increased cell proliferation (14,15).
Furthermore, manipulation of calreticulin levels affected cancer
cell proliferation, angiogenesis and differentiation (15). However, another study performed by
Miglino et al (12)
demonstrated that calreticulin negatively regulates the
proliferation of bronchial (B) SMCs. These controversial findings
inspired our group to investigate the regulatory role of
calreticulin in cell proliferation.
It was demonstrated that the interaction of
calreticulin with stem-loop structures of C/EBP-β and C/EBP-α mRNAs
leads to inhibition of translation of C/EBP proteins, indicating
the potential involvement of calreticulin in the
post-transcriptional processing of certain GC-rich mRNAs via
regulation of C/EBP protein expression (16). In the present study, the expression
of calreticulin and C/EBP-α in a CSE-treated cell model was
examined and the effect of CSE on the proliferation of human ASMCs
was assessed. In addition, the molecular mechanism by which CSE
controls cell proliferation through inhibiting C/EBP-α was
investigated.
Materials and methods
Cell culture and treatments
Normal human ASMCs were purchased from the American
Type Tissue Collection (Manassas, VA, USA). ASMCs were cultured in
Dulbecco's modified Eagle's medium (DMEM, Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco, Thermo Fisher Scientific, Inc.) and
maintained in a humidified atmosphere with 5% CO2 at
37°C. The subcultures of ASMCs between passage 4 and 6 were used in
the experiments. For cell treatment, ASMCs were seeded in 6-well
plates and then stimulated with various concentrations of CSE (0,
2.5, 5, 10, 15 and 20%) for 24, 48 or 72 h. For all experiments,
cells were made quiescent by incubation in serum-free medium
overnight prior to exposure to CSE.
Preparation of CSE
CSE was freshly generated under standardized
conditions as previously described (17). In brief, aqueous CSE was obtained by
combustion of two University of Kentucky 3R4F research cigarettes
(filters removed) purchased from the University of Kentucky
(Lexington, KT, USA) and passing the resulting smoke through 25 ml
DMEM using a peristaltic pump, followed by filtering through a
0.22-µm pore filter. The obtained solution was referred to as
having 100% strength.
Cell proliferation assay
The proliferation of ASMCs was determined by a
colorimetric assay using MTT. Following CSE treatment, the
supernatant was removed and 150 µl MTT (AMRESCO, LLC., Cleveland,
OH, USA) was added to each well. Following incubation at 37°C for 4
h, the reaction product of MTT was extracted with dimethyl
sulfoxide (DMSO). The absorbance was measured at 570 nm using the
multiskan MK3 (Thermo Fisher Scientific, Inc.) with DMSO as a
blank.
Cell apoptosis assay
For detection of apoptosis, human ASMCs from each
group were stained with Annexin V conjugated to fluorescein
isothiocyanate (FITC) as well as propidium iodide (PI) using the
FITC Annexin V/Dead Cell Apoptosis kit (Life Technologies; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. In brief, subsequent to treatment, human ASMCs were
suspended and incubated in buffer containing Annexin V and PI for 5
min at room temperature in the dark. The cells were then assessed
using a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes,
NJ, USA) and the data were analyzed using FlowJo software 7.6 (Tree
Star, Inc., Ashland, OR, USA).
Immunofluorescence
ASMCs were prepared on chambered slides (BD Falcon;
BD Biosciences) and exposed to 10% CSE for 24 h. Cells were washed
with phosphate-buffered saline (PBS) and fixed with 4%
paraformaldehyde for 10 min. Subsequently, cells were permeabilized
with methanol and then blocked with 3% bovine serum albumin (BSA,
Gibco; Thermo Fisher Scientific, Inc.) in PBS at room temperature
for 1 h. The cells were incubated with anti-C/EBP-α primary
antibody (dilution, 1:300; no. ab40761; Abcam, Cambridge, UK) at
4°C overnight. Detection was performed with goat anti-rabbit
immunoglobulin (Ig) G secondary antibody (dilution, 1:500; no.
ab175471; Abcam) labeled with Alexa Fluor 568 (red) at 37°C for 1
h, using ProLong Gold anti-fade with DAPI (blue). Images were
obtained using a fluorescence microscope (Axiovert 200; Carl Zeiss
AG, Oberkochen, Germany).
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from cells of each group
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.).
Complementary (c)DNA was synthesized using the PrimeScript™ RT
reagent kit (Takara Bio, Inc., Otsu, Japan), according to the
manufacturer's instructions. The cDNA obtained served as a template
for PCR using PCR Master Mix (Promega Corp., Madison, WI, USA) and
the following gene-specific primers: C/EBP-α forward,
5′-GGCGGCGACTTTGACTACC-3′ and reverse, 5′-CTGCTTGGCTTCATCCTCCTC-3′;
β-actin forward, 5′-ACACTGTGCCCATCTACGACG-3′ and reverse,
5′-AGGGGCCGGACTCCTCATACT-3′ (Shanghai GenePharma Co., Ltd.,
Shanghai, China). PCR conditions were set as follows: 5 min at
95°C, followed by 35 cycles of 30 sec at 94°C, 40 sec at 54°C and
40 sec at 72°C, and a final extension at 72°C for 6 min (on an
ABI9700. PCR products were separated on a 1.5% agarose gel, mRNA
levels were captured and then calculated using the comparative
cycle threshold method and normalized to the expression of β-actin
mRNA as a control (18).
Western blot analysis
Following treatment with CSE, the medium was removed
and cell protein was extracted using the Total Protein Extraction
kit (cat. no. AR0103; Boster Biological Technology, Wuhan, China)
and then quantified using the BSA method (17). Whole-cell lysate (20 µg) was loaded
onto an 8% Tris-glycine gel (Invitrogen; Thermo Fisher Scientific,
Inc.). Following electrophoresis, proteins were transferred to a
nitrocellulose membrane. The membrane was blocked with 5% BSA in
Tris-buffered saline containing Tween-20 for 1 h at room
temperature. Blots were then incubated at 4°C overnight with
primary antibodies against calreticulin (dilution, 1:1,000; no.
ab2908), C/EBP-α (dilution, 1:1,000; no. ab40761) and β-actin
(dilution, 1:5,000; no. ab8227), followed by the secondary antibody
anti-mouse IgG (dilution, 1:1,000; no. ab131368; Abcam) at 37°C for
1 h. All antibodies were purchased from Abcam. An enhanced
chemiluminescence detection system (ECL) western blot reagent
(RPN210; GE Healthcare, Chalfont, UK) was used to detect the
signals on the membranes with the aid of a Benchtop Ultraviolet
Transilluminator (VWR, Radnor, PA, USA).
RNA interference
ASMCs were seeded into 6-well plates and incubated
for 24 h prior to transfection with 50 nM control small interfering
siRNA vector, siRNA calreticulin (5′-GGAGGAUGAUGAGGACAAATT-3′) or
siRNA C/EBP-α (5′-GACAAGAACAGCAACGAGUTT-3′; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA). The sequence of siRNA used
for negative control was 5′- UUC UCC GAA CGU GUC ACG UTT-3.
Transfected cells were cultured in DMEM medium and incubated at
37°C for 24 h. Following treatment with 10% CSE for 24 h, ASMCs
were collected for analysis using the aforementioned assays.
Statistical analysis
Values are expressed as the mean ± standard
deviation. Statistical analyses were performed using one-way
analysis of variance (for multiple-group comparisons) or Student's
t-test (for comparison between two groups). SPSS 13.0 software
(SPSS, Inc., Chicago, IL, USA) was used for all statistical
analyses. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of CSE on ASMC
proliferation
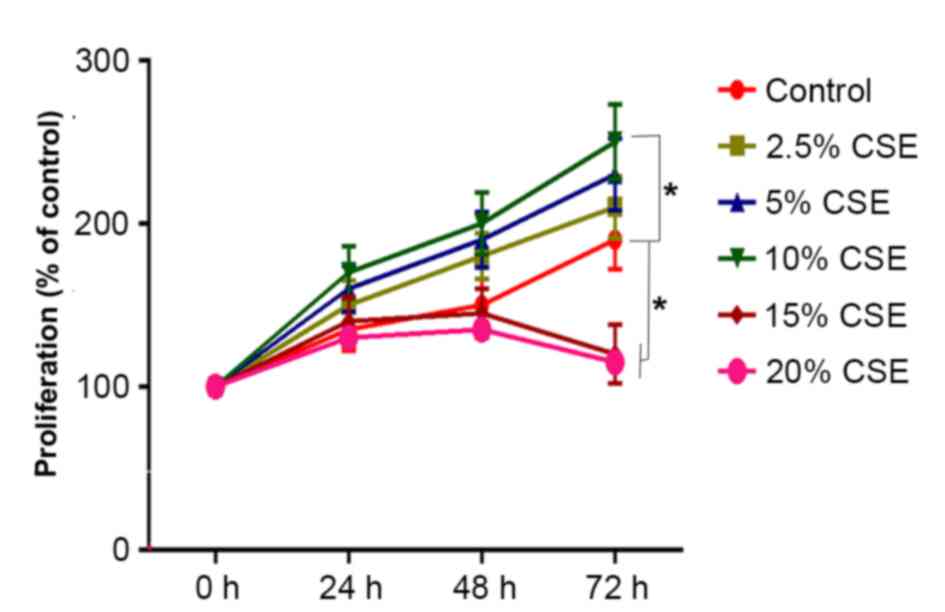
To investigate the effect of CSE on cell
proliferation, ASMCs were treated with 0, 2.5, 5, 10, 15 or 20% CSE
for 24, 48 or 72 h and subjected to the MTT colorimetric assay. As
shown in Fig. 1, treatment with
2.5–10% CSE increased ASMC proliferation compared with the control
in a dose-dependent manner, with the increase being significant
with 10% CSE (P<0.05) compared with 0–5% CSE. By contrast, CSE
at 15 and 20% significantly decreased ASMC proliferation compared
with the control (P<0.05), which may have been due to the
cytotoxic effect of high concentrations of CSE (Fig. 1).
Effects of CSE on expression of
calreticulin and C/EBP-α
Since the stimulating effect of CSE on cell
proliferation was greatest at the concentration of 10% of CSE, this
concentration was used for the subsequent experiments. When
stimulated with 10% CSE, the proliferation of human ASMCs was
significantly increased from 24 h onwards as compared with that in
the control group (P<0.05; Fig.
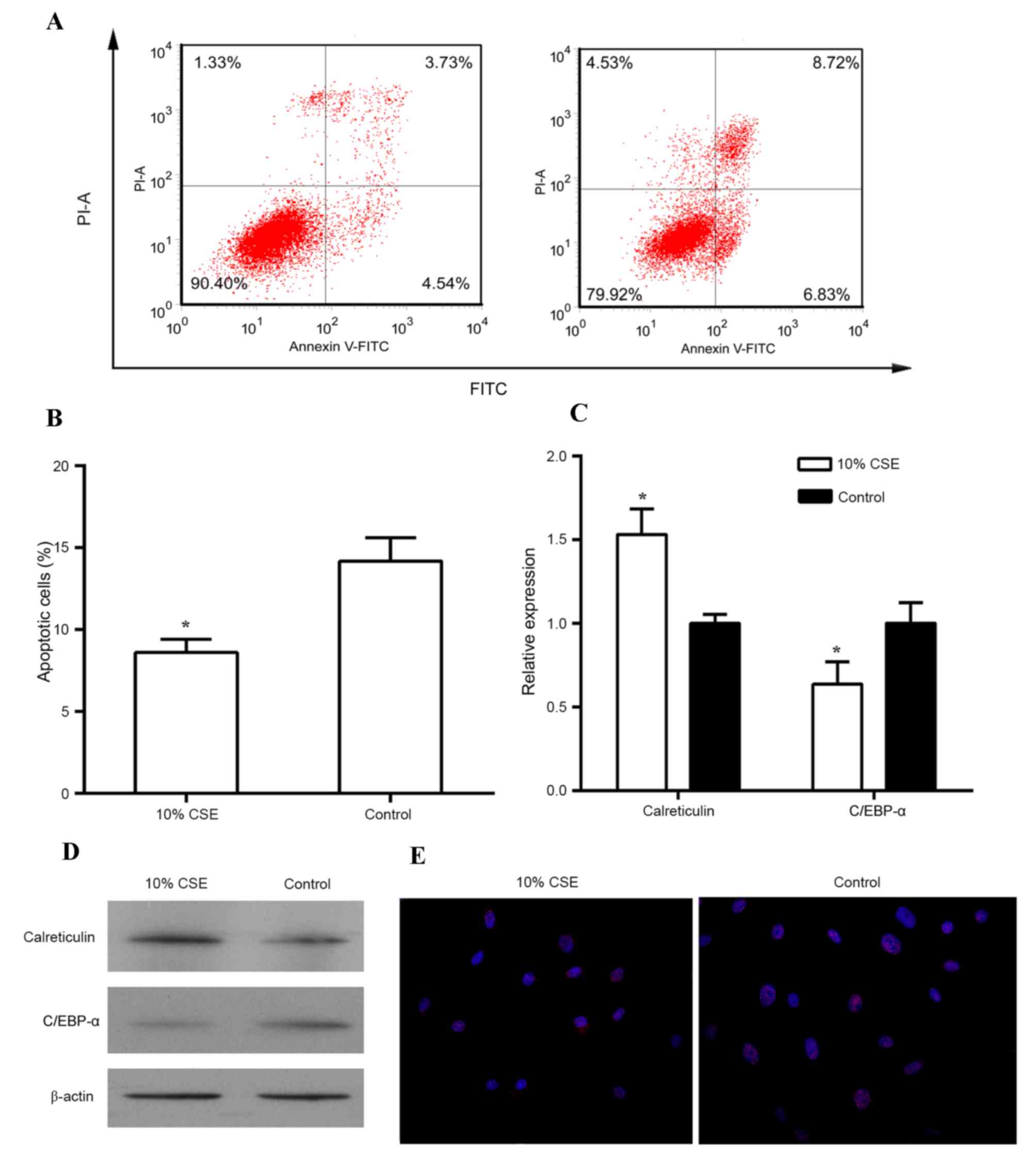
1B). In addition, treatment with 10% CSE reduced the apoptotic
rate of human ASMCs in comparison to that of untreated cells
(Fig. 2A and B). A previous study
demonstrated that C/EBP-α and its regulator calreticulin are
implicated in the control of cell proliferation (16). Thus, the expression of calreticulin
and C/EBP-α was then examined in these cells treated with CSE. As
shown in Fig. 2C and D, treatment of
human ASMCs with 10% CSE resulted in an increase in mRNA and
protein levels of calreticulin but caused a decrease in mRNA and
protein levels of C/EBP-α as compared to those in the control cells
(all P<0.05). In addition, immunostaining with antibodies
against C/EBP-α revealed that 10% CSE suppressed the expression of
C/EBP-α in the nuclei of human ASMCs (Fig. 2E).
Knockdown of calreticulin suppresses
the proliferation of human ASMCs
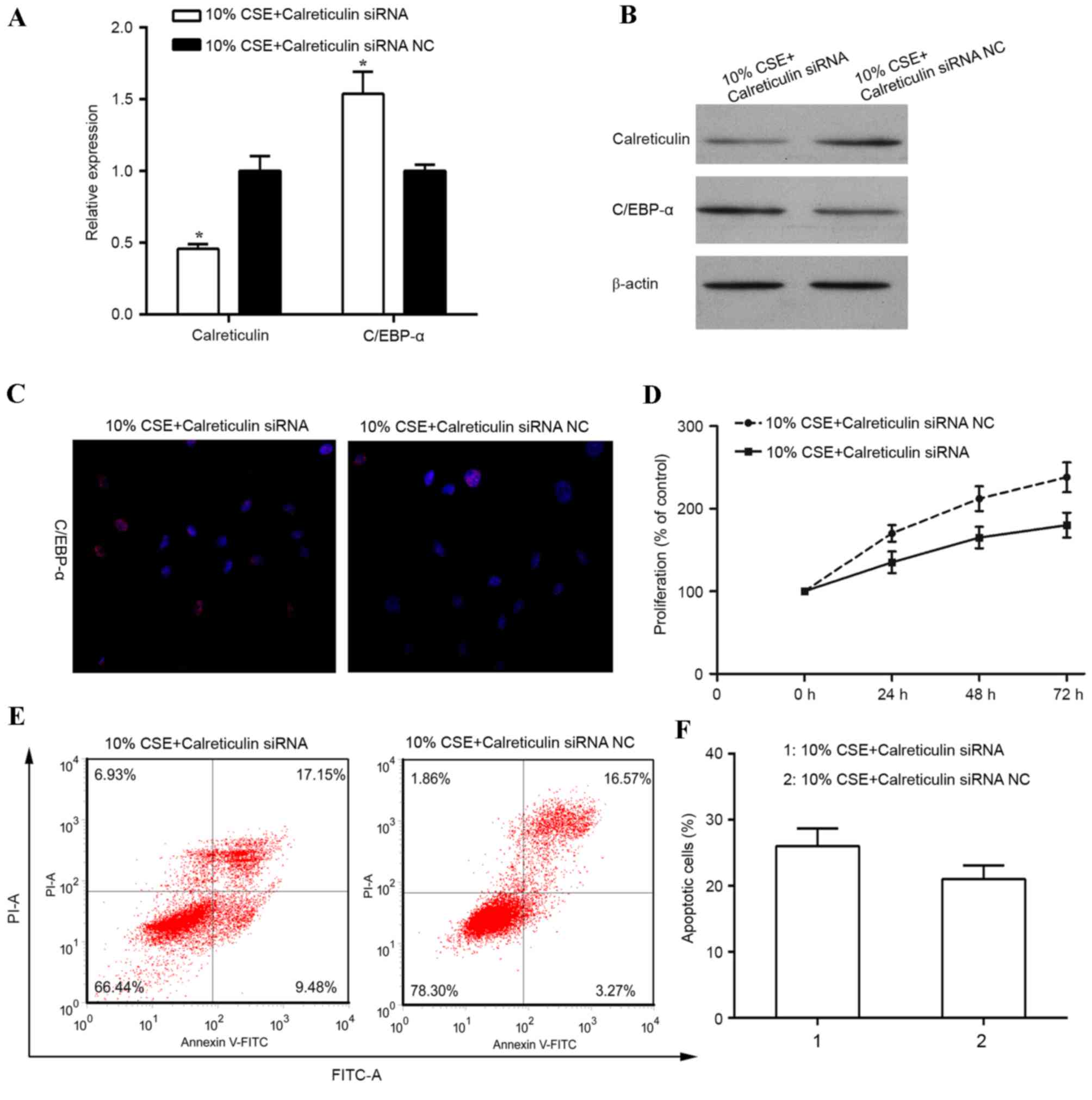
To investigate the role of calreticulin in the
regulation of cell proliferation in human ASMCs treated with CSE,
knockdown of calreticulin was performed and the effect on cell
proliferation and apoptosis was examined. RT-qPCR and western blot
analysis revealed the calreticulin knockdown efficiency (Fig. 3A and B). When ASMCs were transfected
with calreticulin siRNA, the expression of calreticulin was
significantly diminished at the mRNA level (P<0.05) and also
suppressed at the protein level, compared with that in control
siRNA-transfected ASMCs stimulated with 10% CSE. A previous study
reported that calreticulin transcriptionally regulates C/EBP-α
through a cis-regulatory CNG-rich loop in the mRNA of C/EBP-α
(19). In the present study,
knockdown of calreticulin resulted in an upregulation of the mRNA
(P<0.05) and protein levels of C/EBP-α, and an increase in
C/EBP-α expression in the nucleus in human ASMCs stimulated with
10% CSE (Fig. 3A-C). In addition,
knockdown of calreticulin was found to suppress ASMC proliferation
induced by 10% CSE (Fig. 3D).
Furthermore, the percentage of apoptotic cells was increased in
CSE-stimulated human ASMCs with calreticulin knockdown (Fig. 3E and F).
Simultaneous knockdown of calreticulin
and C/EBP-α increases the proliferation of human ASMCs
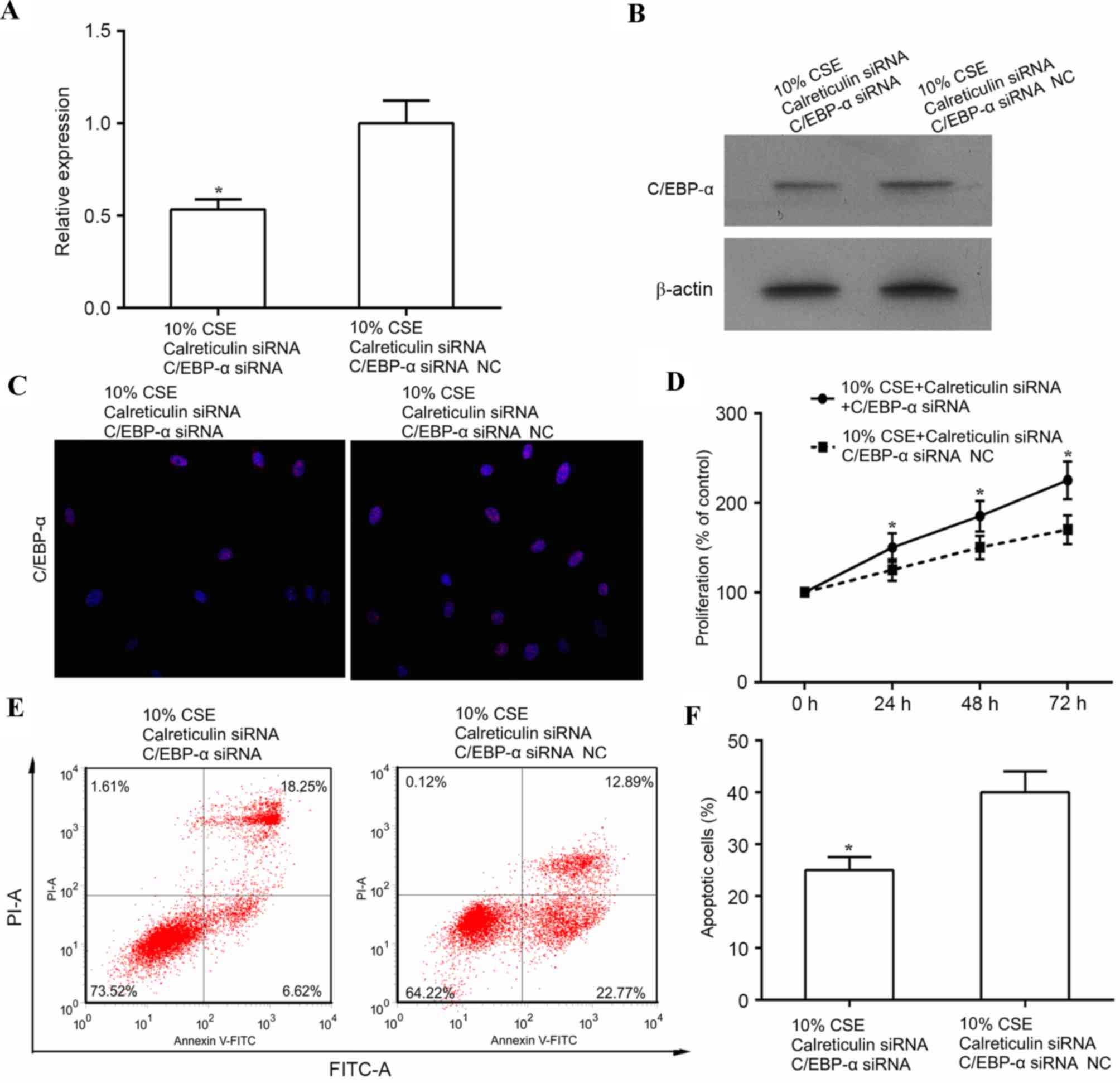
The aforementioned results indicated that CSE (10%)
promoted cell proliferation, which was associated with increased
expression of calreticulin and decreased expression of C/EBP-α. In
addition, knockdown of calreticulin upregulated C/EBP-α and
suppressed cell proliferation. This evidence led to the hypothesis
that CSE promotes ASMC proliferation through suppressing of C/EBP-α
via upregulation of calreticulin. To further test this, human ASMCs
were co-transfected with calreticulin siRNA, and either C/EBP-α
siRNA or control siRNA, and cell proliferation and apoptosis was
examined in these cells following treatment with 10% CSE. As shown
in Fig. 4A and B, the expression of
C/EBP-α was significantly decreased at the mRNA level (P<0.05)
and markedly suppressed at the protein level by simultaneous
knockdown of calreticulin and C/EBP-α as compared with that in the
control siRNA (knockdown of calreticulin only) in CSE-treated human
ASMCs. In addition, double knockdown of calreticulin and C/EBP-α
reduced the expression of C/EBP-α in the nuclei of ASMCs compared
with that in ASMCs subjected to knockdown of calreticulin alone
(Fig. 4C). Of note, double knockdown
of calreticulin and C/EBP-α led to increased cell proliferation
(P<0.05) and reduced apoptosis (P<0.05) in the calreticulin
knockdown only group (Fig.
4D-F).
Discussion
Cigarette smoke has been considered a major factor
in the pathogenesis of COPD, which causes inflammatory injury
(20,21). The present study showed that CSE
promoted ASMC proliferation, which is a key factor of airway
remodeling in COPD (3,4,22). The
effect of CSE at 0–10% on ASMC proliferation was
concentration-dependent; however, CSE at >10% had a cytotoxic
effect. This observation is in agreement with that of previous
studies reporting that CSE caused necrosis of neonatal vascular
SMCs and this toxicity was mainly mediated by volatile components
such as acrolein and acetaldehyde, possibly in association with
nitric oxide and carbon monoxide (23). In the present study, C/EBP-α was
identified as a molecular target of CSE. Treatment with CSE
significantly decreased C/EBP-α protein levels, particularly the
biologically reactive form, which resides in the nucleus. These
findings were consistent with those of other studies suggesting
that CSE promoted ASMC proliferation (5,6,24) and inhibited C/EBP-α (9,10).
Additionally, a previous study indicated that CSE can significantly
induce proliferation, with C/EBP-α and C/EBP-β proteins upregulated
simultaneously (25). However, to
the best of our knowledge, the present study was the first to
propose that CSE stimulated ASMC proliferation through C/EBP-α.
The present study further investigated the upstream
signaling of C/EBP-α. C/EBP-α can be detected in the liver, adipose
tissue, intestine, lung, adrenal gland as well as myeloid and
placental cells. Studies on BSMCs and abiogenesis revealed that
C/EBP-α is regulated by calreticulin (12,26).
Thus, in the present system, calreticulin levels were assessed
after CSE treatment its association with C/EBP-α was assessed.
Following treatment with 10% CSE calreticulin was significantly
increased at the RNA and protein level. Induction of calreticulin
was inversely correlated with C/EBP-α. Thus, the present study
hypothesized that calreticulin, which was upregulated by CSE,
functioned as a negative regulator of C/EBP-α. To verify this
hypothesis, siRNA-mediated knockdown of calreticulin and C/EBP-α
was performed. Cells' proliferation capacity impaired by
calreticulin knockdown was restored by simultaneous knockdown of
C/EBP-α. These results further supported the finding that CSE
promotes ASMC proliferation through inhibition of C/EBP-α. By
contrast, knockdown of calreticulin only suppressed ASMC
proliferation. Furthermore, cells with calreticulin knockdown had
significant higher C/EBP-α mRNA and protein levels. This result
provided evidence that calreticulin functions as negative regulator
of C/EBP-α.
The results of the present study revealed that the
induction of C/EBP-α is more profound at the protein level than at
the RNA level. This may be explained by the mechanism of
regulation, as C/EBP-α is predominantly regulated at the
translational level. A previous study reported that calreticulin
transcriptionally regulated C/EBP-α through a cis-regulatory
CNG-rich loop in the mRNA of C/EBP-α (16). Calreticulin binds to a stem loop
within the C/EBP-α mRNA, which is formed by internal base-pairing
of the (GC)n repeat motif. When calreticulin is bound to this loop,
translation of the full-length C/EBP-α (p42) is inhibited.
It has been demonstrated that intervention with
anisodamine, an antagonist of muscarinic acetylcholine receptors,
via increasing the expression of cyclin D1, prevents
smoking-induced ASMC proliferation, exerting a protective and
reversing effect regarding CSE-induced changes (27,28). It
has been suggested that inhibition of four and a half LIM domains
protein 1 limited the CSE-induced proliferation of pulmonary
arterial SMCs and may represent a potential therapeutic target for
pulmonary hypertension (19).
Furthermore, the results of the present study revealed that CSE
promoted the proliferation of ASMCs via induction of calreticulin,
which inhibits the expression of C/EBP-α. This result provided the
molecular link for airway remodeling in COPD and may provide
insight into the molecular pathogenetic mechanism as well as
possible therapeutic approaches with calreticulin or C/EBP-α as key
targets.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Hainan Province (grant no. 807081).
References
|
1
|
Gorska K, Krenke R, Kosciuch J, Korczynski
P, Zukowska M, Domagala-Kulawik J, Maskey-Warzechowska M and Chazan
R: Relationship between airway inflammation and remodeling in
patients with asthma and chronic obstructive pulmonary disease. Eur
J Med Res. 14 Suppl 4:S90–S96. 2009.
|
|
2
|
Yawn BP: Differential assessment and
management of asthma vs chronic obstructive pulmonary disease.
Medscape J Med. 11:202009.PubMed/NCBI
|
|
3
|
Hogg JC, Chu F, Utokaparch S, Woods R,
Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson
HO and Paré PD: The nature of small-airway obstruction in chronic
obstructive pulmonary disease. N Engl J Med. 350:2645–2653. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pini L, Pinelli V, Modina D, Bezzi M,
Tiberio L and Tantucci C: Central airways remodeling in COPD
patients. Int J Chron Obstruct Pulmon Dis. 9:927–932. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pera T, Gosens R, Lesterhuis AH, Sami R,
van der Toorn M, Zaagsma J and Meurs H: Cigarette smoke and
lipopolysaccharide induce a proliferative airway smooth muscle
phenotype. Respir Res. 11:482010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He F, Li B, Zhao Z, Zhou Y, Hu G, Zou W,
Hong W, Zou Y, Jiang C, Zhao D and Ran P: The pro-proliferative
effects of nicotine and its underlying mechanism on rat airway
smooth muscle cells. PLoS One. 9:e935082014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu J, Wu YN, Zhang W, Zhang XM, Ding X,
Li HQ, Geng M, Xie ZQ and Wu HM: Monocarboxylate transporter 4
facilitates cell proliferation and migration and is associated with
poor prognosis in oral squamous cell carcinoma patients. PLoS One.
9:e879042014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seow CY, Schellenberg RR and Paré PD:
Structural and functional changes in the airway smooth muscle of
asthmatic subjects. Am J Respir Crit Care Med. 158:S179–S186. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Didon L, Roos AB, Elmberger GP, Gonzalez
FJ and Nord M: Lung-specific inactivation of CCAAT/enhancer binding
protein alpha causes a pathological pattern characteristic of COPD.
Eur Respir J. 35:186–197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roth M, Johnson PR, Borger P, Bihl MP,
Rüdiger JJ, King GG, Ge Q, Hostettler K, Burgess JK, Black JL and
Tamm M: Dysfunctional interaction of C/EBPalpha and the
glucocorticoid receptor in asthmatic bronchial smooth-muscle cells.
N Engl J Med. 351:560–574. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang H, Iakova P, Wilde M, Welm A, Goode
T, Roesler WJ and Timchenko NA: C/EBPalpha arrests cell
proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell.
8:817–828. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miglino N, Roth M, Lardinois D, Tamm M and
Borger P: Calreticulin is a negative regulator of bronchial smooth
muscle cell proliferation. J Allergy (Cairo).
2012:7832902012.PubMed/NCBI
|
|
13
|
Wang WA, Groenendyk J and Michalak M:
Calreticulin signaling in health and disease. Int J Biochem Cell
Biol. 44:842–846. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiang WF, Hwang TZ, Hour TC, Wang LH,
Chiu CC, Chen HR, Wu YJ, Wang CC, Wang LF, Chien CY, et al:
Calreticulin, an endoplasmic reticulum-resident protein, is highly
expressed and essential for cell proliferation and migration in
oral squamous cell carcinoma. Oral Oncol. 49:534–541. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu YC, Weng WC and Lee H: Functional roles
of calreticulin in cancer biology. Biomed Res Int. 2015:5265242015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Timchenko LT, Iakova P, Welm AL, Cai ZJ
and Timchenko NA: Calreticulin interacts with C/EBPalpha and
C/EBPbeta mRNAs and represses translation of C/EBP proteins. Mol
Cell Biol. 22:7242–7257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pera T, Atmaj C, van der Vegt M, Halayko
AJ, Zaagsma J and Meurs H: Role for TAK1 in cigarette smoke-induced
proinflammatory signaling and IL-8 release by human airway smooth
muscle cells. Am J Physiol Lung Cell Mol Physiol. 303:L272–L278.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Y, Pu G, Chen C and Yang L: Inhibition
of FHL1 inhibits cigarette smoke extract-induced proliferation in
pulmonary arterial smooth muscle cells. Mol Med Rep. 12:3801–3808.
2015.PubMed/NCBI
|
|
20
|
Mortaz E, Kraneveld AD, Smit JJ, Kool M,
Lambrecht BN, Kunkel SL, Lukacs NW, Nijkamp FP and Folkerts G:
Effect of cigarette smoke extract on dendritic cells and their
impact on T-cell proliferation. PLoS One. 4:e49462009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu XJ, Luo GX, Zeng X, Lan LL, Ning Q, Xu
YJ, Zhao JP and Xie JG: Genotoxicity and reduced heat shock protein
70 in human airway smooth muscle cells exposed to cigarette smoke
extract. J Huazhong Univ Sci Technolog Med Sci. 33:827–833. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen QW, Edvinsson L and Xu CB: Cigarette
smoke extract promotes human vascular smooth muscle cell
proliferation and survival through ERK1/2- and NF-κB-dependent
pathways. ScientificWorldJournal. 10:2139–2156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ambalavanan N, Carlo WF, Bulger A, Shi J
and Philips JB III: Effect of cigarette smoke extract on neonatal
porcine vascular smooth muscle cells. Toxicol Appl Pharmacol.
170:130–136. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang XY, Xu YJ, Liu XS and Zhang ZX:
Cigarette smoke extract promotes proliferation of airway smooth
muscle cells in asthmatic rats via regulating cyclin D1 expression.
Chin Med J (Engl). 123:1709–1714. 2010.PubMed/NCBI
|
|
25
|
Helbling D, Mueller BU, Timchenko NA,
Schardt J, Eyer M, Betts DR, Jotterand M, Meyer-Monard S, Fey MF
and Pabst T: CBFB-SMMHC is correlated with increased calreticulin
expression and suppresses the granulocytic differentiation factor
CEBPA in AML with inv (16). Blood. 106:1369–1375. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miglino N, Roth M, Lardinois D, Sadowski
C, Tamm P and Borger P: Cigarette smoke inhibits lung fibroblast
proliferation by translational mechanisms. Eur Respir J.
39:705–711. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu GN, Yang K, Xu ZP, Zhu L, Hou LN, Qi H,
Chen HZ and Cui YY: Protective effects of anisodamine on cigarette
smoke extract-induced airway smooth muscle cell proliferation and
tracheal contractility. Toxicol Appl Pharmacol. 262:70–79. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zeng DX, Xu YJ, Liu XS, Wang R and Xiang
M: Cigarette smoke extract induced rat pulmonary artery smooth
muscle cells proliferation via PKCα-mediated cyclin D1 expression.
J Cell Biochem. 112:2082–2088. 2011. View Article : Google Scholar : PubMed/NCBI
|