Introduction
Human embryonic stem cells (hESC) were originally
derived and maintained in culture systems containing inactivated
mouse embryonic fibroblast (MEF) feeder layers to permit them to
grow continuously in an undifferentiated state (1,2). Induced
pluripotent stem cells (iPSCs) generated from somatic cells were
also cultured on MEF (3). The
possibility of differentiating these pluripotent cells into cells
with a defined phenotype is of great importance in scientific
research. However, effective alternatives to xenogenic-based
culture systems must be developed if therapies using cell types
derived from pluripotent cells are to become a clinical reality
(4,5). In this clinical scenario, feeder-free
culture systems have been developed to avoid the use of MEF
(4–7). However, despite the availability of
these alternative techniques, feeder layers are still considered to
be the best culture method owing to their ability to preserve
certain critical properties such as cell growth and viability,
stemness phenotype and clonogenic potential (8). Several previous studies have reported
that human-derived cells and decellularized cellular matrices are
feasible as feeder layers to support hESCs in culture (9–13);
however, there is no consensus on the best cell type to use.
Therefore, human hair follicle-derived mesenchymal stem cells
(hHFDCs) have emerged as a promising source of feeder-layer cells
for hESCs as they can be obtained from both males and females, they
have the capability of long-term proliferation, there is a
plentiful supply of starting material and the possibility of
autologous matching, and they are compatible with clinical
applications (14).
In the present study, hHFDCs were isolated, cultured
and characterized in order to evaluate whether they can be used as
a substitute for MEFs as a feeder layer for hESCs.
Materials and methods
Isolation and culture of mouse
embryonic fibroblasts
The present study was approved by the Ethics
Committee of the Health Sciences Center of the Federal University
of Rio de Janeiro (Protocol 026; Rio de Janeiro, RJ, Brazil) and
was conducted in compliance with National Institutes of Health
policies (15).
A total of 5 8-week-old pregnant female C57/BL6 mice
(20–30 g) were obtained from the Institute of Biophysics Carlos
Chagas Filho (Rio de Janeiro, Brazil). The animal protocol was
designed to minimize pain or discomfort to the animals. Animals
were housed at a controlled temperature (23°C) and humidity (55%)
under a 12:12 h light-dark cycle and received standard mouse chow
and water ad libitum.
MEFs were derived from C57Bl/6 mouse
strain embryos
Pregnant females were euthanized by barbiturate
overdose (150 mg/kg thiopental; Thiopentax; Cristália Produtos
Químicos Farmacêuticos, Itapira, Sao Paulo, SP, Brazil) at day 13.5
of gestation. The embryos were placed in 100-mm plastic culture
dishes containing sterile ice-cold PBS (LGC Biotecnologia, Cotia,
Sao Paulo, SP, Brazil). Using a magnifying glass and tweezers, the
viscera were removed and the remaining cells were transferred to
culture dishes containing Dulbecco's modified Eagle's medium
(DMEM)/F12 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) with 0.25% trypsin-EDTA solution (Gibco; Thermo Fisher
Scientific, Inc.) for enzymatic dissociation. All material was
poured into 15 ml conic tubes, and 3 ml DMEM-F12 supplemented with
200 ml/l fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.), 2 mmol/l L-glutamine (Gibco; Thermo Fisher Scientific,
Inc.), 50 U/ml penicillin (Gibco; Thermo Fisher Scientific, Inc.)
and 50 µg/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc.)
were added to inactivate trypsin. The solution was subsequently
centrifuged at 300 × g for 5 min at room temperature. The
supernatant was discarded and the pellet resuspended in 10 ml of
the same medium. Finally, cells were seeded at a density of
approximately 105 cells/ml in 75 cm2 culture
flasks (Corning Incorporated, Corning, NY, USA) and placed in an
incubator for 3–4 days at 37°C with an atmosphere containing 5%
CO2 and 95% humidity.
Isolation and culture of human hair
follicle derived cells
Human skin tissue from the temporal region of the
scalp was obtained during rejuvenation facial plastic surgery on 3
patients between 40 and 60 years old from the Luiz Pimentel Plastic
Surgery Clinic in Niterói, Rio de Janeiro in 2008, with approval of
the Institutional Ethics and Research Committee. All patients gave
informed consent prior to the study. Skin tissue was washed
extensively in cold PBS, and fat was removed using scissors,
forceps and a magnifying glass in a 35 mm culture dish (Corning
Incorporated) containing cold PBS. The tissue was sectioned into 2
mm slices, transferred to an Erlenmeyer flask and subjected to
enzymatic dissociation with collagenase type II (360 U/mg;
Worthington Biochemical Corporation, Lakewood, NJ, USA) diluted in
DMEM, with gentle agitation overnight at 37°C. The digested
material was poured into 50 ml conical tubes and centrifuged at 380
× g for 5 min at room temperature. The supernatant was
discarded and the pellet was resuspended in low-glucose DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 150 ml/l
FBS, 50 U/ml penicillin and 50 µg/ml streptomycin. Cells were
seeded in 25 cm2 culture flasks (Corning Incorporated)
coated with 10 ml/l gelatin (Sigma-Aldrich; Merck kGaA, Darmstadt,
Germany) and cultured for 5–7 days in an incubator at 37°C, in an
atmosphere containing 5% CO2 and 95% humidity. The medium was
replenished every 2 days.
Feeder layer preparation
hHFDC (3–10 passages) and MEF (3 passages) were used
as feeder cells. These cells were mitotically inactivated using 10
µg/ml mitomycin C (Sigma-Aldrich; Merck kGaA) for 3 h and washed
three times with PBS. Inactivated hHFDC and MEF were then seeded on
100 ml/l gelatin (Sigma-Aldrich; Merck kGaA) coated 6-well plates
(Corning Incorporated) at 8×104 cells/well (35 mm).
Feeder cells were grown to confluence in their respective growth
media (as described above) and then the medium was changed to hESC
medium.
hESC culture on MEF and hHFD feeder
layers
The hESC line H9 (2)
was donated to the Federal University of Rio de Janeiro by Dr James
Thomson from the University of Wisconsin (Madison, WI, USA). At
passages 56–65, all the colonies of H9-hESC, which had until then
been cultured on MEF, were subsequently sub-cultured on inactivated
feeder cells (hHFDC and MEF) at 37°C for 4–5 days. Morphologically
undifferentiated, colony-forming cells were selected for each
passage and dissociated mechanically into small clumps with a
needle and micropipette tip, visualized using light microscopy. The
culture medium for H9 cells consisted of DMEM/F12 supplemented with
200 ml/l knockout serum (KSR; Gibco; Thermo Fisher Scientific,
Inc.), 10 ml/l non-essential amino acids (Gibco; Thermo Fisher
Scientific, Inc.), 0.1 mmol/l β-mercaptoethanol (Gibco; Thermo
Fisher Scientific, Inc.), 2 mmol/l L-glutamine, 55 µg/ml garamycin
(Schering-Plough Corporation, Kenilworth, NJ, USA) and 10 ng/ml
basic fibroblast growth factor (bFGF; Gibco; Thermo Fisher
Scientific, Inc.). The culture medium was replenished daily. Cells
were then frozen in liquid nitrogen using a freezing solution
consisting of 100 ml/l dimethyl sulfoxide (DMSO; Sigma-Aldrich;
Merck Millipore) and 900 ml/l FBS.
Flow cytometric analysis
hHFDCs at passage 3 were dissociated with 0.25%
trypsin-EDTA and counted for immunophenotypic analysis. Samples
were blocked with PBS supplemented with 50 ml/l FBS for 20 min at
4°C. Then, 0.5–1×106 cells were stained using monoclonal
antibodies (dilution according to the manufacturer's protocol) for
20 min in the dark at 4°C. The following monoclonal antibodies were
used, all at a dilution of 1:30: Cluster of differentiation
(CD)45-APC (cat. no. 340942), CD117-PercP-Cy5.5 (cat. no. 333947),
CD73-PE (cat. no. 550257), CD31-FITC (cat. no. 555445),
CD54-PercP-Cy5.5 (cat. no. 555512), CD166-PE (cat. no. 559263),
CD44-FITC (cat. no. 347943), CD146-PE (cat. no. 550315), CD19-FITC
(cat. no. 347543), CD14-PE (cat. no. 555398), human leukocyte
antigen-antigen D related (HLA-DR)-PercP-Cy5.5 (cat. no. 551375),
CD90-PE-Cy5 (cat. no. 555597; all BD Pharmingen, San Diego, CA,
USA), CD34-PE-Cy7 (cat. no. 348801; BD Biosciences, Franklin Lakes,
NJ, USA), CD105-FITC (cat. no. 2CHF11; Immunostep, Salamanca,
Spain), and CD133-PE (cat. no. 29303; Miltenyi Biotec, Inc.,
Cambridge, MA, USA). Following staining, the cells were washed with
PBS, centrifuged at 4°C and 250 × g for 5 min and resuspended in
PBS in preparation for processing. Samples were processed in a BD
FACSAria II instrument (BD Biosciences) and the results were
analyzed using BD FACSDiva 6.0.1 software (BD Biosciences). These
characterization experiments were repeated in three independent
hHFDC lines.
Population-doubling time (PDT)
assay
hHFDCs at passage 3 were seeded at a density of
~2×104 cells in 35 mm plates with a 2 mm grid (Nalge
Nunc International, Penfield, NY, USA). The next day, 4 quadrants
were randomly chosen and cells were counted each day for 7 days,
when the cells reached confluence and an exponential curve of cell
growth was constructed. By applying a base 2 logarithm to the
cell/mm2 axis a linear regression was performed and the
inverse of the angular coefficient α was used to calculate the
PDT.
In vitro hHFDC differentiation
hHFDCs were seeded at a density of 1×104
cells/cm2 in six-well plates. The culture medium was
then replaced by the specific differentiation medium. For
adipogenic differentiation, cells were cultured at 37°C in
adipogenic medium consisting of 4.5 g/l DMEM-high glucose (Gibco;
Thermo Fisher Scientific, Inc.) with 200 ml/l FBS, 2 mmol/l
L-glutamine, 50 U/ml penicillin and 50 µg/ml streptomycin,
10−7 M dexamethasone (Sigma-Aldrich; Merck kGaA), 2.07
µM insulin (Sigma-Aldrich; Merck kGaA) and 0.45 mmol/l
3-isobutyl-1-methylxanthine (Sigma-Aldrich; Merck kGaA) for 3
weeks. Differentiated cells were stained with 20 ml/l Oil Red O
(Sigma-Aldrich; Merck kGaA) to detect cytoplasmic lipid vacuoles.
For osteogenic differentiation, cells were cultured in osteogenic
medium consisting of DMEM-High glucose 4.5 g/l with 200 ml/l FBS, 2
mmol/l L-glutamine, 50 U/ml penicillin and 50 µg/ml streptomycin,
10−7 M dexamethasone, 0.5 µM ascorbic acid
(Sigma-Aldrich; Merck kGaA) and 10 mmol/l Na-β-glycerolphosphate
(Sigma-Aldrich; Merck kGaA)) for 3 weeks. Differentiated cells were
stained with 10 ml/l Alizarin Red (Sigma-Aldrich; Merck kGaA) to
detect extracellular calcium deposits. For chondrogenic
differentiation, cells were cultured at 37°C in chondrogenic medium
consisting of DMEM-high glucose 4.5 g/l with 200 ml/l FBS, 10 ng/ml
transforming growth factor β (TGF-β; Sigma-Aldrich; kGaA), 0.5
µg/ml insulin, 50 µM ascorbic acid and 10 ml/l goat serum for 3
weeks. Differentiated cells were processed for histology and
stained with 10 ml/l toluidine blue, which stains connective
tissue.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from cells using an RNeasy
micro kit (Qiagen, GmbH, Hilden, Germany) according to the
manufacturer's protocol. cDNA was synthesized from total RNA using
a High-Capacity Reverse Transcription kit (Applied Biosystems;
Thermo Fisher Scientific, Inc.) following the manufacturer's
protocol. Genomic DNA contamination was removed using the gDNA
Eliminator Spin Column that was part of the kit. Aliquots (500 ng)
of each DNA sample were amplified in a Peltier Thermal Cycler
PTC-200 (MJ; Bio-Rad Laboratories, Inc., Hercules, CA, USA) in a
20-µl reaction mixture containing 1X PCR Buffer (Promega
Corporation, Madison, WI, USA), 2.5 mM MgCl2, 0.2 mM of
each deoxynucleotide triphosphate (dNTP), 0.2 mM each of sense and
antisense primers, and 1.25 units of Go TaqR DNA Polymerase
(Promega Corporation). PCR was performed using the following
parameters: Denaturation at 95°C for 5 min, 30 cycles at 95°C for 1
min, primer-specific annealing temperature at 56–62°C (Tables I and II) for 1 min, extension at 72°C for 1 min
and a final extension step at 72°C for 10 min. Negative control
RT-PCRs were conducted under the same conditions without reverse
transcriptase. Internal control PCRs were conducted using primers
for GAPDH or β-actin. The sequences of primers and sizes of the
expected products are listed in Tables
I and II. The PCR product (n=3)
was electrophoresed using a 2% agarose gel (Roche Diagnostics,
Basel, Switzerland) and stained with ethidium bromide (E1510;
Sigma-Aldrich; Merck kGaA). Gels were visualized on a UV
transilluminator (UVVIS-20 Mighty Bright; Hoefer, Inc., Holliston,
MA, USA).
 | Table I.Primer sequences and polymerase chain
reaction conditions for hESC pluripotency and differentiation gene
expression. |
Table I.
Primer sequences and polymerase chain
reaction conditions for hESC pluripotency and differentiation gene
expression.
| Gene product | Sense primer
sequence Orientation: 5′-3′ | Anti-sense primer
sequence Orientation: 3′-5′ | Annealing
temperature (°C) | Size (base
pairs) |
|---|
| Oct3/4 |
AGCCTGAGGGCGAAGCAGGA |
CCCCAGGGTGAGCCCCACAT | 56 | 236 |
| Sox2 |
AGCTACAGCATGATGCAGGA |
GGTCATGGAGTTGTACTGCA | 56 | 126 |
| Klf4 |
TCTCAAGGCACACCTGCGAA |
TAGTGCCTGGTCAGTTCATC | 56 | 105 |
| Nanog |
CAGCCCTGATTCTTCCACCAGTCCC |
TGGAAGGTTCCCAGTCGGGTTCACC | 56 | 391 |
| Afp |
GAATGCTGCAAACTGACCACGCTGGAAC |
TGGCATTCAAGAGGGTTTTCAGTCTGGA | 62 | 281 |
| Gata6 |
GCCAACTGTCACACCACAAC |
TGGGGGAAGTATTTTTGCTG | 60 | 265 |
|
Brachyury |
GCCCTCTCCCTCCCCTCCACGCACAG |
CGGCGCCGTTGCTCACAGACCACAGG | 62 | 274 |
| Msx1 |
CGAGAGGACCCCGTGGATGCAGAG |
GGCGGCCATCTTCAGCTTCTCCAG | 62 | 305 |
| Nestin |
CACCTCAAGATGTCCCTCAG |
AGCAAAGATCCAAGACGCC | 59 | 176 |
| Gapdh |
ACCATGGGGAAGGTGAAGGT |
CATGGGTGGAATCATATTGG | 62 | 163 |
| Table II.Primer sequences and polymerase chain
reaction conditions for growth factor expression in MEF and hHFDC
feeders. |
Table II.
Primer sequences and polymerase chain
reaction conditions for growth factor expression in MEF and hHFDC
feeders.
| Gene product | Sense primer
sequence Orientation: 5′-3′ | Anti-sense primer
sequence Orientation: 3′-5′ | Anneling
temperature (°C) | Size (base
pairs) |
|---|
| qmBmp4 |
ATTGCAGCTTTCTAGAGGTCC |
GGGAGCCAATCTTGAACAAAC | 59 | 149 |
| qhBmp4 |
GCACTGGTCTTGAGTATCCTG |
TGCTGAGGTTAAAGAGGAAACG | 59 | 135 |
| qmFgf2 |
GTCTATCAAGGGAGTGTGTGC |
CTGGAGTATTTCCGTGACCG | 58 | 150 |
| qhFgf2 |
ACCCTCACATCAAGCTACAAC |
AAAAGAAACACTCATCCGTAACAC | 58 | 141 |
| qmTgfb |
CCTGAGTGGCTGTCTTTTGA |
CGTGGAGTTTGTTATCTTTGCTG | 61 | 124 |
| qhTgfb |
GCCTTTCCTGCTTCTCATGG |
TCCTTGCGGAAGTCAATGTAC | 61 | 150 |
| qmPedf |
ACATCCACAGCACCTACAAG |
TCCCATAGGACTTCTCCAGAG | 59 | 141 |
| qhPedf |
AGCTGAGTTATGAAGGCGAAG |
ATCCTCGTTCCACTCAAAGC | 59 | 149 |
| qmVegf |
GAACTTTCTGCTCTCTTGGG |
CTTCGCTGGTAGACATCCATG | 60 | 144 |
| qhVegf |
AGGGCAGAATCATCACGAAG |
GGATGGCTTGAAGATGTACTCG | 60 | 130 |
| Gapdh |
ACCATGGGGAAGGTGAAGGT |
CATGGGTGGAATCATATTGG | 62 | 163 |
| β-actin |
CATCACTATTGGCAACGAGCG |
ATGGATGCCACAGGATTCCA | 58 | 85 |
Alkaline phosphatase activity
hESCs at passages 4 and 10 were fixed in 40 ml/l
paraformaldehyde for 2 min at room temperature, permeabilized with
50 ml/l Triton™ X-100 (T9284; Sigma-Aldrich, Merck kGaA) and
stained using an alkaline phosphatase detection kit (SCR004; Merck
kGaA), containing Fast Red Violet (0.8 g/l), Naphthol phosphate
solution (4 mg/ml) and water in a 2:1:1 ratio at room temperature
away from light for 15 min. Red color reactions in cells were
visualized using phase contrast microscopy (Olympus Corporation,
Tokyo, Japan).
Immunohistochemistry
For immunofluorescence analysis, hESCs and hHFDCs
were washed with PBS and fixed with 40 ml/l paraformaldehyde in PBS
(pH 7.4) for 15 min at room temperature, permeabilized with three
washes with 30 ml/l Triton™ X-100 in PBS for 10 min and then
blocked with 20 ml/l bovine serum albumin (Sigma-Aldrich; Merck
kGaA) in PBS for 30 min at room temperature to prevent non-specific
binding. Incubation was carried out overnight at 4°C with the
following primary antibodies: Rabbit anti-human octamer-binding
transcription factor (Oct)4 polyclonal antibody (1:200; ab-19857;
Abcam, Cambridge, MA, USA) and mouse anti-human stage-specific
embryonic antigen (SSEA)-4 monoclonal antibody (1:100; MAB4304;
Merck kGaA). Cells were subsequently washed three times with PBS
for 10 min and incubated with the following secondary antibodies
for 1 h at room temperature: Alexa Fluor 488 conjugate goat
anti-mouse polyclonal antibody (1:400; A-21151; Thermo Fisher
Scientific, Inc.) and Cy3-AffinePure donkey anti-rabbit polyclonal
antibody (1:1,000; 711-165-152; Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA). The specificity of each
antibody was verified by negative controls included in each
experiment. In addition, nuclear DNA was stained with
4′-6-diamidino-2-phenylindole (DAPI; D9542; Sigma-Aldrich; Merck
kGaA) and coverslips were mounted in an anti-fade solution,
VECTASHIELD H-1000 (Vector Laboratories, Inc., Burlingame, CA,
USA). Fluorescence was observed using an inverted fluorescence
Zeiss Axiovert 130 microscope (Carl Zeiss Microscopy, GmbH,
Germany) coupled to an AxioVision version 4.6 software system (Carl
Zeiss AG, Oberkochen, Germany) and digital photomicrographs were
captured.
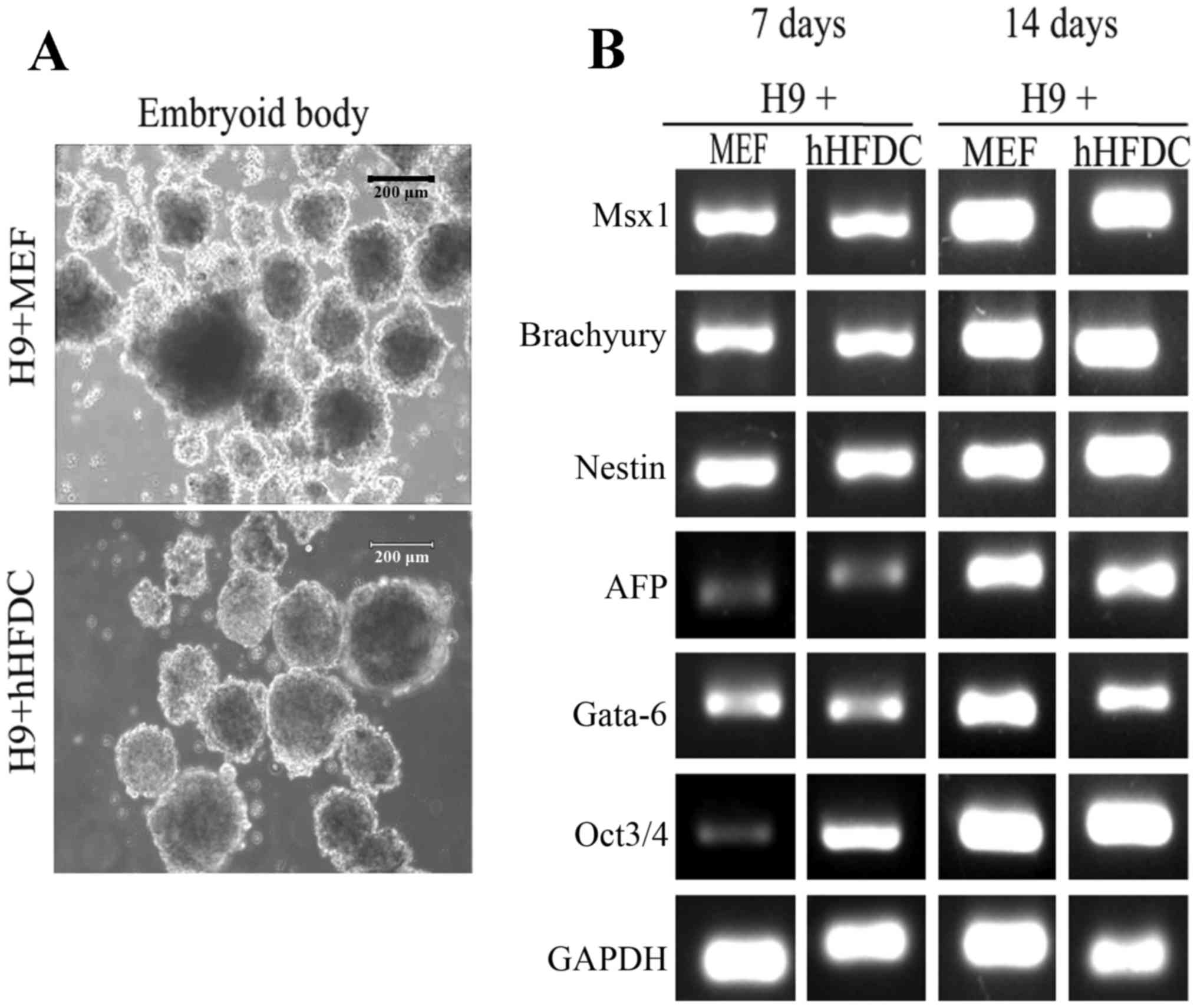
Embryoid body formation
For embryoid body (EB) formation, cell aggregates of
hESCs were grown in suspension in non-adherent plates without
feeder layers in medium without bFGF. After 1 week, EBs derived
from hESCs cultured over hHFDC or MEF were collected and
transferred to 2% gelatin-coated dishes for 2 more weeks and
subsequently analyzed using RT-PCR (as described above). The
markers used to test hESC differentiation into embryonic germ
layers were: Ectodermal (Nestin), mesodermal (Msx1 and Brachyury)
and endodermal (alpha fetoprotein and Gata 6). The sequences of
primers used are listed in Table
I.
Results
Characterization of hHFDC
For the long-term use of these cells as feeder
layers, primary cultured cells were cryopreserved at early passages
and maintained under the same culture conditions as MEF. hHFDCs
were efficiently cultured in vitro with high proliferation
rates and exhibiting elliptical nuclei and fibroblast-like
morphology (thin and elongated adherent cells) similar to MEF
(Fig. 1A). Immunophenotypical
analysis detected mesenchymal stem cells markers (CD105, CD90,
CD73, CD44, CD146 and CD166). Furthermore, hHFDCs exhibited low
expression of hematopoietic surface markers (<2%), such as CD14,
CD45, CD19 and HLA-DR, and endothelial or progenitor markers
including CD31, CD34, CD133 and CD117 (Fig. 1B). To further characterize the cells
and to assess the time to confluence during routine culture, the
doubling time of hHFDCs was measured. These cells exhibited rapid
proliferation, with a mean PDT of 34.5±0.02 h (n=3; Fig. 1C). To evaluate differentiation,
hHFDCs were induced to differentiate into adipogenic, osteogenic
and chondrogenic lineages (Fig. 1D).
Adipogenic differentiation of hHFDC was apparent following 3 weeks
of incubation in adipogenic medium. The formation of lipid vacuoles
was evident, as detected by positive Oil Red O staining. Similarly,
differentiation in osteogenic medium induced an osteoblastic
phenotype, with the cells being strongly stained by Alizarin Red,
which indicates the deposition of calcium in the extracellular
matrix. Connective tissue was observed in hHFDCs submitted to
chondrogenic induction via staining with toluidine blue.
Non-treated control cultures did not show spontaneous adipocyte,
osteoblast or chondroblast formation following 3 weeks of
culture.
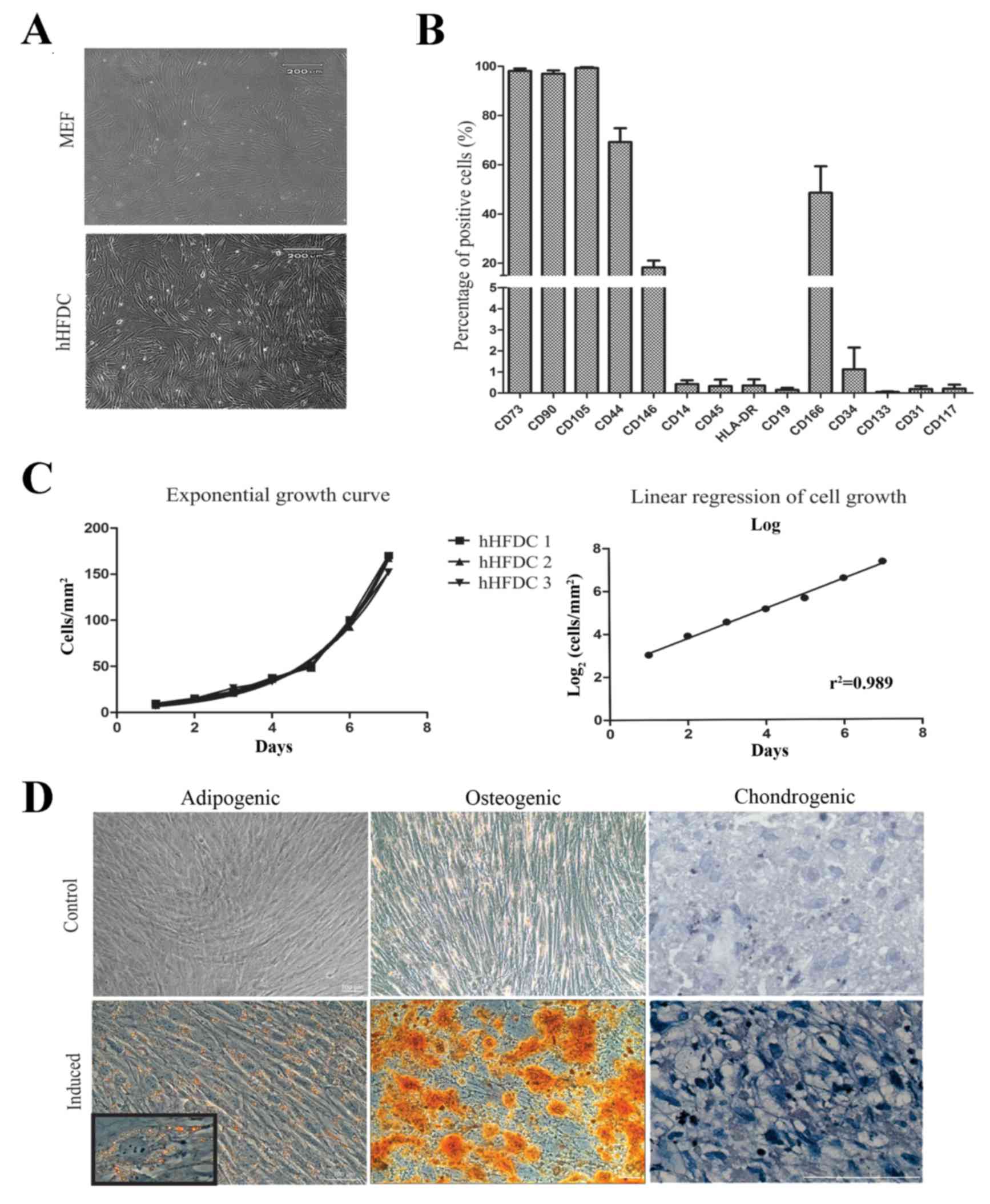 | Figure 1.hHFDC characterization. (A)
Morphology of MEF and hHFDC feeder layers. (B) Immunophenotypic
profile of hHFDC: Percentage of cells that express molecular
characteristics of mesenchymal, hematopoietic, endothelial and
progenitor cells, respectively. (C) PDT: hHFDCs show rapid
proliferation, with an average PDT of 34.5±0.02 h (n=3). (D)
Differentiation of hHFDCs into adipocytes, osteoblasts and
chondroblasts. Staining with Oil Red O for fat vacuoles
demonstrated differentiation into adipogenic tissue in induced and
non-induced controls (inset magnification, ×40); staining with
Alizarin Red in induced cells revealed calcium deposits compared
with non-induced controls. Staining with toluidine blue for
sulfated proteoglycans demonstrated the differentiation into
chondrogenic tissue types in induced compared with non-induced
hHFDC controls. Scale bar: 100 µm. hHFDC, human hair
follicle-derived mesenchymal stem cell; MEF, mouse embryonic
fibroblast; PDT, population doubling time; CD, cluster of
differentiation; HLA-DR, human leukocyte antigen-antigen D
related. |
hESC morphology
hESC colonies maintained on hHFDC feeder layers
exhibited typical morphology of undifferentiated hESC, including a
high nucleus-to-cytoplasm ratio, 1 to 3 nucleoli, rounded shape and
typical spacing between the cells, similar to hESCs cultured on MEF
(Fig. 2A and B). Continuous
proliferation of hESCs on the human feeder layer was normally
maintained for >10 passages with preservation of the
undifferentiated state (data not shown).
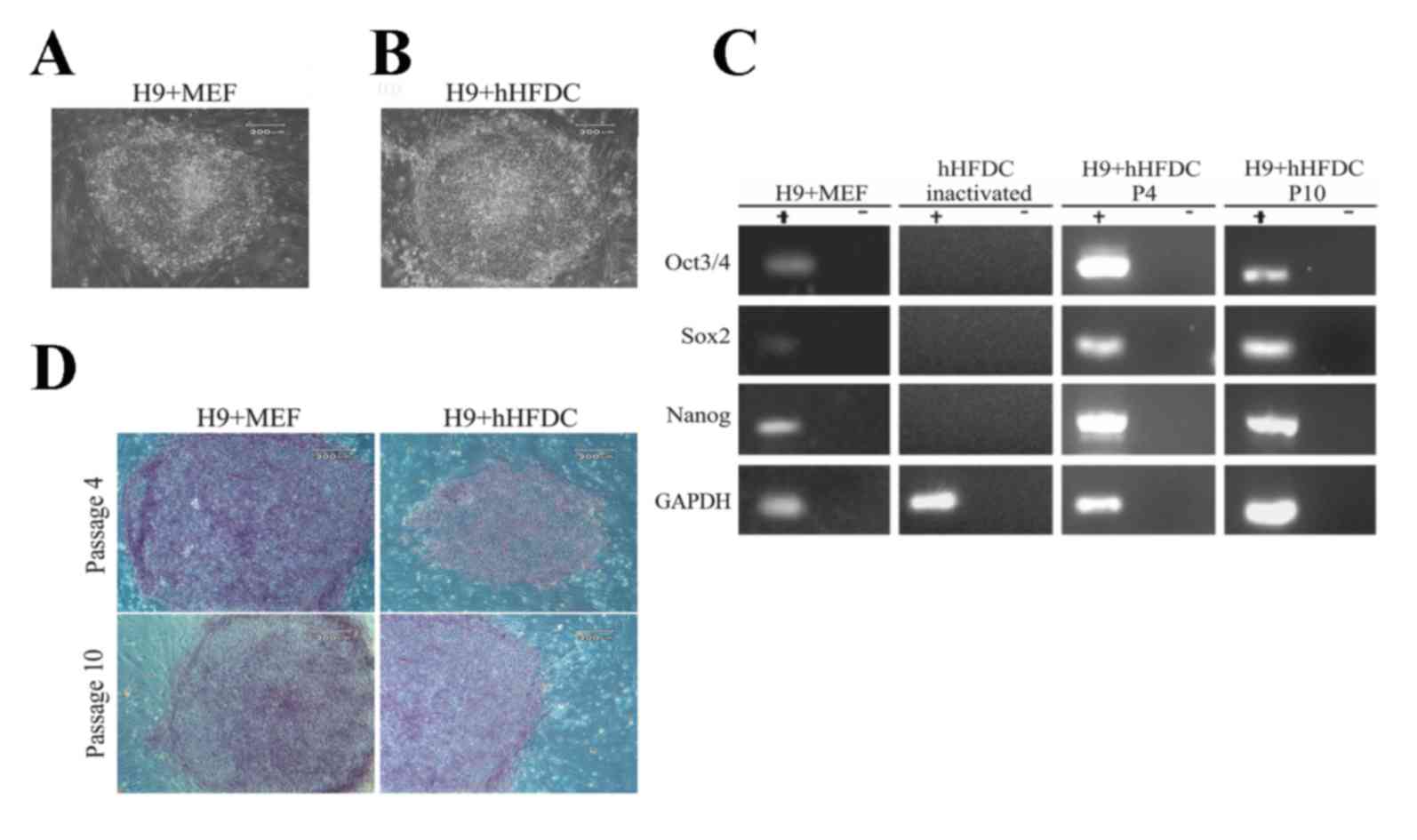 | Figure 2.Morphology of cells and colonies of
hESCs showing the expression of AP, transcription factors
characteristic of undifferentiated cells and pluripotency makers.
H9 cells grown over (A) MEF and (B) hHFDC. (C) Expression of
transcription factors characteristic of undifferentiated hESCs. MEF
as a positive control, inactivated HFDCS as a negative control, and
H9 cells in P4 and P10 grown over hHFDC. (D) Expression of AP in
hESCs grown over MEF and hHFDC in P4 and P10. Scale bar: (A-C) 200
µm. hESC, human embryonic stem cell; MEF, mouse embryonic
fibroblast; hHFDC, human hair follicle-derived mesenchymal stem
cell; AP, alkaline phosphatase; P, passage; Oct, octamer-binding
transcription factor; Sox2, sex determining region Y box 2. |
Pluripotency maintenance of hESC on
hHFDC
RT-PCR demonstrated that hESC colonies cultured on
hHFDC expressed pluripotency-associated genes, including
transcription factors Oct3/4, Sox2 and Nanog at passages 4 and 10,
similar to what is observed when these cells are cultured on MEF
(Fig. 2C). Inactivated hHFDCs were
used as a negative control for the PCR. Positive staining for
alkaline phosphatase was strongly detected in hESCs cultured on
both MEF and hHFDC feeders at passages 4 and 10 (Fig. 2D). Additionally, immunofluorescence
staining demonstrated the nuclear localization of Oct3/4 protein
and cytoplasmic localization of SSEA-4 in hESC colonies cultured on
hHFDC or MEF layers (Fig. 3).
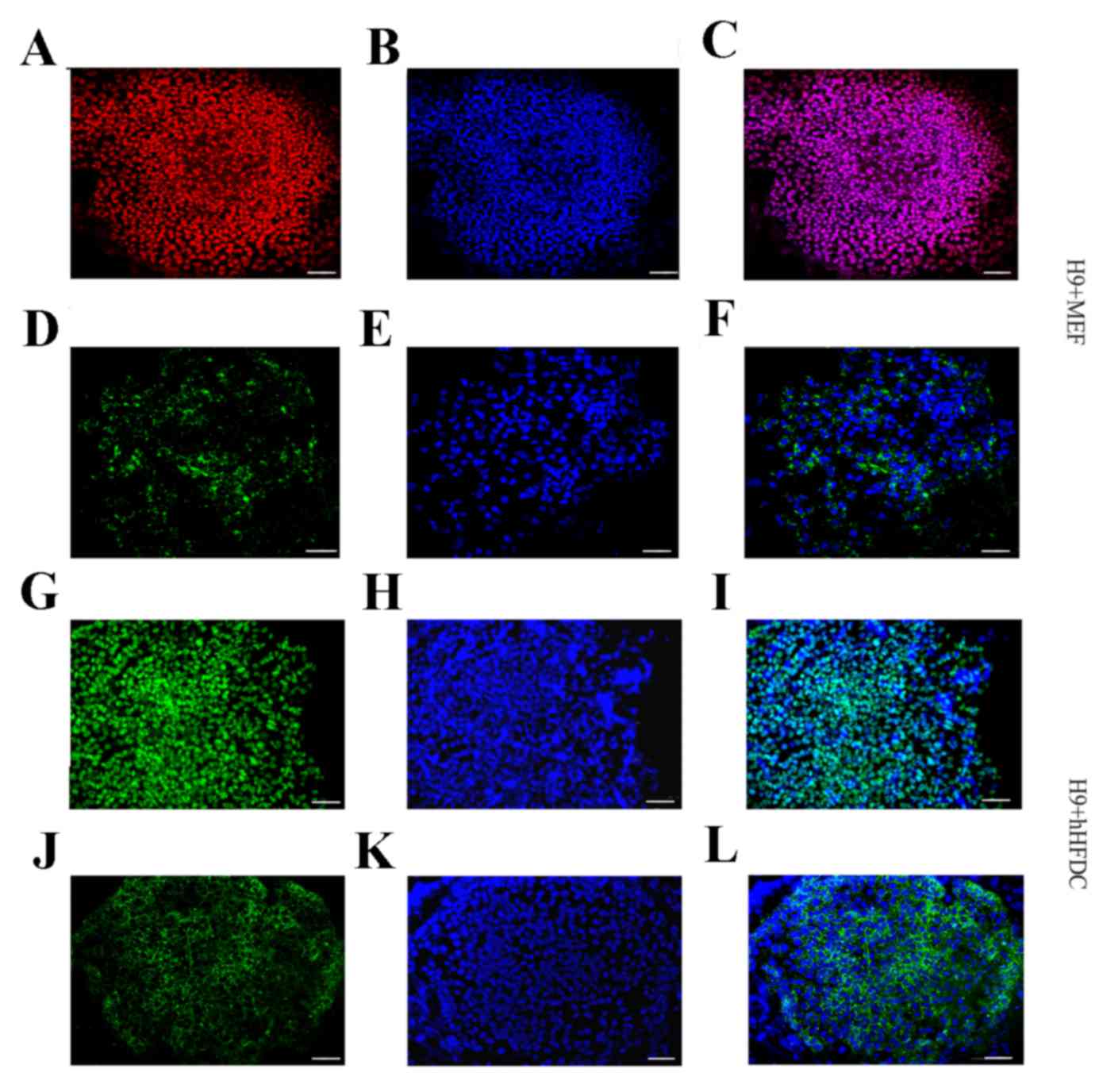 | Figure 3.Expression of pluripotency molecules
in undifferentiated hESCS grown over MEF and hHFDC. The cells were
positive for Oct3/4 (red and green; A and G, respectively) and
SSEA-4 (green; D and J). The nuclei were labeled with
4′,6-diamidino-2-phenylindole (blue; B, E, H and K). Merged images
(C, F, I and L). Scale bar: 20 µm. hESC, human embryonic stem cell;
MEF, mouse embryonic fibroblast; hHFDC, human hair follicle-derived
mesenchymal stem cell; P, passage; SSEA, stage-specific embryonic
antigen; Oct, octamer-binding transcription factor. |
Growth factor analysis
Growth factor analysis revealed similar
amplification of transcripts for bone morphogenetic protein
(BMP)-4, FGF-2, vascular endothelial growth factor (VEGF), pigment
epithelial-derived factor and TGF-β in hHFDCs and MEFs prior to and
following inactivation. Expression of all of these growth factors
was maintained when hESCs were cultured on both feeder layers
(Fig. 4).
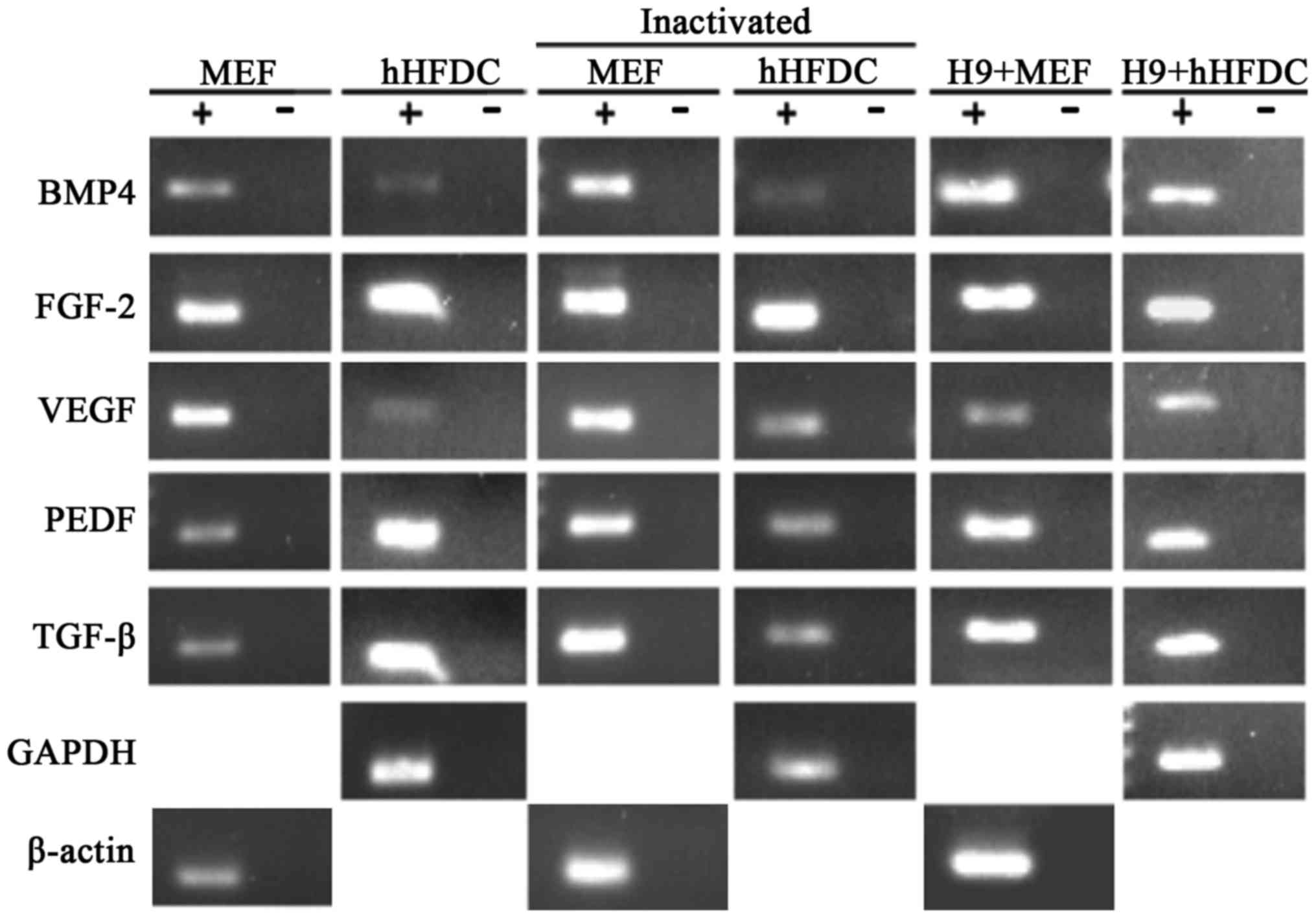 | Figure 4.Expression of growth factors in
feeder cells. Reverse transcription polymerase chain reaction
revealed amplification of transcripts in MEFs, hHFDCs, inactivated
MEFs, inactivated hHFDCs, H9 grown over MEF and H9 grown over
hHFDC. GAPDH and β-actin were used as internal controls. MEF, mouse
embryonic fibroblast; hHFDC, human hair follicle-derived
mesenchymal cell; BMP, bone morphogenetic protein; FGF, fibroblast
growth factor; VEGF, vascular endothelial growth factor; PEDF,
pigment epithelium-derived factor; TGF, transforming growth
factor. |
Differentiation capacity of hESC
To determine the differentiation capacity of
pluripotent hESC in vitro, EBs were cultured in suspension.
Similar to EBs grown on MEF feeders, hESCs on hHFDC underwent
spontaneous differentiation into the three germ layers when
cultured without a feeder layer and without bFGF in vitro
(Fig. 5A). The EBs cultured for 1
and 2 weeks expressed markers for ectoderm (Nestin), mesoderm
(Brachyury and Msx1) and endoderm (alpha fetoprotein and GATA 6),
confirming the pluripotent state of hESC grown on hHFDC and MEF
(Fig. 5B). Notably, Oct3/4 was
detected in EBs cultured on both feeder layers (Fig. 5B).
Discussion
Mesenchymal stem cells show great potential for use
in cellular therapies. In hair follicles, populations of transit
amplifying cells form bulbs that encase clusters of mesenchymal
cells known as dermal papilla (DP) cells (16). DP cells are the focus of intense
interest as they regulate hair follicle development and growth, and
are also thought to be a reservoir of multipotent stem cells
(17). Previous studies have
demonstrated that the hair follicle DP contains mesenchymal stem
cells (14,18–20);
however, the multipotentiality of hHFDCs has not been fully
investigated.
In the present study, a population of hHFDCs from
the scalp of patients undergoing facelift surgery were isolated and
characterized, and found to have features of mesenchymal stem
cells. Immunophenotypic analysis of hHFDCs cultured in vitro
was performed, and the results demonstrated that >95% of cells
expressed surface molecules characteristic of mesenchymal stem
cells (CD90, CD73, CD105), whereas <2% expressed hematopoietic
markers (CD45, CD34, CD14, CD19 and HLA-DR). These results are
similar to those of Liu et al (18), who also investigated human hair
follicle cells; however, a lower expression of CD105 (33.77%) was
reported than in the present study (99%). This difference may be
due to the cell type used (only cells that migrated from specific
regions of the hair follicle) and/or the shorter time (4 h) used in
the previous study for collagenase digestion (18). The results of the present study
demonstrates that this culture method is effective for expanding a
homogeneous population of mesenchymal stem cells, as prescribed by
the International Society for Cellular Therapy in 2006 (21). Furthermore, the adhesion molecules
CD44 and CD166 were expressed, which confirms that the cells
retained mesenchymal characteristics, again in consonance with the
results of Liu et al (18).
The present study also confirmed the presence of CD146, which is
considered a pericyte marker present in mesenchymal cells (22,23). The
results demonstrated a low expression of the molecules commonly
found in endothelial cells (CD31 and CD133) and progenitor cells
(c-kit), corroborating the results of a previous study by Wang
et al (24) investigating
umbilical cord cells.
The PDT recorded in the present study was also
consistent with the characteristics of stem cells, specifically a
high proliferation potential of 34.5 h. Similar results have been
demonstrated in previous studies utilizing cells of the outer root
sheath of the human hair follicle (PDT=33 h) (25) and human hair follicle-derived
mesenchymal cells (PDT=36 h) (14).
In addition, several studies have reported similar results for
mesenchymal cells from other sources, including menstrual blood
cells (PDT=24–37 h) (26,27) and umbilical cord blood cells (PDT=36
h) (28).
Furthermore, the present study determined that
hHFDCs are able to differentiate into several mesenchymal lineages
including osteoblasts, adipocytes and chondroblasts (14,18,20).
However, Bajpai et al (14)
reported a faster rate of adipogenic differentiation (14 days) than
that observed in the present study, in which lipid vacuoles in the
cytoplasm of hHFDCs were only observed 21 days following adipogenic
differentiation. This difference may be due to the supplementation
of bFGF in the culture medium used by Bajpai et al (14). Notably, the culture method used in
the present study was effective at producing human hair follicle
mesenchymal stem cells that maintained the expression of surface
molecules characteristic of mesenchymal cells and were able to
differentiate into three mesodermal lineages.
The hHFDC culture was established and it was
subsequently evaluated whether these cells could replace MEF as a
feeder layer for hESCs. hHFDCs are an attractive cell source in
regenerative medicine for several reasons: Cell isolation is
feasible, cells may potentially be used in both males and females
and they may allow autologous matching compatible with clinical
applications. hESCs grown over MEF and hHFDC layers were maintained
under the same culture conditions and hHFDCs displayed a
fibroblast-like morphology similar to MEF, as described in the
literature (14,18). No morphological changes were observed
when these cells were inactivated with mitomycin C (data not
shown), in agreement with the results of Lee et al (29). In previous studies the morphology of
hESCs maintained on human feeder layers differed slightly from
hESCs maintained on MEF layers (30,31).
However, hESCs grown on hHFDC were found to have typical hESC
morphology, with a high nucleus/cytoplasm ratio, prominent nucleoli
and close spacing between cells that were maintained for 10
passages (1,2). Similar results were found when hESCs
were cultured on foreskin cells (30) and menstrual blood cells (9).
Many authors use enzymatic digestion for hESCs
cultured on human feeder layers (30,32–34).
However, the commonly used enzymes including trypsin, collagenase
type IV and dispase (35), are
animal derived. Additionally, it has been observed that manual
dissection of colonies is better for maintaining genetic stability
compared with enzymatic disaggregation as a method of passaging
hESCs (36–39). To avoid genetic alterations and
minimize the risk of contamination with products of animal origin,
hESCs on hHFDC were passaged every 5 days by mechanical
dissociation in the present study, as reported by Cho et al
(40). Furthermore, an increase in
the number of differentiated colonies was observed when the
enzymatic digestion method was used (data not shown), as described
in the literature (41).
The pluripotency of hESCs on hHFDC was confirmed by
RT-PCR, which detected the expression of genes encoding
transcription factors Oct3/4, Sox2 and Nanog in passages 4 and 10.
Inactivated HFDCs did not express these genes, demonstrating that
expression only occurs in hESCs. Similar results were obtained by
Lee et al (33), who reported
that Oct3/4 was expressed in hESCs grown over placental cells.
Furthermore, hESCs grown over hHFDC expressed the surface protein
SSEA-4, the transcription factor Oct3/4, and alkaline phosphatase,
in accordance with results reported by Zhan et al (32) and Cho et al (40), who used umbilical cord cells as
feeder layers. It should be noted that none of the feeder-layer
cells expressed these proteins.
The results of the present study characterize these
cells as pluripotent stem cells, demonstrating that hHFDCs readily
retain the undifferentiated state of hESCs. It is important to
mention that previous studies have not evaluated the gene
expression or alkaline phosphatase of hESCs cultured on foreskin
cells (30,42,43) or
human umbilical cord blood cells (32).
Recently, several groups have demonstrated that
growth factors produced by fibroblasts, including FGF2, TGF-β,
BMP-4 and PDGF (41,44–48), may
serve a role in maintaining the undifferentiated growth of hESCs.
The most commonly used fibroblasts are MEF (1,2).
However, these primary cells senesce after 5 to 6 passages, thus
limiting their continued use (49)
and the need to derive new feeder cells may result in culture
variation. In the present study, feeder cells were established from
human hair follicles, which can be used for at least 16 passages as
feeders for hESCs. Previous studies have demonstrated that TGFβ and
BMP4 induce the catagen phase in hair follicles (50,51).
Conversely, VEGF is thought to be important for anagen maintenance
(52) and FGF-2 has been identified
as the major regulator in determining the patterning of hair
pigmentation (53). Growth factor
analysis revealed that all these transcripts were amplified in
hHFDCs before and after inactivation and when hESCs were grown on
hHFDC and MEF. Future studies investigating longer-term cultures
and the role and pathways for each of these factors in
undifferentiated hESC growth may provide more information regarding
clinical applications.
The differentiation capacity of hESC was confirmed
when cells were grown in suspension following continuous culture on
both types of feeder layers. The cells were able to differentiate,
spontaneously, forming EBs containing the cell types representative
of the three germ layer; this is, similar to results found in
studies using feeder layers of breast parenchymal cells,
endometrial cells and human fibroblast cells (33), placental cells (54), foreskin cells (30), umbilical cord blood cells (32), and umbilical cord stromal cells
(40). Following 14 days culture,
the expression of pluripotency transcription factors was still
detected, suggesting that 14 days was insufficient to induce the
differentiation of all EBs. These results are consistent with those
of a previous report in which undifferentiated cells grown on
foreskin cells expressed Oct3/4 even after one month of the EB
culture protocol (30).
In summary, the present study demonstrates that
hHFDCs are a suitable for use in feeder layers for human
pluripotent cells due to their high proliferative capacity and
availability, and the possibility of autologous use when used with
iPSC derived from the same patient hHFDCs are therefore a more
effective as feeder layers compared with other types of human
feeder cells.
Acknowledgements
The present study was supported by the Conselho
Nacional de Desenvolvimento Científico e Tecnológico (CNPq
308702/2010-7), Fundação Coordenação de Aperfeiçoamento de Pessoal
de Nível Superior (CAPES 504488/2010-4), Fundação de Apoio à
Pesquisa do Estado do Rio de Janeiro (FAPERJ E26/110.272/2014) and
DECIT Ministério da Saúde (465656/2014-5), Brazil.
References
|
1
|
Reubinoff BE, Pera MF, Fong CY, Trounson A
and Bongso A: Embryonic stem cell lines from human blastocysts:
Somatic differentiation in vitro. Nat Biotechnol. 18:399–404. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thomson JA, Itskovitz-Eldor J, Shapiro SS,
Waknitz MA, Swiergiel JJ, Marshall VS and Jones JM: Embryonic stem
cell lines derived from human blastocysts. Science. 282:1145–1147.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takahashi K, Tanabe K, Ohnuki M, Narita M,
Ichisaka T, Tomoda K and Yamanaka S: Induction of pluripotent stem
cells from adult human fibroblasts by defined factors. Cell.
131:861–872. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakagawa M, Taniguchi Y, Senda S, Takizawa
N, Ichisaka T, Asano K, Morizane A, Doi D, Takahashi J, Nishizawa
M, et al: A novel efficient feeder-free culture system for the
derivation of human induced pluripotent stem cells. Sci Rep.
4:35942014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu HF, Chai C, Lim TC, Leong MF, Lim JK,
Gao S, Lim JK and Wan AC: A defined xeno-free and feeder-free
culture system for the derivation, expansion and direct
differentiation of transgene-free patient-specific induced
pluripotent stem cells. Biomaterials. 35:2816–2826. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vuoristo S, Toivonen S, Weltner J, Mikkola
M, Ustinov J, Trokovic R, Palgi J, Lund R, Tuuri T and Otonkoski T:
A novel feeder-free culture system for human pluripotent stem cell
culture and induced pluripotent stem cell derivation. PLoS One.
8:e762052013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jang M, Lee ST, Kim JW, Yang JH, Yoon JK,
Park JC, Ryoo HM, van der Vlies AJ, Ahn JY, Hubbell JA, et al: A
feeder-free, defined three-dimensional polyethylene glycol-based
extracellular matrix niche for culture of human embryonic stem
cells. Biomaterials. 34:3571–3580. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scafetta G, Siciliano C, Frati G and De
Falco E: Culture of human limbal epithelial stem cells on tenon's
fibroblast feeder-layers: A translational approach. Methods Mol
Biol. 1283:187–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
dos Silva Santos D, de Coelho Oliveira VC,
Asensi KD, Vairo L, Carvalho AB, de Campos Carvalho AC and
Goldenberg RC: Human menstrual blood-derived mesenchymal cells as
new human feeder layer system for human embryonic stem cells. Cell
Med. 7:25–35. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma X, Li H, Xin S, Ma Y and Ouyang T:
Human amniotic fluid stem cells support undifferentiated
propagation and pluripotency of human embryonic stem cell without
b-FGF in a density dependent manner. Int J Clin Exp Pathol.
7:4661–4673. 2014.PubMed/NCBI
|
|
11
|
Chen Q, Qiu C, Huang Y, Jiang L, Huang Q,
Guo L and Liu T: Human amniotic epithelial cell feeder layers
maintain iPS cell pluripotency by inhibiting endogenous DNA
methyltransferase 1. Exp Ther Med. 6:1145–1154. 2013.PubMed/NCBI
|
|
12
|
Lim ML, Jungebluth P, Sjöqvist S, Nikdin
H, Kjartansdóttir KR, Unger C, Vassliev I and Macchiarini P:
Decellularized feeders: An optimized method for culturing
pluripotent cells. Stem Cells Transl Med. 2:975–982. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abraham S, Sheridan SD, Miller B and Rao
RR: Stable propagation of human embryonic and induced pluripotent
stem cells on decellularized human substrates. Biotechnol Prog.
26:1126–1134. 2010.PubMed/NCBI
|
|
14
|
Bajpai VK, Mistriotis P and Andreadis ST:
Clonal multipotency and effect of long-term in vitro expansion on
differentiation potential of human hair follicle derived
mesenchymal stem cells. Stem Cell Res. 8:74–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
National Research Council: Guide for the
Care and Use of Laboratory Animals. 8th. The National Academies
Press; Washington, DC: pp. 1–220. 2011
|
|
16
|
Pasolli HA: The hair follicle bulge: A
niche for adult stem cells. Microsc Microanal. 17:513–519. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Driskell RR, Clavel C, Rendi M and Watt
FM: Hair follicle dermal papilla cells at a glance. J Cell Sci.
124:1179–1182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu JY, Peng HF, Gopinath S, Tian J and
Andreadis ST: Derivation of functional smooth muscle cells from
multipotent human hair follicle mesenchymal stem cells. Tissue Eng
Part A. 16:2553–2564. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hoogduijn MJ, Gorjup E and Genever PG:
Comparative characterization of hair follicle dermal stem cells and
bone marrow mesenchymal stem cells. Stem Cells Dev. 15:49–60. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jahoda CA, Whitehouse J, Reynolds AJ and
Hole N: Hair follicle dermal cells differentiate into adipogenic
and osteogenic lineages. Exp Dermatol. 12:849–859. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dominici M, Le Blanc K, Mueller I,
Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A,
Prockop DJ and Horwitz E: Minimal criteria for defining multipotent
mesenchymal stromal cells. The International Society for Cellular
Therapy position statement. Cytotherapy. 8:315–317. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Buhring HJ, Treml S, Cerabona F, de Zwart
P, Kanz L and Sobiesiak M: Phenotypic characterization of distinct
human bone marrow derived MSC subsets. Ann N Y Acad Sci.
1176:124–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gronthos S, Franklin DM, Leddy HA, Robey
PG, Storms RW and Gimble JM: Surface protein characterization of
human adipose tissue-derived stromal cells. J Cell Physiol.
189:54–63. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang H, Hung S, Peng S, Huang C, Wei H,
Guo Y, Fu YS, Lai MC and Chen CC: Mesenchymal stem cells in the
wharton's jelly of the human umbilical cord. Stem Cells.
22:1330–1337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Na GY, Paek SH, Park BC, Kim DW, Lee WJ,
Kim MK and Kim JC: Isolation and characterization of outer root
sheath melanocytes of human hair follicles. Br J Dermatol.
155:902–909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Asensi KD, Fortunato RS, dos Santos DS,
Pacheco TS, de Rezende DF, Rodrigues DC, Mesquita FC,
Kasai-Brunswick TH, de Carvalho AC and Goldenberg RC: Reprogramming
to pluripotent state modifies mesenchymal stem cell resistance to
oxidative stress. J Cell Mol Med. 18:824–831. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Patel AN, Park E, Kuzman M, Benetti F,
Silva FJ and Allickson JG: Multipotent menstrual blood stromal stem
cells: Isolation, characterization, and differentiation. Cell
Transplant. 17:303–311. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nekanti U, Dastidar S, Venugopal P, Totey
S and Ta M: Increased proliferation and analysis of differential
gene expression in human Wharton's jelly-derived mesenchymal
stromal cells under hipoxya. Int J Biol Sci. 9:499–512. 2010.
View Article : Google Scholar
|
|
29
|
Lee CH, Park JH, Lee JH, Ahn JY, Park JH,
Lee BR, Kim DY and Lim JM: Replacement of mouse embryonic
fibroblasts with bone marrow stromal cells for use in establishing
and maintaining embryonic stem cells in mice. Cell Biol Int.
36:537–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Amit M, Margulets V, Segev H, Sharik K,
Laevsky I, Coleman R and Itskovitz-Eldor J: Human feeder layers for
human embryonic stem cells. Biol Reprod. 68:2150–2156. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheng L, Hammond H, Ye Z, Zhan X and
Dravid G: Human adult marrow cells support prolonged expansion of
human embryonic stem cells in culture. Stem Cells. 21:131–142.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhan X, Hill C, Brayton CF and Shamblott
MJ: Cells derived from human umbilical cord blood support the
long-term growth of undifferentiated human embryonic stem cells.
Cloning Stem Cells. 10:513–522. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee JB, Song JM, Lee JE, Park JH, Kim SJ,
Kang SM, Know JN, Kim MK, Roh SI and Yoon HS: Available human
feeder cells for the maintenance of human embryonic stem cells.
Reproduction. 128:727–735. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Richards M, Tan S, Fong CY, Biswas A, Chan
WK and Bongso A: Comparative evaluation of various human feeders
for prolonged undifferentiated growth of human embryonic stem
cells. Stem Cells. 21:546–556. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoffman LM and Carpenter MK:
Characterization and culture of human embryonic stem cells. Nat
Biotechnol. 23:699–708. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baker DE, Harrison NJ, Maltby E, Smith K,
Moore HD, Shaw PJ, Heath PR, Holden H and Andrews PW: Adaptation to
culture of human embryonic stem cells and oncogenesis in vivo. Nat
Biotechnol. 25:207–215. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mitalipova MM, Rao RR, Hoyer DM, Johnson
JA, Meisner LF, Jones KL, Dalton S and Stice SL: Preserving the
genetic integrity of human embryonic stem cells. Nat Biotechnol.
23:19–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brimble SN, Zeng X, Weiler DA, Luo Y, Liu
Y, Lyons IG, Freed WJ, Robins AJ, Rao MS and Schulz TC: Karyotypic
stability, genotyping, differentiation, feeder-free maintenance and
gene expression sampling in three human embryonic stem cell lines
derived prior to August 9, 2001. Stem Cells Dev. 13:585–597. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Draper JS, Smith K, Gokhale P, Moore HD,
Maltby E, Johnson J, Meisner L, Zwaka TP, Thomson JA and Andrews
PW: Recurrent gain of chromosomes 17q and 12 in cultured human
embryonic stem cells. Nat Biotechnol. 22:53–44. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cho M, Lee EJ, Nam H, Yang JH, Cho J, Lim
JM and Lee G: Human feeder layer system derived from umbilical cord
stromal cells for human embryonic stem cells. Fertil Steril.
93:2525–2531. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Villa-Diaz LG, Pacut C, Slawny NA, Ding J,
O'shea KS and Smith GD: Analysis of the factors that limit the
ability of feeder cells to maintain the undifferentiated state of
human embryonic stem cells. Stem Cells Dev. 18:641–651. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pekkanen-Mattila M, Ojala M, Kerkelä E,
Rajala K, Skottman H and Aalto-Setälä K: The effect of human and
mouse fibroblast feeder cells on cardiac differentiation of human
pluripotent stem cells. Stem Cells Int. 2012:8750592012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hovatta O, Mikkola M, Gertow K, Strömberg
AM, Inzunza J, Hreinsson J, Rozell B, Blennow E, Andäng M and
Ahrlund-Richter L: A culture system using human foreskin
fibroblasts as feeder cells allows production of human embryonic
stem cells. Hum Reprod. 18:1404–1409. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sánchez L, Gutierrez-Aranda I, Ligero G,
Martín M, Ayllón V, Real PJ, Ramos-Mejía V, Bueno C and Menendez P:
Maintenance of human embryonic stem cells in media conditioned by
human mesenchymal stem cells obviates the requirement of exogenous
basic fibroblast growth factor supplementation. Tissue Eng Part C
Methods. 18:387–396. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Anisimov SV, Christophersen NS, Correia
AS, Hall VJ, Sandelin I, Li JY and Brundin P: Identification of
molecules derived from human fibroblast feeder cells that support
the proliferation of human embryonic stem cells. Cell Mol Biol
Lett. 16:79–88. 2010.PubMed/NCBI
|
|
46
|
Xi J, Wang Y, Zhang P, He L, Nan X, Yue W
and Pei X: Human fetal liver stromal cells that overexpress bFGF
support growth and maintenance of human embryonic stem cells. PLoS
One. 5:1–10. 2010. View Article : Google Scholar
|
|
47
|
Kumar N, Pethe P and Bhartiya D: Role of
TGFbeta and myofibroblasts in supporting the propagation of human
embryonic stem cells in vitro. Int J Dev Biol. 54:1329–1336. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eiselleova L, Peterkova I, Neradil J,
Slaninova I, Hampl A and Dvorak P: Comparative study of mouse and
human feeder cells for human embryonic stem cells. Int J Dev Biol.
52:353–363. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Amand MM, Hanover JA and Shiloach J: A
comparison of strategies for immortalizating mouse embryonic
fibroblasts. J Biol Methods. 3:e412016. View Article : Google Scholar
|
|
50
|
Paus R and Foitzik K: In search of the
hair ‘hair cycle clock’: A guided tour. Differentiation.
72:489–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Stenn KS and Paus R: Controls of hair
follicle cycling. Physiol Rev. 81:449–494. 2001.PubMed/NCBI
|
|
52
|
Tong X and Coulombe PA: Keratin 17
modulates hair follicle cycling in a TNF alpha-dependent fahion.
Genes Dev. 20:1353–1364. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Weiner L, Han R, Scichitano BM, Li J,
Hasegawa K, Grossi M, Lee D and Brissette JL: Dedicated ephitelial
recipient cells determine pigmentation patterns. Cell. 130:932–942.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mckay TR, Camarasa MV, Iskender B, Ye J,
Bates N, Miller D, Fitzsimmons JC, Foxler D, Mee M, Sharp TV, et
al: Human feeder cell line for derivation and culture of
hESCs/hiPSc. Stem Cell Res. 7:154–162. 2011. View Article : Google Scholar : PubMed/NCBI
|