Introduction
Prostate cancer is the second most common cancer and
the sixth leading cause of cancer mortality worldwide (1). According to estimates of the American
Cancer Society, the expected number of new cases of prostate cancer
is 220,800, which accounted for ~25% of new cancer diagnoses in
2015. However, the expected number of prostate cancer deaths in
2015 was ~27,540, which was slightly less compared with the
expected mortality rate of 29,480 in 2014 (2). Although patients with hormone-sensitive
localized prostate cancer may experience successful outcomes with
the application of surgery, radiotherapy or hormonal therapy, the
disease inevitably progresses into castration-resistant and
metastatic prostate cancer, negatively affecting quality of life
and markedly reducing the survival rate. Therefore, it is necessary
to investigate the underlying mechanisms of the onset and
progression of prostate cancer in order to develop updated
therapeutics, as well as preventative strategies. Increasing
research has indicated that autophagy has an important role in
cancer, which may become an effective drug target in anticancer
therapy (3,4).
Cyclooxygenase-2 (COX-2), an inducible iso-enzyme
that converts arachidonic acid to prostaglandins, is involved in
cancer angiogenesis, apoptosis and invasion (5). Celecoxib is a specific COX-2 inhibitor
that competes with arachidonic acid for the active site of
cyclooxygenase. Numerous trials have indicated that the regular use
of nonsteroidal anti-inflammatory drugs may provide benefits
against malignancies (6,7).
In the present study, whether celecoxib was able to
induce apoptosis and autophagy in PC3 cells, an
androgen-independent cell line, was investigated. Furthermore,
whether celecoxib-induced autophagy exerted protective effects in
PC3 cells and the underlying mechanism for the induction of
autophagy were investigated. The present study also investigated
whether the inhibition of autophagy by targeting the activated
molecule was able to enhance apoptosis induced by celecoxib.
Materials and methods
Cells and cell culture
Prostate cancer PC3 cells were obtained from
Chongqing Key Laboratory of Molecular Oncology and Epigenetics
(Chongqing, China). Cells were cultured in RPMI 1640 medium
(Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc. Waltham, MA, USA), penicillin (100 µg/ml) and streptomycin
(100 µg/ml) at 37°C in an atmosphere of 95% air and 5%
CO2.
Reagents and antibodies
Antibodies for phospho-JNK (cat. no. 4668; 1:1,000),
JNK1 (cat. no. 3708; 1:1,000) and cleaved-caspase 3 (cat. no. 9664;
1:1,000) were purchased from Cell Signaling Technology, Inc.,
(Danvers, MA, USA). Anti-poly (ADP-ribose) polymerase (PARP; cat.
no. 556494; 1:1,000) antibody and P62 (cat. no. 610497; 1:1,000)
were purchased from BD Biosciences (San Jose, CA, USA). Anti-LC3B
(cat. no. L7543; 1:1,000) antibody was purchased from Sigma-Aldrich
(Merck KGaG, Darmstadt, Germany). Antibodies against β-actin (cat.
no. ABM-0001; 1:1,000) were purchased from Zoonbio Biotecnology
Co., Ltd. (Nanjing, China) and both secondary antibodies including
goat anti-rabbit IgG-HRP secondary antibody (cat. no. ASS1006;
1:2,000) and goat anti-mouse IgG-HRP secondary antibody (cat. no.
ASS1007; 1:2,000) were purchased from Abgent, Inc., (San Diego, CA,
USA). The JNK inhibitor SP600125 (cat. no. s1460) and pan-caspase
inhibitor z-VAD (cat. no. S7023) were purchased from Selleck
Chemicals (Houston, TX, USA). Chloroquine diphosphate (CQ; cat. no.
50-63-5) was purchased from Sigma-Aldrich (Merck Millipore). The
autophagy inhibitor 3-methyladenine (3MA; cat. no. sc-205596) was
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) and
dissolved in dimethyl sulfoxide (DMSO; 10 mM) to pretreat the cells
prior to the MTT assay, flow cytometry and western blot analysis.
Celecoxib (Pfizer Inc., New York, NY, USA) was dissolved in
dimethyl sulfoxide (DMSO) and used within 1 month.
Cell viability assay
The effect of celecoxib on cell viability was
measured using 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl
tetrazoliumbromide (MTT) assay. Cells were seeded at 5,000–10,000
cells/well in 96-well plates and incubated overnight at 37°C.
Following this, different concentrations of celecoxib were added
and the plates were incubated for an additional 24 or 48 h at 37°C.
Subsequently, 20 µl of MTT solution (5 mg/ml) was added to each
well and the cells were incubated for 4 h at 37°C. After removal of
the culture medium, 100 µl dimethyl sulfoxide (DMSO) was added per
well to dissolve the formazan crystals and the optical density (OD)
was measured at 460 nm by a microplate reader. Changes in
percentage viability were calculated using the following formula:
Cell viability (%) = (OD of the experimental sample / OD of the
control group) × 100.
Measurement of apoptosis by flow
cytometry
Cell apoptosis was detected using an annexin
V-fluorescein isothiocyanate/propidium iodide (PI) kit (Beyotime
Institute of Biotechnology, Haimen, China). Cells were collected
and suspended in 200 µl medium buffer. Subsequently, ~10 µl annexin
V solution was added to the cell suspension solution and the
solution was incubated for 15 min in the dark at room temperature.
Following this, 300 µl medium buffer and 5 µl PI were added and the
cell suspension was immediately analyzed using a flow cytometric
machine. Annexin V-negative/PI-negative represented viable cells,
annexin V-positive/PI-negative represented early apoptotic cells,
annexin V-positive/PI-positive represented terminal apoptotic cells
and annexin V-negative/PI-positive cells represented necrotic
cells. Flow cytometry acquired ~104 cells and the data
were acquired using a FACSCanto 6-color flow cytometer (BD
Biosciences, San Jose, CA, USA) and analyzed using BD FACSDiva™
software version 6 (BD Biosciences).
Western blot analysis
Cells were washed twice with ice-cold
phosphate-buffered saline (PBS), lysed in lysis buffer (1% Triton
X-100, 50 mM Tris-Cl, pH 7.4, 150 mM Nacl, 10 mM EDTA, 100 mM NaF,
1 mM Ma3VO4, 1 mM PMSF and 2 µg/ml aprotinin) and quantified using
bicin-choninic acid (BCA) assay kit (Beyotime Institute of
Biotechnology). Equal amounts of denatured protein lysates (40
µg/lane) were separated by 12% SDS-PAGE and transferred to a
nitro-cellulose membrane (cat. no. CS011-0001; ExCell Biology,
Inc., Shanghai, China). Following this, the membranes were blocked
with 5% skim milk in Tris-buffered saline-Tween 20 buffer (TBST; 10
mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% Tween) at room temperature
for 2 h and the blots were incubated with the indicated primary
antibodies (anti-phospho-JNK, JNK1, LC3B, P62, cleaved-caspase 3,
PARP, β-actin; 1:1,000) overnight at 4°C. After three 10 min washes
with TBST buffer, the membranes were incubated with secondary
antibodies (goat anti-rabbit IgG-HRP secondary antibody, goat
anti-mouse IgG-HRP secondary antibody; both 1:2,000) for 30 min at
room temperature. Following the use of HRP-conjugated IgG secondary
antibodies, proteins were detected with an enhanced
chemiluminescence reagent (EMD Millipore, Billercia, MA, USA) and
visualized using an electrophoresis gel imaging analysis
system.
Transmission electron microscopy
(TEM)
For visualization of the cellular ultrastructure
using TEM, celecoxib-treated cells were treated with trypsin,
rinsed twice with warm PBS (37°C) and fixed for 1 h in 2.5%
glutaraldehyde in 0.1 M cacodylate buffer with 1% sucrose. After
washing with PBS, the cells were fixed in 1% osmium tetroxide and
embedded in Epon resin. Sections of 0.1-mm thickness were cut and
stained with uranyl acetate/lead citrate and visualized under a
Hitachi-7,500 TEM (Hitchi, Ltd., Tokyo, Japan).
Statistical analysis
Statistical analysis was conducted using SPSS v.16.0
(SPSS, Inc., Chicago, IL, USA). Data was confirmed in three
independent experiments and expressed as the mean + standard
deviation. Differences between groups were evaluated using
Student's t-tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
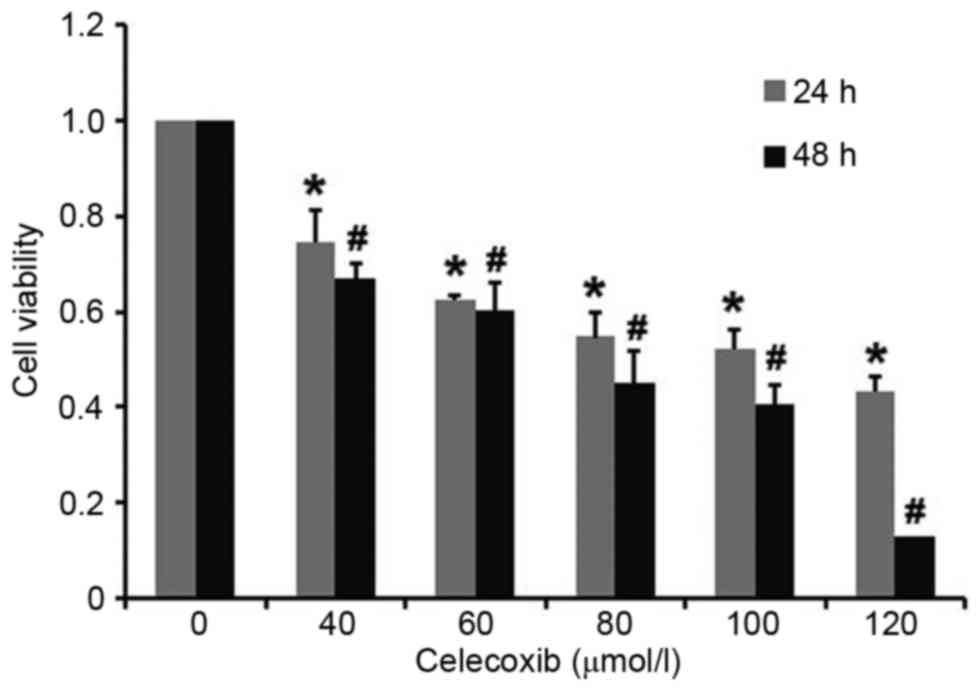
Celecoxib decreases cell viability in
a dose-dependent and time-dependent manner
PC3 cells were exposed to celecoxib for 24 and 48 h,
respectively, and cell viability was measured using an MTT assay.
Results demonstrated that cytotoxicity in PC3 cells was induced by
celecoxib in a dose-dependent and time-dependent manner, with all
doses of celecoxib resulting in a significant decrease in cell
viability compared with the untreated control group at 24 and 48 h
(P<0.05; Fig. 1). The 50%
inhibitory concentration of celecoxib for 24 h was 100 µmol/l,
which was chosen for subsequent experiments.
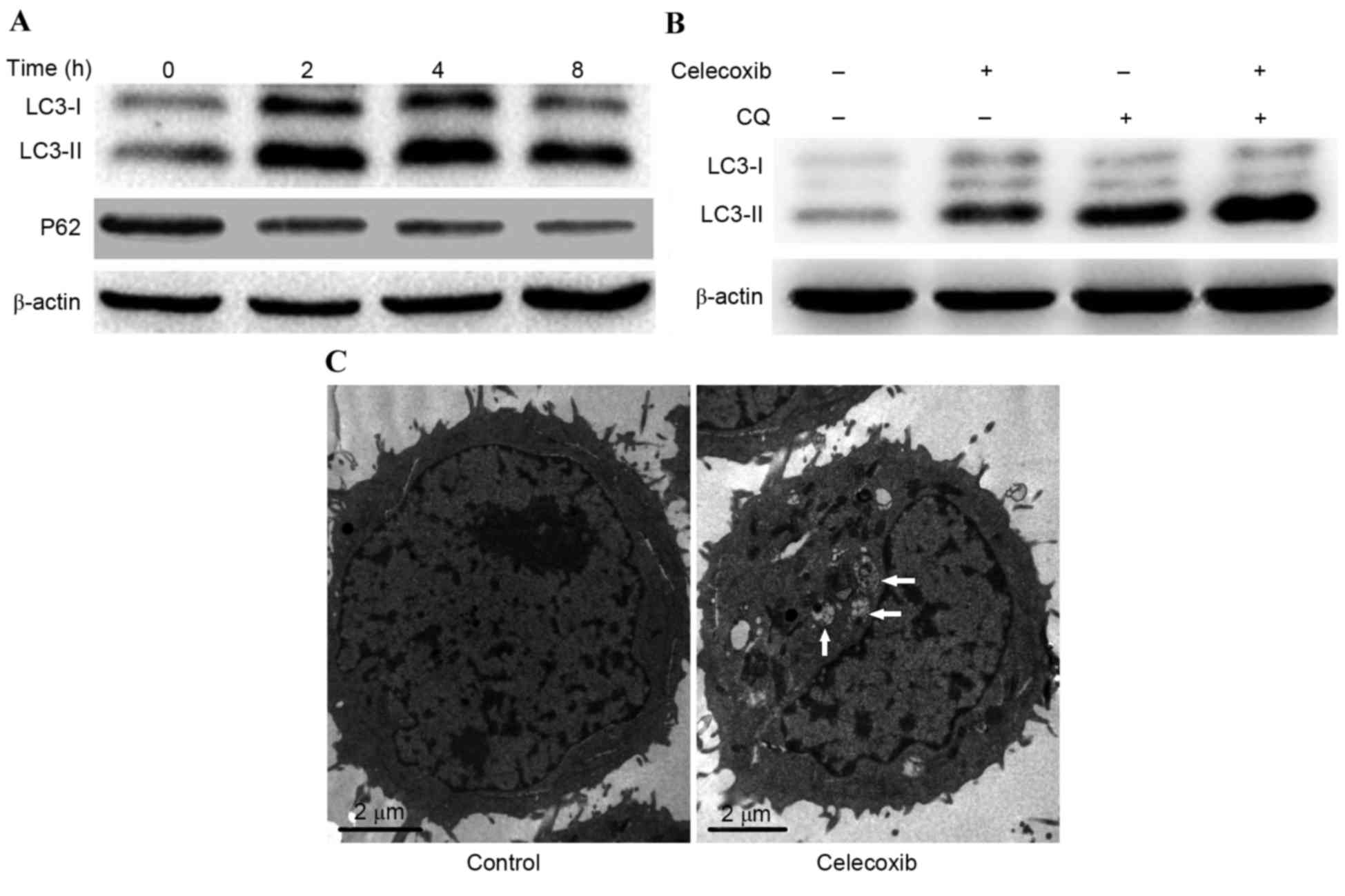
Celecoxib induces autophagy in PC3
cells
To explore whether celecoxib was able to induce
autophagy in PC3 cells, the conversion of LC3-I to LC3-II and the
degradation of P62 was detected by western blot analysis. Results
demonstrated that the conversion of LC3-I to LC3-II increased
following treatment with celecoxib; however, P62 degraded gradually
as PC3 cells were incubated with celecoxib (100 µmol/l) for 0, 2, 4
and 8 h (Fig. 2A). In order to
determine if the observed change in LC3 was due to autophagy, CQ, a
lysosome inhibitor for LC3-II turnover, was combined with celecoxib
to detect autophagy flux. Results demonstrated that combined
exposure to celecoxib and CQ induced a marked increase in LC3-II
expression levels compared with celecoxib or CQ alone (Fig. 2B). Furthermore, TEM was used to
detect autophagosomes (autophagic vacuoles), which is considered to
be the morphological hallmark of autophagy. Results demonstrated
that celecoxib induced marked autophagosome formation compared with
the control group (Fig. 2C). These
results demonstrated that autophagy was induced by celecoxib
treatment.
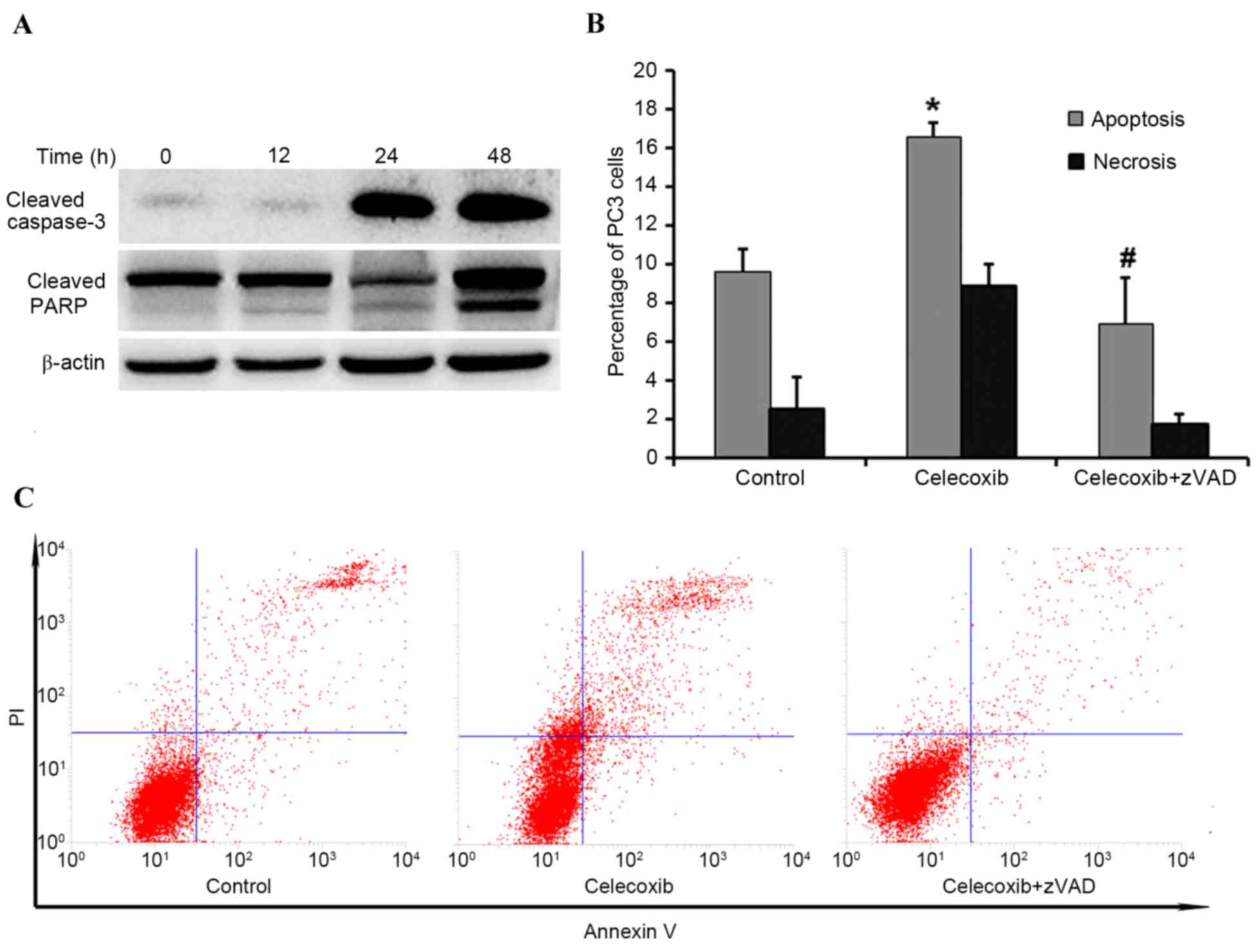
Celecoxib induces apoptosis in PC3
cells
In order to determine whether celecoxib-induced
cytotoxicity was due to activated apoptosis, western blotting was
performed to evaluate expression levels of PARP and caspase 3.
Compared with the control group (0 h), the expression levels of
PARP and caspase 3 markedly increased in a time-dependent manner
(Fig. 3A). To further elucidate
whether PC3 cell apoptosis was induced by celecoxib, flow cytometry
was applied to compare the following groups: Control group;
celecoxib-treated for 8 h; and celecoxib plus the pan-caspase
inhibitor zVAD. Flow cytometry indicated that celecoxib was able to
induce significant apoptosis (P<0.05) compared with the control
group, and zVAD was able to significantly inhibit (P<0.05)
celecoxib-induced apoptosis (Fig. 3B and
C, respectively).
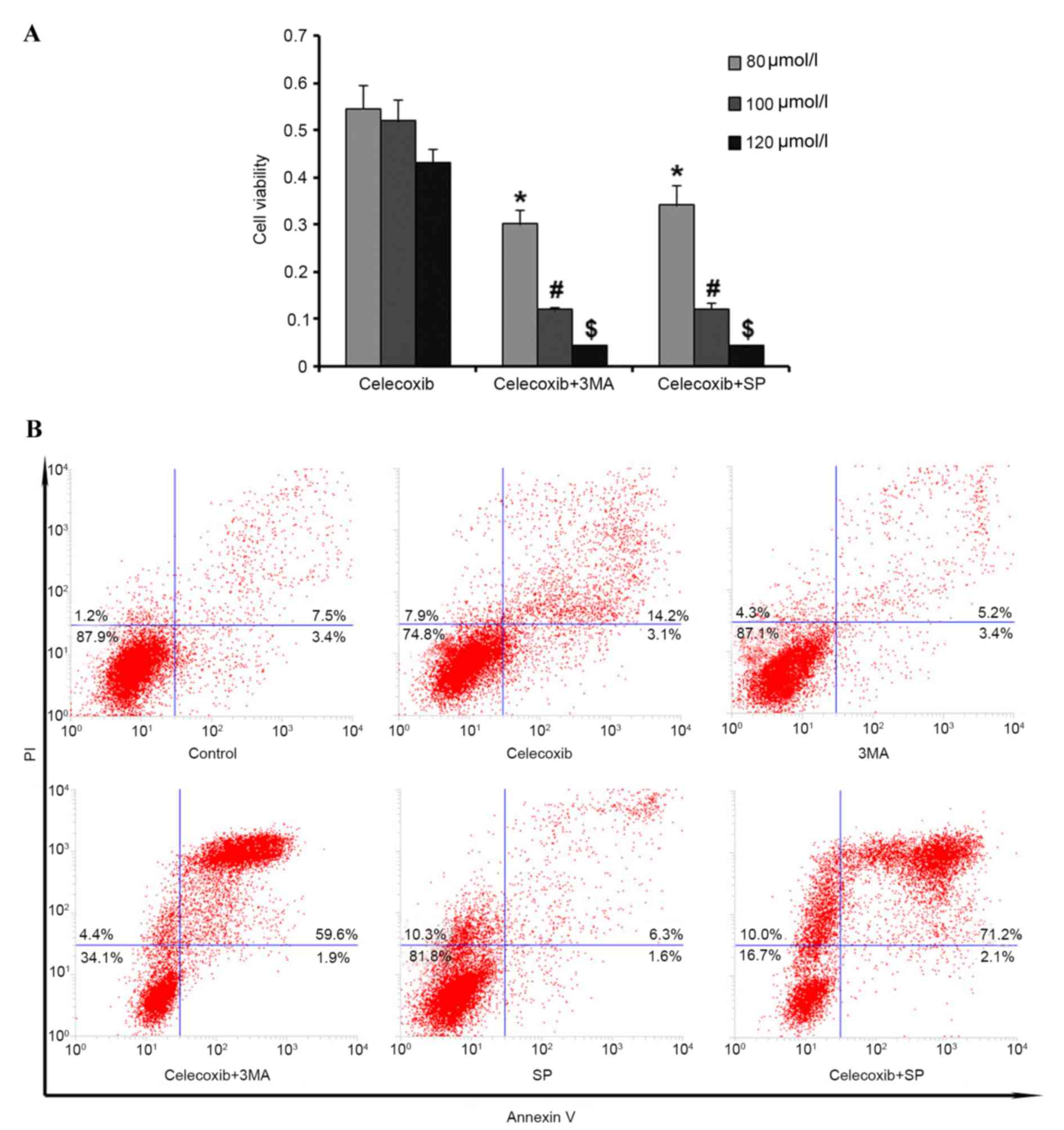
Autophagy has a cytoprotective role in
celecoxib-induced apoptosis
To investigate the role of autophagy in
celecoxib-induced apoptosis in PC3 cells, cells were pretreated
with 3-methyladenine (3MA) to inhibit autophagy before celecoxib
exposure (80, 100 and 120 µmol/l). MTT assay results demonstrated
that cell viability was significantly reduced (P<0.05) by
inhibition of autophagy compared with the celecoxib-only group
(Fig. 4A). Furthermore, flow
cytometry was used to detect cell apoptosis following pretreatment
with 3MA and the results were consistent with the MTT assay results
(Fig. 4B). Altogether, these results
demonstrated that autophagy may have a protective role in PC3 cells
under celecoxib-induced apoptosis.
Celecoxib-induced apoptosis is
mediated by JNK activation
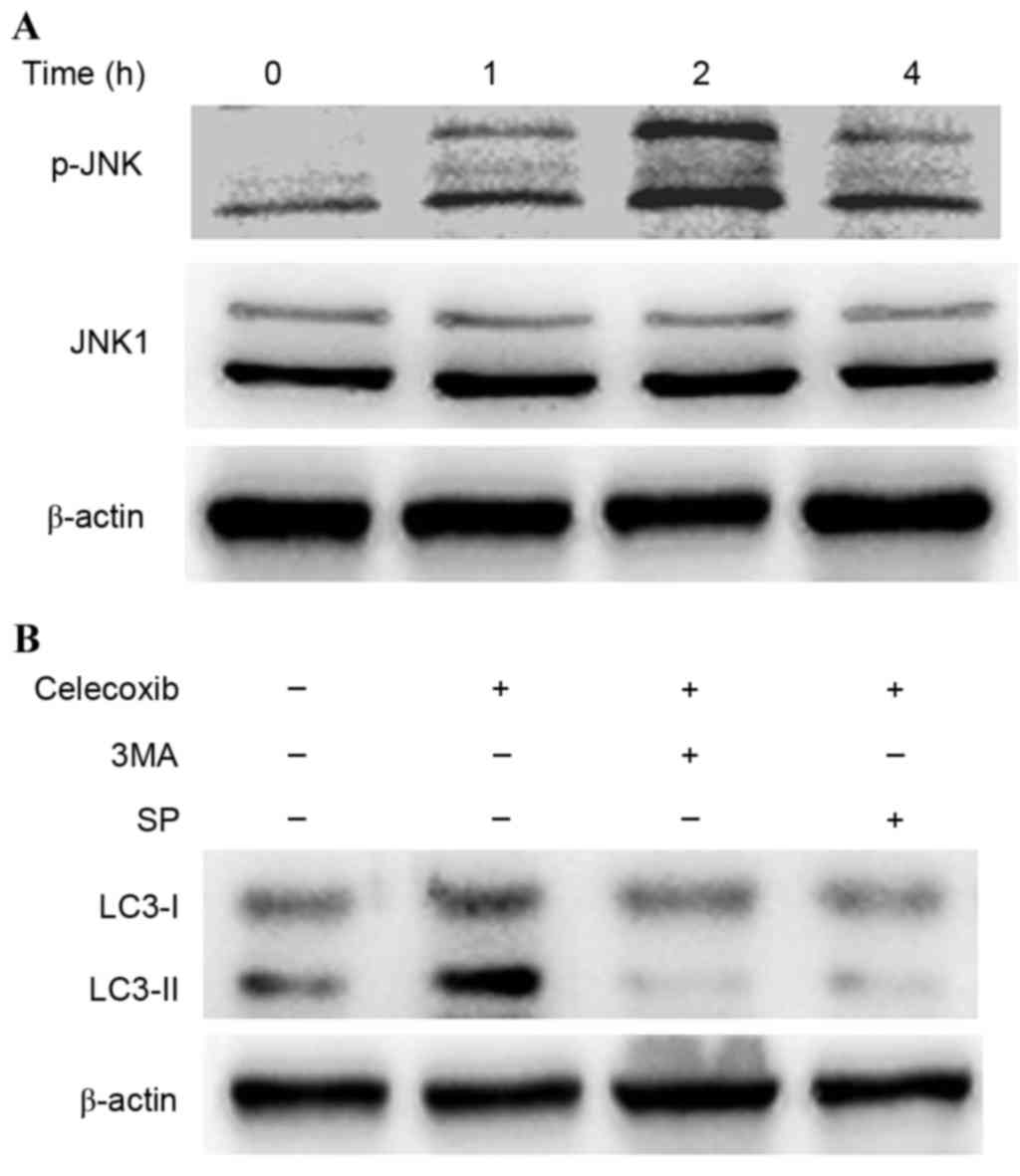
JNK activation has been demonstrated to be involved
in autophagy due to various stresses (8). To investigate whether JNK was activated
in celecoxib-induced autophagy, western blot analysis was conducted
to detect the phosphorylation of JNK in PC3 cells following
exposure to celecoxib (Fig. 5). JNK
was activated into pJNK following incubation with celecoxib (100
µmol/l) for 0, 1, 2 and 4 h (Fig.
5A). Furthermore, SP600125, a specific inhibitor of JNK, was
able to inhibit the conversion of LC3-I to LC3-II (Fig. 5B) and reduce cell viability by
increasing cell apoptosis (Fig. 4).
These results demonstrated that JNK was related to
celecoxib-induced apoptosis.
Discussion
In the present study it was demonstrated that
celecoxib was able to induce apoptosis and autophagy in PC3 cells.
COX-2, an enzyme involved in cancer angiogenesis, apoptosis and
invasiveness, has been associated with poor prognosis in various
types of cancer, including rectal and cervical cancer (9,10).
Celecoxib is a specific COX-2 inhibitor that is widely used to
treat acute pain, rheumatoid arthritis and familial adenomatous
polyposis. Previous studies have reported that celecoxib may exert
anticancer effects in various human neoplasms, such as lung cancer,
breast carcinomas, colorectal cancer and prostate cancer (11–13). A
meta-analysis of 11 randomized clinical trials by Chen et al
(14) reported that celecoxib was
beneficial in the treatment of various types of advanced cancer.
Considering the significant role of COX-2 in the regulation of
cancer angiogenesis, apoptosis and invasiveness, the molecular
mechanisms of COX-2 have been intensively studied in prostate
cancer (15,16). A study by Khor et al (17) investigated the relationship between
COX-2 overexpression and outcomes in patients with prostate cancer
following radiation treatment and the results demonstrated that
COX-2 expression levels were positively associated with biochemical
failure and distant metastasis. Furthermore, a study by Patel et
al (6) revealed that celecoxib
possesses different COX-2-independent anticancer properties, which
exert synergic effects with COX-2-dependent effects against
prostate cancer growth.
Autophagy, an evolutionarily conserved adaptive
cellular process, enables cells to engulf dysfunctional cellular
proteins and damaged organelles, which eventually fuse with
lysosomes for degradation when exposed to metabolic, toxic, hypoxic
and infectious stresses (18).
Accumulating evidence suggests that autophagy exerts potent
anti-cancer mechanisms to suppress tumor initiation (19,20).
However, at the advanced stage of cancer, autophagy enables cancer
cells to enhance nutrient utilization and improve growth in the
context of therapeutic intervention, and so the role of autophagy
in cancer is controversial (21,22).
Research on beclin 1 autophagy gene deletion in prostate cancer has
indicated that autophagy suppresses prostatic tumorigenesis
(23,24), whereas other studies have
demonstrated that autophagy is a survival mechanism to overcome
stresses in prostate cancer (3,25).
Androgen deprivation is considered the backbone of
therapy for hormone-sensitive or castration-resistant prostate
cancer (26). The underlying
mechanism of androgen deprivation and autophagy activation has been
demonstrated in vivo as well as in vitro models of
prostate cancer (27,28). Specifically, AMP-dependent protein
kinase activation leading to the suppression of mammalian target of
rapamycin is one of the most investigated mechanisms for autophagy
induction in androgen-dependent or androgen-independent models of
prostate cancer (29,30).
JNK, a mitogen-activated protein kinase, contributes
to apoptotic signal transduction when exposed to various forms of
stress (31). JNK target proteins
include the transcription factor c-Jun, apoptotic regulatory
proteins, such as B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X
protein (Bax), and forkhead transcription factor (32). A study by Kurinna et al
(33) demonstrated that ceramide was
able to induce translocation of active JNK from the nucleus to the
cytoplasm and mitochondrial fraction to promote apoptosis in lung
cancer-derived A549 cells. A study by Deng et al (32) indicated that activation of JNK was
able to upregulate acetylcholinesterase expression levels through a
c-Jun-dependent mechanism during apoptosis in colon cancer cells.
Furthermore, previous research has highlighted that the
JNK-mediated pathway is associated with autophagy activation. A
study by Wei et al (34)
demonstrated that, during starvation, autophagy is activated by
JNK-1 mediated Bcl-2 phosphorylation and disruption of the
Bcl-2/beclin 1 complex. Reactive oxygen species may also induce
autophagy to resist apoptosis by JNK activation in mesenchymal stem
cells, while some natural agents, such as matrine, mediate
crosstalk between autophagy and apoptosis via the
JNK-Bcl-2/Bcl-xL-Bax/Bcl-2 homologous antagonist killer pathway to
interplay with beclin 1 (6,13,35–37). In
the present study, it was demonstrated that JNK is also involved in
autophagy stimulation due to celecoxib treatment in
hormone-insensitive prostate cancer cells. Furthermore, suppression
of JNK by the specific inhibitor, SP600125, was able to inhibit
autophagy and enhance celecoxib-induced cytotoxicity in PC3
cells.
In conclusion, the present study demonstrated that
celecoxib induces apoptosis in PC3 cells; however, it also
activates autophagy, which exerts cytoprotective effects in
prostate cancer PC3 cells. JNK is activated in celecoxib-treated
cells and, when considering the dual roles of JNK in autophagy and
apoptosis, further investigation is required to illustrate the
precise underlying mechanisms of JNK. Meanwhile, the blockade of
autophagy via the JNK-mediated pathway may provide a promising
strategy for prostate cancer therapy.
Acknowledgments
The present study was financially supported by
grants from the Natural Science Foundation of China (grant nos.
81370706 and 81372758).
Glossary
Abbreviations
Abbreviations:
|
Bcl-2
|
B-cell lymphoma 2
|
|
CQ
|
chloroquine
|
|
COX-2
|
cyclooxygenase-2
|
|
FCM
|
flow cytometric
|
|
DMSO
|
dimethyl sulfoxide
|
|
JNK
|
c-Jun N-terminal kinase
|
|
TEM
|
transmission electron microscopy
|
|
LC3
|
microtubule associated protein 1 light
chain 3
|
|
3MA
|
3-methyladenine
|
|
PMSF
|
phenylmethylsulphonyl fluoride
|
|
PVDF
|
polyvinylidene fluoride
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
MTT
|
methyl thiazolyl tetrazolium
|
|
OD value
|
optical density value
|
|
PBS
|
phosphate-buffered saline
|
|
PARP
|
poly (ADP-ribose) polymerase
|
References
|
1
|
Center MM, Jemal A, Lortet-Tieulent J,
Ward E, Ferlay J, Brawley O and Bray F: International variation in
prostate cancer incidence and mortality rates. Eur Urol.
61:1079–1092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Amaravadi RK and Thompson CB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taketo MM: Cyclooxygenase-2 inhibitors in
tumorigenesis (part I). J Natl Cancer Inst. 90:1529–1536. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patel MI, Subbaramaiah K, Du B, Chang M,
Yang P, Newman RA, Cordon-Cardo C, Thaler HT and Dannenberg AJ:
Celecoxib inhibits prostate cancer growth: Evidence of a
cyclooxygenase-2-independent mechanism. Clin Cancer Res.
11:1999–2007. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang S and Sinicrope FA:
Celecoxib-induced apoptosis is enhanced by ABT-737 and by
inhibition of autophagy in human colorectal cancer cells.
Autophagy. 6:256–269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li DD, Wang LL, Deng R, Tang J, Shen Y,
Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al: The pivotal role of
c-Jun NH2-terminal kinase-mediated Beclin 1 expression during
anticancer agents-induced autophagy in cancer cells. Oncogene.
28:886–898. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith FM, Reynolds JV, Kay EW, Crotty P,
Murphy JO, Hollywood D, Gaffney EF, Stephens RB and Kennedy MJ:
COX-2 overexpression in pretreatment biopsies predicts response of
rectal cancers to neoadjuvant radiochemotherapy. Int J Radiat Oncol
Biol Phys. 64:466–472. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ishikawa H, Ohno T, Kato S, Wakatsuki M,
Iwakawa M, Ohta T, Imai T, Mitsuhashi N, Noda SE, Nakano T and
Tsujii H: Cyclooxygenase-2 impairs treatment effects of
radiotherapy for cervical cancer by inhibition of radiation-induced
apoptosis. Int J Radiat Oncol Biol Phys. 66:1347–1355. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mao JT, Roth MD, Fishbein MC, Aberle DR,
Zhang ZF, Rao JY, Tashkin DP, Goodglick L, Holmes EC, Cameron RB,
et al: Lung cancer chemoprevention with celecoxib in former
smokers. Cancer Prev Res (Phila). 4:984–993. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Perroud HA, Rico MJ, Alasino CM, Queralt
F, Mainetti LE, Pezzotto SM, Rozados VR and Scharovsky OG: Safety
and therapeutic effect of metronomic chemotherapy with
cyclophosphamide and celecoxib in advanced breast cancer patients.
Future Oncol. 9:451–462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Basler JW and Piazza GA: Nonsteroidal
anti-inflammatory drugs and cyclooxygenase-2 selective inhibitors
for prostate cancer chemoprevention. J Urol. 171:S59–S63. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen J, Shen P, Zhang XC, Zhao MD, Zhang
XG and Yang L: Efficacy and safety profile of celecoxib for
treating advanced cancers: A meta-analysis of 11 randomized
clinical trials. Clin Ther. 36:1253–1263. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hussain T, Gupta S and Mukhtar H:
Cyclooxygenase-2 and prostate carcinogenesis. Cancer Lett.
191:125–135. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gallego G Aparicio, Díaz Prado S, Jiménez
Fonseca P, García Campelo R, Cassinello Espinosa J and Antón
Aparicio LM: Cyclooxygenase-2 (COX-2): A molecular target in
prostate cancer. Clin Transl Oncol. 9:694–702. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Khor LY, Bae K, Pollack A, Hammond ME,
Grignon DJ, Venkatesan VM, Rosenthal SA, Ritter MA, Sandler HM,
Hanks GE, et al: COX-2 expression predicts prostate-cancer outcome:
Analysis of data from the RTOG 92-02 trial. Lancet Oncol.
8:912–920. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ziparo E, Petrungaro S, Marini ES, Starace
D, Conti S, Facchiano A, Filippini A and Giampietri C: Autophagy in
prostate cancer and androgen suppression therapy. Int J Mol Sci.
14:12090–12106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jin S: P53, Autophagy and tumor
suppression. Autophagy. 1:171–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ozpolat B and Benbrook DM: Targeting
autophagy in cancer management-strategies and developments. Cancer
Manag Res. 7:291–299. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen L, Jiang K, Jiang H and Wei P:
MiR-155 mediates drug resistance in osteosarcoma cells via inducing
autophagy. Exp Ther Med. 8:527–532. 2014.PubMed/NCBI
|
|
23
|
Aita VM, Liang XH, Murty VV, Pincus DL, Yu
W, Cayanis E, Kalachikov S, Gilliam TC and Levine B: Cloning and
genomic organization of beclin 1, a candidate tumor suppressor gene
on chromosome 17q21. Genomics. 59:59–65. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh
H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al:
Promotion of tumorigenesis by heterozygous disruption of the beclin
1 autophagy gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kung HJ: Targeting tyrosine kinases and
autophagy in prostate cancer. Horm Cancer. 2:38–46. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merseburger AS, Hammerer P, Rozet F,
Roumeguère T, Caffo O, da Silva FC and Alcaraz A: Androgen
deprivation therapy in castrate-resistant prostate cancer: How
important is GnRH agonist backbone therapy? World J Urol.
33:1079–1085. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki
D, Lara PN Jr..White RW DeVere, Gao AC and Evans CP: Targeting
autophagy overcomes enzalutamide resistance in castration-resistant
prostate cancer cells and improves therapeutic response in a
xenograft model. Oncogene. 33:4521–4530. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bennett HL, Stockley J, Fleming JT, Mandal
R, O'Prey J, Ryan KM, Robson CN and Leung HY: Does
androgen-ablation therapy (AAT) associated autophagy have a
pro-survival effect in LNCaP human prostate cancer cells? BJU Int.
111:672–682. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chhipa RR, Wu Y and Ip C: AMPK-mediated
autophagy is a survival mechanism in androgen-dependent prostate
cancer cells subjected to androgen deprivation and hypoxia. Cell
Signal. 23:1466–1472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chhipa RR, Wu Y, Mohler JL and Ip C:
Survival advantage of AMPK activation to androgen-independent
prostate cancer cells during energy stress. Cell Signal.
22:1554–1561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Verheij M, Bose R, Lin XH, Yao B, Jarvis
WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, et al:
Requirement for ceramide-initiated SAPK/JNK signalling in
stress-induced apoptosis. Nature. 380:75–79. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deng R, Li W, Guan Z, Zhou JM, Wang Y, Mei
YP, Li MT, Feng GK, Huang W, Liu ZC, et al: Acetylcholinesterase
expression mediated by c-Jun-NH2-terminal kinase pathway during
anticancer drug-induced apoptosis. Oncogene. 25:7070–7077. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kurinna SM, Tsao CC, Nica AF, Jiffar T and
Ruvolo PP: Ceramide promotes apoptosis in lung cancer-derived A549
cells by a mechanism involving c-Jun NH2-terminal kinase. Cancer
Res. 64:7852–7856. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu GY, Jiang XX, Zhu X, He WY, Kuang YL,
Ren K, Lin Y and Gou X: ROS activates JNK-mediated autophagy to
counteract apoptosis in mouse mesenchymal stem cells in vitro. Acta
Pharmacol Sin. 36:1473–1479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang J and Yao S: JNK-Bcl-2/Bcl-xL-Bax/Bak
pathway mediates the crosstalk between matrine-induced autophagy
and apoptosis via interplay with beclin 1. Int J Mol Sci.
16:25744–25758. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
García Rodríguez LA and González-Pérez A:
Inverse association between nonsteroidal anti-inflammatory drugs
and prostate cancer. Cancer Epidemiol Biomarkers Prev. 13:649–653.
2004.PubMed/NCBI
|